634901 SMART cDNA 文库构建实验流程(clontech)
- 格式:pdf
- 大小:782.30 KB
- 文档页数:9

cDNA文库构建的具体步骤及详细说明cDNA文库构建的具体步骤及详细说明cDNA 文库是指某生物某发育时期所转录的全部mRNA 经反转录形成的cDNA 片段与某种载体连接而形成的克隆的集合。
经典cDNA 文库构建的基本原理是用Oligo(dT) 作逆转录引物,或者用随机引物,给所合成的cDNA 加上适当的连接接头,连接到适当的载体中获得文库。
其基本步骤包括:(1)mRNA的提纯获取高质量的mRNA是构建高质量的cDNA 文库的关键步骤之一。
(2)cDNA第一条链的合成。
(3)cDNA第二条链的合成。
(4)双链cDNA的修饰。
(5)双链cDNA的分子克隆。
(6)cDNA文库的扩增。
(7)cDNA文库鉴定评价。
一、Superscipt II—RT合成第一链1. 在一RNase-free的0.2 ml PCR管中加入x ul mRNA(大约500 ng) 、1 ul Xho I Primer(1.4 ug/ul)(5’GAGAGAGAGAGAGAGAGAGAAC TAGTCTCGAGTTTTTTTT TTTTTTTTTT…3’)、11-x ul RNase-free water(大于500 ng mRNA 分n管(500 ng/tube)合成第一链,第一链合成完毕后将n管合成一管进行第二链合成。
)。
2. 混匀后,70℃反应10分钟。
3. 反应完成后,立刻将反应体系置于冰上5 min。
4. 稍微离心一下,顺序加入以下试剂:(1)4 ul 5×first strand buffer(2)2 ul 0.1 M DTT(3)1 ul 10 mM dNTP(自己配制)5. 混匀,稍微离心反应物之后,42℃放置2分钟。
6. 反应完成,趁热加入1 ul Superscipt II—RT,混匀。
7. 42℃反应50分钟,然后70℃,15分钟灭活反转录酶。
二、cDNA第二链的合成1. 第一链反应完成后,取2ul一链产物-20℃冰箱中保存,待电泳检测。
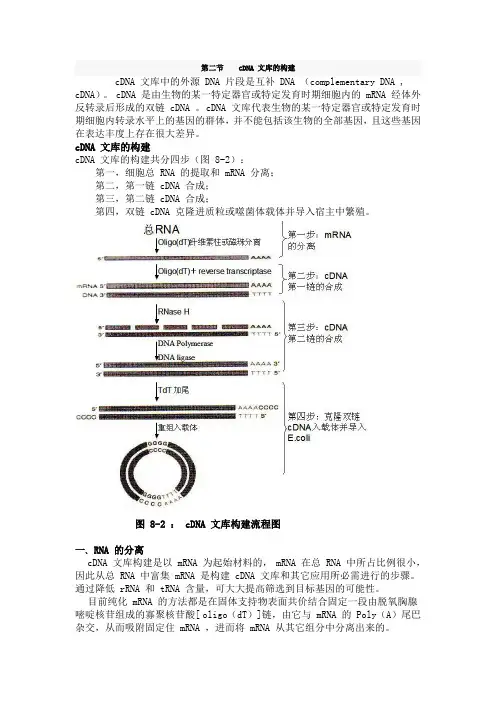
第二节cDNA 文库的构建cDNA 文库中的外源 DNA 片段是互补 DNA (complementary DNA , cDNA)。
cDNA 是由生物的某一特定器官或特定发育时期细胞内的 mRNA 经体外反转录后形成的双链 cDNA 。
cDNA 文库代表生物的某一特定器官或特定发育时期细胞内转录水平上的基因的群体,并不能包括该生物的全部基因,且这些基因在表达丰度上存在很大差异。
cDNA 文库的构建cDNA 文库的构建共分四步(图 8-2):第一,细胞总 RNA 的提取和 mRNA 分离;第二,第一链 cDNA 合成;第三,第二链 cDNA 合成;第四,双链 cDNA 克隆进质粒或噬菌体载体并导入宿主中繁殖。
图 8-2 : cDNA 文库构建流程图一、RNA 的分离cDNA 文库构建是以 mRNA 为起始材料的, mRNA 在总RNA 中所占比例很小,因此从总RNA 中富集mRNA 是构建cDNA 文库和其它应用所必需进行的步骤。
通过降低rRNA和tRNA 含量,可大大提高筛选到目标基因的可能性。
目前纯化mRNA 的方法都是在固体支持物表面共价结合固定一段由脱氧胸腺嘧啶核苷组成的寡聚核苷酸[ oligo(dT)]链,由它与mRNA 的Poly(A)尾巴杂交,从而吸附固定住mRNA ,进而将mRNA从其它组分中分离出来的。
1.mRNA 的完整性指导合成高分子量蛋白质的能力,指导合成目的多肽的能力,mRNA 的大小,总mRNA 制剂指导合成cDNA 第一链长分子的能力。
2.mRNA 的丰度高丰度mRNA :珠蛋白,免疫球蛋白,卵清蛋白,在特定细胞中占50-90% 。
低丰度mRNA :含量 0.5% 被称为低丰度或稀有mRNA 。
3.mRNA 的富集3.1按大小对 mRNA 进行分级分离3.2 cDNA 的分离级分离近年来多采用此方法,特别是大mRNA ,可避免降解mRNA , agarose 分离大小易辨。

SMART技术红曲霉全长cDNA文库构建 【编者按】医药论文是科技论文的一种是用来进行医药科学研究和描述研究成果的论说性文章。
论文网为您提供医药论文范文参考,以及论文写作指导和格式排版要求,解决您在论文写作中的难题。
SMART技术红曲霉全长cDNA文库构建 作者:朱碧云,高蓝,黄欣,李浩明 【摘要】目的构建红曲霉cDNA文库,为红曲霉功能基因的研究以及筛选、克隆红曲霉次生代谢产物合成途径相关基因奠定基础。
方法采用SMART(switching mechanism at 5 end of RNA transcript)技术构建红曲霉全长cDNA文库,经涂平板和酶切反应测定文库的克隆数、重组率,PCR测定插入片断的大小。
结果原始文库的库容为2.03 105,重组率达94%。
插入片段大小多在0.7~2.0 kb,平均大小在1 kb左右。
结论所构建文库的代表性和重组片段的序列完整性达到了用于目的基因的分离筛选和克隆表达的建库要求。
【关键词】红曲霉菌cDNA文库SMART Abstract:Objective Construction of Monascus cDNA Library provide foundations for the research of Monascus functional genes and screening,cloning genes related to the synthesis pathway of secondary metabolites in Monascus.Methods A cDNA library was constructed based on SMART system. The number of the clones and the recombination rate of the library were determined by plate coating and restrictive digestion,and the size of inserted fragment was determined by PCR. Results The primary library capacity was 2.03 105,average inserted fragment size was about 1 000 bp and the percentage of recombination were 94%. Conclusion The library met the requirement for cloning target genes and expressing target proteins. Key words: Monascus; cDNA Library; SMART 红曲霉菌(Monascus)是我国重要的微生物资源,其应用已有上千年的历史。

CDNA文库1. CDNA文库中重组DNA片断得原始供体来源与细胞中表达出得mRNA,将某一特定类型细胞表达得mrna经反转录酶催化形成与之互补得CDNA,重组克隆后得到得CDNA文库有各自不同得适合范围。
CDNA文库在研究具体某类特定细胞中基因组得表达状态以及表达基因得功能鉴定方面具有特殊得优势,从而使它在个体发育,细胞分化,细胞周期调控2. CDNA文库得质量(1)文库的代表性CDNA文库的代表性是指文库中包含得重组CDNA分子是否能完整地反映出来原细胞中表达地全部信息(即mrna种类),它是体现文库质量地最重要标本。
文库地库容量,它是指构建建出地原始CDNA文库中包含地独立地重组子克隆数。
具备完全好代表性地CDNA文库需要满足地库容量取决与来源细胞中表达出地基因序列地总复杂程度。
具体来就是来源细胞中表达出地mrna种类和每种MRNA序列地拷贝数N=ln(1-p)/ln(1-n/t),P为文库中包含细胞中任何一种mrna序列信息地概率,通常设为99%,N为文库中P概率出现细胞中任何一种mrna序列理论上应具有地最少重组克隆数,n为细胞中最稀少地mrna序列地拷贝数,t为细胞中表达出地所有mrna地总拷贝数,以人类细胞为列,人类基因组携带地遗传基因总数约为100000种,具体到某一特定类型地细胞中,表达出地基因种类仅为基因组全部基因地15%。
因此对于巨大部分地人类细胞,每个细胞内具体表达地mrna种类约为15000种,全体mrna序列地总拷贝约为500000个,而细胞中稀少地mrna种类地拷贝数平均为8个。
因此,用人类细胞来构建CDNA文库时候,要以99%概率保证文库中包含有细胞表达地任何一种mrna地序列信息,构建出地原始CDNA文库理论上应具有地最少独立克隆数为N=ln(1_99%)/ln(1-8/500000)=2.9*10(5)一个具有完好代表性地CDNA文库至少具有10(6)以上的库容量3.MRNA是由5‘端非编码区,中间地编码序列和3’端非翻译区。

cDNA文库组标准流程一. Total RNA的提取 (2)二. mRNA的分离 (7)三.cDNA双链合成 (10)四.载体制备 (15)五. cDNA双链和载体的连接 (18)六.电转化流程 (19)七.快速鉴定、菌落PCR (22)八.pBlueScript cDNA库扩增 (25)一.Total RNA的提取1.试剂配制准备工作:1、研钵、 5ml/10ml/ 25ml移液管、 100ml/250ml量筒、 250ml/100ml容量瓶、药匙、试剂瓶等玻璃制品均用锡纸包裹口部, 置于烤箱内,180℃, 烤6小时。
2、 50ml/1.5ml离心管、枪头等塑料制品用0.1‰DEPC水浸泡过夜后, 121℃20mins 高压灭菌。
3、电泳槽及电泳托、梳子用3%双氧水处理。
4、常见试剂及其配方:▲DEPC水: 在1000ml去离子水中加入100ul DEPC, 静置过夜后高压灭菌。
▲0.78M柠檬酸纳: PH=4~5三水合柠檬酸纳 22.94g加DEPC水定容至100ml, 室温放置备用。
▲10%肌氨酸钠:肌氨酸钠 10g加DEPC水定容至100ml, 室温放置备用。
▲变性裂解液:0.78M柠檬酸钠 8.25ml10%肌氨酸钠 12.375ml异硫氰酸胍 118.05g加DEPC水定容至 250ml, 室温放置备用临用前加β-巯基乙醇使其终浓度为1%( v/v)▲ 2M 醋酸钠 PH=4.5O 13.6gNaAc·3H2加DEPC水定容至50ml,高压灭菌, 室温放置备用▲3M醋酸钠 PH=5O 20.4gNaAc·3H2加DEPC水定容至50ml,高压灭菌, 室温放置备用▲ 4M LiCL:LiCL 24.164g加DEPC水定容至100ml,高压灭菌, 室温放置备用▲0.5M EDTA PH=8.0EDTA 18.61g用NaOH调PH值至8.0, 定容到100ml, 高压灭菌, 室温放置备用▲10X MOPS (3-( N-吗啉代) 丙磺酸):MOPS 41.86gNaAC·3HO 4.10g20.5MEDTA( PH 8.0) 20ml用NaOH调PH值 6.5 , DEPC水定容到1L, 室温避光放置备用。

【共享】全长cDNA文库的构建—SMART技术全长cDNA文库的构建—SMART技术真核细胞的mRNA在加工过程中有一个比喻为“穿鞋戴帽”的过程,因此mRNA 的末端都带有一段Poly A,这是利用逆转录酶制备cDNA文库的基础。
但是由于cDNA的5'端的序列各不相同,如何获得全长的cDNA,如何扩增由微量的mRNA逆转录得到的cDNA文库、如何利用已知片断序列得到全长的cDNA(即RACE),曾经是一个令人困扰的问题。
常见的做法是在合成cDNA的双链后在两端连上接头,利用已知的接头序列再进行扩增,或者是利用末端转移酶在双链cDNA的3'末端加上一连串的G或者C(或者A/T),再通过补齐粘末端,利用已知的两头序列进行扩增。
但是这些方法不同程度的存在一些问题,比如接头的连接效率非常有限,会导致部分信息的丢失,再加上这些方法需要多次用不同的酶处理有限的样品,需要经过反复的纯化,会损失很多有用的信息,特别是少量样品中的低丰度信息,很大程度上会影响结果的准确性。
另外由于mRNA容易部分降解,很难确定得到的cDNA是否就是全长的cDNA,还是cDNA的片断。
SMART技术的出现是一个新的里程碑。
这个称作Switching Mechanism At 5' end of the RNA Transcript(SMART),原理实际上非常简单:在合成cDNA的反应中事先加入的3'末端带Oligo(dG)的SMART引物,由于逆转录酶以mRNA为模板合成cDNA,在到达mRNA的5'末端时碰到真核mRNA特有的“帽子结构”,即甲基化的G时会连续在合成的cDNA末端加上几个(dC),SMART引物的Oligo(dG)与合成cDNA末端突出的几个C配对后形成cDNA的延伸模板,逆转录酶会自动转换模板,以SMART引物作为延伸模板继续延伸cDNA单链直到引物的末端,这样得到的所有cDNA 单链的一端有含Oligo(dT)的起始引物序列,另一端有已知的SMART引物序列,合成第二链后可以利用通用引物进行扩增。

cDNA文库组标准流程一. Total RNA的提取 (2)二. mRNA的分离 (5)三.cDNA双链合成 (8)四.载体制备 (11)五. cDNA双链和载体的连接 (13)六.电转化流程 (14)七.快速鉴定、菌落PCR (16)八.pBlueScript cDNA库扩增 (18)一.Total RNA的提取1.试剂配制准备工作:1、研钵、5ml/10ml/ 25ml移液管、100ml/250ml量筒、250ml/100ml容量瓶、药匙、试剂瓶等玻璃制品均用锡纸包裹口部,置于烤箱内,180℃,烤6小时。
2、50ml/1.5ml离心管、枪头等塑料制品用0.1‰DEPC水浸泡过夜后,121℃20mins 高压灭菌。
3、电泳槽及电泳托、梳子用3%双氧水处理。
4、常用试剂及其配方:▲DEPC水:在1000ml去离子水中加入100ul DEPC, 静置过夜后高压灭菌。
▲0.78M柠檬酸纳:PH=4~5三水合柠檬酸纳22.94g加DEPC水定容至100ml,室温放置备用。
▲10%肌氨酸钠:肌氨酸钠10g加DEPC水定容至100ml,室温放置备用。
▲变性裂解液:0.78M柠檬酸钠8.25ml10%肌氨酸钠12.375ml异硫氰酸胍118.05g加DEPC水定容至250ml,室温放置备用临用前加β-巯基乙醇使其终浓度为1%(v/v)▲2M 醋酸钠PH=4.5NaAc·3H2O 13.6g加DEPC水定容至50ml,高压灭菌,室温放置备用▲3M醋酸钠PH=5NaAc·3H2O 20.4g加DEPC水定容至50ml,高压灭菌,室温放置备用▲4M LiCL:LiCL 24.164g加DEPC水定容至100ml,高压灭菌,室温放置备用▲0.5M EDTA PH=8.0EDTA 18.61g用NaOH调PH值至8.0,定容到100ml,高压灭菌,室温放置备用▲10X MOPS (3-(N-吗啉代)丙磺酸):MOPS 41.86gNaAC·3H2O 4.10g0.5MEDTA(PH 8.0)20ml用NaOH调PH值 6.5 , DEPC水定容到1L,室温避光放置备用。

c D N A文库的构建方法与原理蛋白质是细胞的功能分子:它们构成结构和调控分子,动力和泵蛋白,酶和受体。
然而,如果仅用传统的生化方法确定某一特异蛋白的全序列,或制备足够量的蛋白进行操作和鉴定都是使人厌烦且昂贵的步骤。
基因克隆和遗传工程对生化领域有很大贡献。
如果只限于将基因组DNA作为材料来源,由于其中仅2%被认为可能编码蛋白质,那么确定蛋白质序列仍然是令人生畏的工作。
其他部分包括结构和调节因子、内含子、非编译外显子和重复及功能未知的非编码序列。
如果分析仅局限在编码序列,那么确定基因产物序列所需的努力就会大大的降低。
因此分子生物学主要原则之一是mRNA作为蛋白质合成的模板,所以mRNA是确定蛋白质序列的理想底物。
不幸的是,现有通用的克隆载体没有一个能容纳mRNA分子作为插入片段。
因此,产生表达序列文库的一个基本步骤是将mRNA分子转变成双链DNA。
来自mRNA分子的DNA拷贝称cDNA,由来自细胞或组织mRNA种类的DNA拷贝组成的文库称为cDNA文库。
1.基本原理cDNA(Complementary DNA)是以mRNA为模板,在反转录酶作用下合成的互补DNA,它的顺序可代表mRNA序列。
cDNA文库的构建是指将cDNA与克隆载体DNA体外重组,然后去转化克隆载体DNA 的宿主细胞,从而得到一群含重组DNA的细菌或噬菌体克隆的过程。
这些序列来自并代表一定组织或细胞类型特定发育或分化阶段的整个mRNA群体。
其过程可概括为:(1)通过反转录酶将各种mRNA转变在cDNA;(2)cDNA与合适的载体重组并导入到宿主中。
cDNA基因文库具有许多优点和特殊用途:首先,cDNA克隆以mRNA为起始材料,这对于有些RNA病毒来说非常适用,因为它们的增殖并不经过DNA中间体,研究这样的生物有机体,cDNA克隆是唯一可行的方法。
第二,cDNA基因文库的筛选简单易行,恰当选择mRNA的来源,使所构建的cDNA基因文库中,某一特定序列的克隆达到很高的比例,简化了筛选特定基因序列克隆的工作量。

SMARTer™ PCR cDNA Synthesis Kit User Manual Cat. Nos. 634925 & 634926United States/Canada 800.662.2566Asia Pacific +1.650.919.7300Europe +33.(0)1.3904.6880Japan +81.(0)77.543.6116SMART er™ PCR cDNA Synthesis Kit User ManualT able of ContentsI. List of Components (3)II. Additional Materials Required (4)III. Introduction & Protocol Overview (5)IV. RNA Preparation & Handling (7)A. General Precautions (7)B. RNA Isolation (7)C. RNA Purity (8)D. Assessing the Quality of the RNA Template (8)V. SMART er cDNA Synthesis (9)A. General Considerations (10)B. PRoToCol: First-Strand cDNA Synthesis (10)C. PRoToCol: cDNA Amplification by lD PCR (12)VI. Analysis of cDNA Amplification Results (16)VII. T roubleshooting Guide (17)VIII. References (18)Appendix A: Protocols for PCR-Select™ (19)A. Additional Materials Required (19)B. PRoToCol: cDNA Amplification by lD PCR (19)C. PRoToCol: Column Chromatography (22)D. PRoToCol: RsaI Digestion (23)E. PRoToCol: Purification of Digested cDNA (23)F. Controls for PCR-Select cDNA Subtraction (25)G. Analysis of Results of SMARTer PCR cDNA Synthesis for PCR-Select cDNA Subtraction (25)H. T roubleshooting (27)Appendix B: Virtual Northern Blots (28)Appendix C: Protocol for Non-Directional Cloning of SMART er cDNA (29)A. Additional Materials Required (29)B. PRoToCol: ds cDNA Polishing (29)List of FiguresFigure 1. Flowchart of SMARTer cDNA synthesis (5)Figure 2. Guide to using the SMARTer cDNA synthesis protocol for PCR-Select cDNA Subtraction,Virtual Northerns, Non-Directional Cloning & library Construction, and other applications. (9)Figure 3. optimizing PCR parameters for SMARTer cDNA synthesis. (15)Figure 4. Analysis for optimizing PCR parameters (16)Figure 5. optimizing PCR parameters for SMARTer cDNA synthesis for use withClontech PCR-Select (21)Figure 6. Virtual Northern blot analysis of cDNA fragments expressed in cells producing γ-globin. (28)List of T ablesTable I: Guidelines for Setting Up PCR Reactions (12)Table II: Cycling Guidelines Based on Starting Material (13)Table III: T roubleshooting Guide for First-Strand cDNA Synthesis & SMARTer PCR Amplification (17)Table IV: T roubleshooting Guide for Preparing SMARTer cDNA for Subtraction (27)SMART er™ PCR cDNA Synthesis Kit User Manual I. List of ComponentsSMART er PCR cDNA Synthesis KitCat. No.Cat. No. 634925634926 10 rxns20 rxns Box 110 µl20 µl • SMART er II A Oligonucleotide (12 µM)5'–AAGCAGTGGTATCAACGCAGAGTACXXXXX–3' Rsa I(X = undisclosed base in the proprietary SMARTer oligo sequence) 5 µl5 µl • Control Mouse Liver T otal RNA (1 µg/µl)Box 210 µl20 µl • 3’ SMART CDS Primer II A (12 µM)5’–AAGCAGTGGTATCAACGCAGAGTACT (30)N -1N–3’Rsa I (N = A, C, G, or T ; N -1 = A, G, or C)200 µl 400 µl • 5’ PCR Primer II A (12 µM)40 µl 80 µl • 5X First-Strand Buffer (RNase-Free)250 mM T ris-HCl (pH 8.3)375 mM KCl30 mM MgCl 2100 µl 200 µl • dNTP Mix (dATP , dCTP , dGTP , and dTTP , each at 10 mM)50 µl 50 µl • Dithiothreitol (DTT ; 100 mM)10 µl 10 µl • RNase Inhibitor (40 U/µl)12 µl 25 µl • SMARTScribe™ Reverse T ranscriptase (100 U/µl)1 ml 1 ml • Deionized H 2O Box 310 20 • CHROMA SPIN™+TE-1000 ColumnsStorage ConditionsStore Control Mouse Liver T otal RNA and SMARTer II A Oligonucleotide at –70°C.• Store the CHROMA SPIN +TE-1000 Columns at room temperature.• Store all other reagents at –20°C.• Licensing InformationFor important information about the use of SMART technology, please see the Notice to Purchaser at theend of this user manual.要稀释本页已使用福昕阅读器进行编辑。


cDNA文库的构建基因文库的构建是现代生命科学研究中的一项重要技术。
自70年代初首例cDNA克隆问世以来,已用构建和筛选cDNA文库的方法克隆了很多基因。
通过构建cDNA文库能直接分离到生命活动过程中的一些调控基因及了解这些基因所编码的蛋白质的相互作用关系.因此cDNA文库的构建是基因克隆的重要方法之一,从cDNA文库中可以筛选到所需的目的基因,并直接用于该目的基因的表达。
它是发现新基因和研究基因功能的工具。
1.cDNA文库构建的原理真核生物基因的结构和表达控制元件与原核生物有很大的不同。
真核生物的基因是断裂的,在基因最后产物中表达的编码序列(外显子)被非编码序列(内含子)分隔开,需经RNA转录后加工过程才使编码序列拼接在一起。
真核生物的基因不能直接在原核生物中表达,只有将加工成熟的mRNA经逆转录合成互补的DNA(complementary DNA,cDNA)接上原核生物表达控制元件,才能在原核生物中表达。
而且,真核细胞的基因通常只有一小部分进行表达,由于mRNA的不稳定性,对基因表达和有关mRNA都常通过对其cDNA来进行研究。
为分离cDNA克隆或研究细胞的cDNA 谱,需要先构建cDNA文库。
所谓cDNA文库是指细胞全部mRNA逆转录成cDNA并被克隆的总和。
cDNA文库应包含的克隆数目可由以下公式来计算:N=ln(1-p)/ln(1-1/n)式中:N-cDNA文库所包含的克隆数目;P-低丰度cDNA存在于库中的概率,通常要求其大于99%;1/n-每一种低丰度mRNA占总mRNA的分数。
2.构建cDNA文库的基本步骤构建cDNA文库的基本步骤有五步:①制备mRNA;②合成cDNA;③制备载体DNA;④双链cDN 的分子克隆;⑤对构建的cDNA文库进行鉴定,测定文库包含的克隆数,抽查克隆的质量和异质性,如果需要可适当扩增。
对cDNA的文库的要求:一是希望文库能包括各种稀有mRNA的cDNA克隆;二是克隆的cDNA应是全长的,避免丢掉5’端的序列。
操作一览(PT3000-2)本操作一览仅为实验流程,对于初用者,请在使用操作一览前仔细阅读SMART TM cDNA 文库构建试剂盒(Cat. No. 634901) 的说明书。
A. 第一链cDNA(fs cDNA)合成1. 在0.5-ml 离心管中混匀以下试剂:1–3 μl RNA 样本(对照反应,使用1 μl 的对照RNA)1 μl SMART™ IV Oligonucleotide1 μl CDS III/3' PCR Primer如果总体积<5 μl时,用水补齐至5 μl。
2. 混匀试剂并瞬时离心。
3. 72°C孵育2 min。
冰上冷却2 min。
4. 瞬时短暂离心。
5. 向每管反应中加入以下试剂:2.0 μl 5X First-Strand Buffer1.0 μl DTT (20 mM)1.0 μl dNTP Mix (10 mM)1.0 μl SMARTScribe MMLV Reverse Transcriptase10.0 μl 总体积6. 使用枪轻柔地混匀试剂,并瞬时离心。
7. 42°C孵育1 hr。
之后的LD PCR(步骤B)操作需在冰上进行。
8.引物延伸合成ds cDNA分别向反应管中加入1 μl 氢氧化钠,68°C孵育30 min,然后进行步骤C。
B. LD PCR扩增合成cDNA1. 在一个新的0.5-ml 离心管中混匀以下试剂:2 μl First-Strand cDNA (from Step A.7)80 μl Deionized H2O10 μl 10X Advantage2 PCR Buffer2 μl 50X dNTP Mix2 μl5' PCR Primer2 μl CDS III/3' PCR Primer2 μl 50X Advantage2 Polymerase Mix100 μl总体积2. 使用枪轻柔地混匀试剂,并瞬时离心。
植物生理学与生物化学国家重点实验室(浙江大学)实验室Protocol汇编实验室主任:吴平参编人员:寿惠霞,毛传澡,吴忠长,王首峰,莫肖蓉,刘洪家,易可可,李靖,黄方亮,周洁,何晓薇等文本编辑:陈铭(2005年12月15日)©本实验室版权所有一. cDNA文库构建方案mRNA的纯化(PolyATtract mRNA Isolation systemIII Z5300)准备:65℃水浴或heating block ;灭菌的无Rnase 1.5 ml 塑料试管;灭菌的无Rnase枪头一.Annealing of probe1.在一支干净的1.5ml 管中加入0.1-1.0 mg 总RNA,补无Rnase的水到500 ul;2.65℃,10 min;3.在RNA管中加 3ulBiotinglated-Oligo (dT) Probe 和 13 ul 20×SSC ,在适温下温和混匀,直到冷却,约要10 min;同时准备0.5×SSC和0.1×SSC.二.Stock solution Preparation1.在无Rnase试管中准备1.2 ml 无菌的0.5×SSC(30 ul20×SSC,+1.170ml Rnase-free water);2.在无Rnase试管中准备1.4 ml 无菌的0.1×SSC(7.0 ul20×SSC,+1.393 ml Rnase-free water);三.Washing of streptavitin paramagnetic particles1.温和悬浮塑料试管中的颗粒至颗粒全部被分散,将小管放到Magnetic Stand 使SA-PMPS颗粒层积在管的一边(-30sec)2.小心去除悬液,不要离心!3.用0.5×SSC(每次0.3 ml)洗SA-PMPS颗粒三次,每次用Magnetic Stand 固定,小心移去悬液;4.用0.1 ml 0.5×SSC 悬浮洗过的SA-PMPS颗粒颗粒。
cDNA文库组标准流程一. Total RNA的提取 (2)二. mRNA的分离 (5)三.cDNA双链合成 (8)四.载体制备 (11)五. cDNA双链和载体的连接 (13)六.电转化流程 (14)七.快速鉴定、菌落PCR (16)八.pBlueScript cDNA库扩增 (18)一.Total RNA的提取1.试剂配制准备工作:1、研钵、5ml/10ml/ 25ml移液管、100ml/250ml量筒、250ml/100ml容量瓶、药匙、试剂瓶等玻璃制品均用锡纸包裹口部,置于烤箱内,180℃,烤6小时。
2、50ml/1.5ml离心管、枪头等塑料制品用0.1‰DEPC水浸泡过夜后,121℃20mins 高压灭菌。
3、电泳槽及电泳托、梳子用3%双氧水处理。
4、常用试剂及其配方:▲DEPC水:在1000ml去离子水中加入100ul DEPC, 静置过夜后高压灭菌。
▲0.78M柠檬酸纳:PH=4~5三水合柠檬酸纳22.94g加DEPC水定容至100ml,室温放置备用。
▲10%肌氨酸钠:肌氨酸钠10g加DEPC水定容至100ml,室温放置备用。
▲变性裂解液:0.78M柠檬酸钠8.25ml10%肌氨酸钠12.375ml异硫氰酸胍118.05g加DEPC水定容至250ml,室温放置备用临用前加β-巯基乙醇使其终浓度为1%(v/v)▲2M 醋酸钠PH=4.5NaAc·3H2O 13.6g加DEPC水定容至50ml,高压灭菌,室温放置备用▲3M醋酸钠PH=5NaAc·3H2O 20.4g加DEPC水定容至50ml,高压灭菌,室温放置备用▲4M LiCL:LiCL 24.164g加DEPC水定容至100ml,高压灭菌,室温放置备用▲0.5M EDTA PH=8.0EDTA 18.61g用NaOH调PH值至8.0,定容到100ml,高压灭菌,室温放置备用▲10X MOPS (3-(N-吗啉代)丙磺酸):MOPS 41.86gNaAC·3H2O 4.10g0.5MEDTA(PH 8.0)20ml用NaOH调PH值 6.5 , DEPC水定容到1L,室温避光放置备用。
SMARTer™ RACE cDNA 扩增试剂盒操作一览(PT4096-2)本操作一览仅为实验流程,对于初用者,请在使用操作一览前仔细阅读SMARTer RACE cDNA 扩增试剂盒(Cat. Nos. 634923 & 634924) 的说明书。
一、RACE-Ready cDNA的准备为了获得RACE-Ready cDNA (说明书的Section V),请准备5’- & 3’-RACE-Ready cDNA 合成反应体系,共需要4个反应管,每管均为10µl反应体系。
1. 混匀以下试剂,并瞬时离心。
在进行Step 7前,将反应管室温放置:2.0 µl 5X First-Strand Buffer1.0 µl DTT (20 mM)1.0 µl dNTP Mix (10 mM)4.0 µl 总体积2. 分别向这些管中加入对应的以下试剂:准备5'-RACE-Ready cDNA 准备3'-RACE-Ready cDNA1.0–2.75 µl RNA* 1.0–3.75 µl RNA*1.0 µl 5'-CDS Primer A 1.0 µl 3'-CDS Primer A*使用1 µl 对照小鼠心脏总RNA(1 µg/µl)。
3. 将step2中的5'-RACE-Ready cDNA 定容至3.75 µl;将3'-RACE-Ready cDNA 定容至4.75µl。
4. 混匀试剂,瞬时离心。
5. 72°C 孵育3 min,然后42°C冷却2 min。
最后14,000rpm离心10sec。
注意: 此步骤在PCR仪中进行,在孵育时,准备step7。
6. 向5’RACE反应管中各加入1 µl SMARTer IIA oligo,混匀后瞬时离心。
In-Fusion SMARTer 定向cDNA文库构建实验流程一、第一链cDNA的合成如果材料受限,总RNA最低起始量为50 ng。
如果材料不受限制,最好以1ug总RNA起始实验。
1.For each sample and Control Mouse Liver Total RNA, combine the following reagents in separate Micro centrifuge tubes:1–3.5 μl RNA (50 ng–1 μg total RNA or 50–100 ng poly A+ RNA)*1 μl 3’ In-Fusion SMARTer CDS Primer (12 μM)x μl去离子水4.5 μl 总体积*使用1 µl 对照小鼠肝脏总RNA(1 µg/µl)。
2. 混匀试剂并瞬时离心。
3. 72°C 孵育3 min,42°C室温2 min。
NOTE:反应的起始几步(步骤4-6) 对于第一链cDNA合成很关键。
最好不要中断实验。
在步骤3的孵育时间内准备步骤4。
4. 室温下准备以下试剂:2.0 μl 5X First-Strand Buffer0.25 μl DTT (100 mM)1.0 μl dNTP Mix (10 mM )1.0 μl SMARTer V Oligonucleotide (12 μM)0.25 μl RNase Inhibitor1.0 μl SMARTScribe™ Reverse Transcriptase (100 U)*5.5 μl总体积/每管* 使用前加入反转录酶。
5. 向每个反应管中加入5.5 μl混合液,使用枪轻柔地混匀试剂,瞬时离心。
6. 42°C孵育90 min。
7. 68°C 加热10 min终止第一链反应。
8. 如果您打算直接进行PCR反应(Section C),那么久从一链产物中取出2份2ul分别放入干净无菌的PCR管中,冰上放置这些反应管。
操作一览(PT3000-2)本操作一览仅为实验流程,对于初用者,请在使用操作一览前仔细阅读SMART TM cDNA 文库构建试剂盒(Cat. No. 634901) 的说明书。
A. 第一链cDNA(fs cDNA)合成1. 在0.5-ml 离心管中混匀以下试剂:1–3 μl RNA 样本(对照反应,使用1 μl 的对照RNA)1 μl SMART™ IV Oligonucleotide1 μl CDS III/3' PCR Primer如果总体积<5 μl时,用水补齐至5 μl。
2. 混匀试剂并瞬时离心。
3. 72°C孵育2 min。
冰上冷却2 min。
4. 瞬时短暂离心。
5. 向每管反应中加入以下试剂:2.0 μl 5X First-Strand Buffer1.0 μl DTT (20 mM)1.0 μl dNTP Mix (10 mM)1.0 μl SMARTScribe MMLV Reverse Transcriptase10.0 μl 总体积6. 使用枪轻柔地混匀试剂,并瞬时离心。
7. 42°C孵育1 hr。
之后的LD PCR(步骤B)操作需在冰上进行。
8.引物延伸合成ds cDNA分别向反应管中加入1 μl 氢氧化钠,68°C孵育30 min,然后进行步骤C。
B. LD PCR扩增合成cDNA1. 在一个新的0.5-ml 离心管中混匀以下试剂:2 μl First-Strand cDNA (from Step A.7)80 μl Deionized H2O10 μl 10X Advantage2 PCR Buffer2 μl 50X dNTP Mix2 μl5' PCR Primer2 μl CDS III/3' PCR Primer2 μl 50X Advantage2 Polymerase Mix100 μl总体积2. 使用枪轻柔地混匀试剂,并瞬时离心。
3. 如果需要可以向管中加2滴矿物油,盖上管盖,置于预热(95°C)的PCR仪器中。
4. 以下程序选择一种进行扩增:GeneAmp 480 GeneAmp 2400/9600•95℃ 1 min •95℃20 sec•x cycles*: •x cycles*:95℃15 sec 95℃ 5 sec68℃ 6 min 68℃ 6 min*参考Table I 中最优循环数的使用。
5. 取5μl产物在1.1% 琼脂糖/EtBr 胶上进行检测,若检测结果与预期不符时,请参考疑难解答指南(说明书的Section VIII)。
6. 然后进入步骤D。
C. 引物延伸扩增ds cDNA1. 混匀以下试剂:11 μl First-Strand cDNA (from Step A.8)71 μl Deionized H2O10 μl10X Advantage 2 PCR Buffer2 μl dNTP Mix2 μl5' PCR Primer2 μl CDS III/3' PCR Primer2 μl 50X Advantage 2 Polymerase Mix100 μl 总体积2. 使用枪轻柔地混匀试剂,并瞬时离心。
3. 如果需要可以加2滴矿物油,盖上管盖后置于预热(95°C)的PCR仪器中。
4. 以下程序选择一种进行扩增:GeneAmp 480 GeneAmp 2400/9600• 72°C 10 min • 72°C 10 min• 95°C 1 min • 95°C 20 sec• 3 cycles:• 3 cycles:95°C 15 sec 95°C 5 sec68°C 8 min 68°C 8 min5. 取5μl产物在1.1% 琼脂糖/EtBr 胶上进行检测,如果结果与预期不符,请参考疑难解答指南(说明书的Section VII)。
D. 蛋白酶K 消化1. 向0.5-ml 无菌管加入50 μl 扩增后的ds cDNA(2–3 μg) 和2 μl 蛋白酶K (20 μg/μl)。
将剩余ds cDNA置于–20°C保存。
2. 混匀试剂并瞬时离心。
3. 45°C孵育20 min,瞬时离心。
4. 加入50 μl Deionized H2O。
5. 再加入100 μl 酚:氯仿:异戊醇,轻柔地连续颠倒1–2 min。
6. 14,000 rpm 离心5 min。
7. 将最上层液体(上清液) 转移到0.5-ml干净的管。
8. 加入100 μl 酚:氯仿:异戊醇,轻柔地连续颠倒1–2 min。
9. 14,000 rpm 离心5 min。
10. 将最上层液体(上清液) 转移到0.5-ml干净的管。
11.加入10 μl 3 M醋酸钠, 1.3 μl 糖原(20 μg/μl)和260 μl室温95% 乙醇,立即进行14,000 rpm 室温离心20 min。
12. 小心去除上清液。
13. 100 μl 80%乙醇清洗。
14. 空气干燥(~10 min)来挥发残留的乙醇。
15. 加入79 μl Deionized H2O 重悬DNA颗粒。
E. SfiI 酶切1. 在0.5-ml离心管中混匀以下试剂:79 μl cDNA (Step D.15)10 μl 10X Sfi Buffer10 μl SfiI Enzyme1 μl 100X BSA100 μl总体积2.混匀并在50°C 孵育2 hr。
3. 加入2 μl 1%二甲苯腈蓝,混匀。
F. CHROMA SPIN TM-400 cDNA分选1. 标记16个1.5-ml离心管,按顺序摆放在管架中。
2. 准备CHROMA SPIN-400纯化柱:a. 室温放置CHROMA SPIN 纯化柱1 hr。
然后颠倒数次混匀胶质。
b. 赶走纯化柱中的气泡,使用1000-μl 移液器轻柔地重悬胶质。
去除底盖让液体滴落。
c. 将纯化柱放置在环形支架上。
d. 借助重力将柱中存储液排干(胶质的顶端应该处于柱外表面1.0-ml标记处。
)e. 流速~1滴/40–60 sec,每滴体积~40 μl。
3. 当存储液排干后,加入700 μl 柱纯化缓冲液并将其排干。
4. 向胶质中央加入~100 μl SfiI消化后的cDNA和二甲苯腈蓝混合液(Step E.3 above)。
5. 当样本被胶质完全吸收后再进行下一步骤。
6. 向胶质中央加入100 μl缓冲液清洗纯化柱。
7. 排干缓冲液,胶质表面不能有液体残留。
当无液体滴落时,再进行下一步骤。
8. 将步骤1中离心管放置在纯化柱下方,使标记1的管对准纯化柱下方液体出口处。
9. 加入600 μl 缓冲液后立即使用管1–16分别收集连续的1滴液体(每管~35 μl)。
10. 从16管中各取3 μl 在1.1% agarose/EtBr gel上150 V.电泳10 min检测分析。
将最能代表实验样本cDNA大小范围的头3-4滴所对应管中的样本合并到一管。
11. 向管中加入以下试剂:1/10 vol. Sodium Acetate (3 M; pH 4.8)1.3 μl Glycogen (20 mg/ml)2.5 vol. 95% ethanol (–20°C)12. 前后轻柔地摇晃管使试剂混匀。
13. –20°C或干冰/乙醇中放置1 hr。
14. 14,000 rpm室温离心20 min。
15. 小心吸掉液体。
16. 短暂瞬时离心。
17. 小心洗掉液体,然后空气干燥~10 min。
18. 加入7 μl Deionized H2O重悬DNA颗粒。
G. cDNA 连接载体1. 标记3个0.5-ml 离心管,并向管中分别加入表(Table II)中所列试剂。
混匀并瞬时离心。
2. 16°C孵育过夜。
3. 分别进行λ-phage 包装。
4. 对文库进行滴度测定(Section H)。
3个连接体系可获得1–2x10e6个单克隆。
未扩增的文库在4°C 可保存2 weeks。
5. [可选]如果文库<1–2x10e6克隆,可将剩余的cDNA进行连接载体。
使用表中cDNA和载体比例重复连接反应能够收获最好的结果;也可以根据cDNA的使用量来扩大体系来做连接。
再进行包装反应和滴度测定实验。
若这时候滴度还是低的话,请参考说明书中Section VIII的疑难解答指南。
6. 为了提高文库的稳定性,首先将步骤G.4中包装反应物合并,然后参照步骤J对文库进行扩增。
扩增后的文库4°C可放置6–7 months,或在–70°C (in 7% DMSO) 可放置1年。
H. 测定未扩增文库的滴度1. 从存储培养板板中选取单个宿主菌株,接种到50-ml含15 ml LB/maltose/MgSO4液体培养基的实验管。
37°C震荡(140 rpm)孵育过夜。
当OD600 o=2.0时,5,000 rpm离心5 min收集菌体。
弃上清,用7.5 ml 10 mM MgSO4.重悬菌体。
2. 准备90-mm LB/MgSO4 固体培养板,37°C预热培养板。
3. 使用1X lambda dilution buffer(稀释度=1:5-1:20)稀释未扩增细胞裂解物。
4. 向过夜培养的200 μl XL1-Blue中加入1 μl 稀释后的噬菌体,37°C作用10–15 min。
5. 加入2 ml 融化的LB/MgSO4 顶层琼脂,快速混匀,立即铺于90-mm 37°C预热的LB琼脂板上。
6. 将培养板室温冷却10 min,37°C 倒置培养6–18 hr。
7.记数菌斑,并计算噬菌体滴度(pfu/ml):I. 鉴定重组率通过感染合适的宿主菌株(例如,E. coli XL1-Blue)和蓝白斑筛选,可鉴定含有插入片段的噬菌体。
参照未扩增文库滴度测定的步骤(Step.H)来进行蓝白斑筛选,步骤中还需增加的实验是:在铺板噬菌体+细菌混合物时,向融化的顶层琼脂中加入IPTG 和X-gal。
2 ml融化的顶层琼脂使用50 μl IPTG、50 μl X-gal存储液。
为了获得500–1,000菌斑/90-mm板,将培养板在37°C 孵育6–18 hr。
白斑和蓝斑的比例可以快速直观的估算重组效率。
成功的连接实验的重组率为80%。
如果您获得的重组率小于这个值,请参考说明书Section VIII的疑难解答指南。
[可选]SMART™ PCR cDNA 文库的PCR插入片段筛选检测连接效率,推荐使用Clontech的Advantage 2 cDNA PCR Kit和λTriplEx LD-Insert Screening Amplimers 检测cDNA插入片段。