酒石酸美托洛尔片一致性评价参比制剂溶出曲线测定
- 格式:pdf
- 大小:168.76 KB
- 文档页数:3

普通口服固体制剂溶出曲线测定与比较指导原则本指导原则适用于仿制药质量一致性评价中普通口服固体制剂溶出曲线测定方法的建立和溶出曲线相似性的比较。
一、背景固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透等,因此,药物的体内溶出和溶解对吸收具有重要影响。
体外溶出试验常用于指导药物制剂的研发、评价制剂批内批间质量的一致性、评价药品处方工艺变更前后质量和疗效的一致性等。
普通口服固体制剂,可采用比较仿制制剂与参比制剂体外多条溶出曲线相似性的方法,评价仿制制剂的质量。
溶出曲线的相似并不意味着两者一定具有生物等效,但该法可降低两者出现临床疗效差异的风险。
二、溶出试验方法的建立溶出试验方法应能客观反映制剂特点、具有适当的灵敏度和区分力。
可参考有关文献,了解药物的溶解性、渗透性、pKa常数等理化性质,考察溶出装置、介质、搅拌速率和取样间隔期等试验条件,确定适宜的试验方法。
(一)溶出仪溶出仪需满足相关的技术要求,应能够通过机械验证及性能验证试验。
必要时,可对溶出仪进行适当改装,但需充分评价其必要性和可行性。
溶出试验推荐使用桨法、篮法,一般桨法选择50—75转/分钟,篮法选择50—100转/分钟。
在溶出试验方法建立的过程中,转速的选择推荐由低到高。
若转速超出上述规定应提供充分说明。
(二)溶出介质溶出介质的研究应根据药物的性质,充分考虑药物在体内的环境,选择多种溶出介质进行,必要时可考虑加入适量表面活性剂、酶等添加物。
1.介质的选择应考察药物在不同pH值溶出介质中的溶解度,推荐绘制药物的pH-溶解度曲线。
在确定药物主成分稳定性满足测定方法要求的前提下,推荐选择不少于3种pH值的溶出介质进行溶出曲线考察,如选择pH值1.2、4.5和6.8的溶出介质。
对于溶解度受pH值影响大的药物,可能需在更多种pH值的溶出介质中进行考察。
推荐使用的各种pH值溶出介质的制备方法见附件。
当采用pH7.5以上溶出介质进行试验时,应提供充分的依据。

盐酸拉贝洛尔片仿制制剂与参比制剂溶出曲线相似性研究郝丽娟;徐艳梅;苗会娟;王春霞;高燕霞【期刊名称】《中国药业》【年(卷),期】2022(31)24【摘要】目的建立盐酸拉贝洛尔片溶出度的测定方法,评价盐酸拉贝洛尔片仿制制剂与参比制剂溶出曲线的相似性。
方法采用桨法,以高效液相色谱(HPLC)法测定制剂在4种溶出介质(pH1.2盐酸溶液、水、pH 4.0醋酸盐缓冲液和pH 6.8磷酸盐缓冲液)中的溶出量,色谱柱为Agilent Eclipse XDB-C_(18)柱(250 mm×4.6 mm,5μm),流动相为乙腈-0.1%磷酸溶液(25∶75,V/V),流速为1.0 mL/min,检测波长为230 nm,柱温为40℃,进样量为20μL;计算累积溶出度,绘制溶出曲线,并采用相似因子(f_(2))法评价溶出曲线的相似性。
结果盐酸拉贝洛尔在4种溶出介质中的质量浓度均在10.11-1 011.00μg/mL(r=0.999 7,0.999 8,0.999 6,0.999 9)范围内与峰面积线性关系良好;精密度、稳定性、重复性试验结果的RSD均小于1.5%。
以参比制剂为对照,B厂仿制制剂样品以pH1.2盐酸溶液和pH 6.8磷酸盐缓冲液为溶出介质时所得溶出曲线与参比制剂具有相似性,C厂仿制制剂在4种溶出介质中均无此相似性。
结论本方法适用于测定盐酸拉贝洛尔片的溶出度,可为盐酸拉贝洛尔片仿制制剂的质量一致性评价提供参考。
【总页数】5页(P51-55)【作者】郝丽娟;徐艳梅;苗会娟;王春霞;高燕霞【作者单位】河北省药品医疗器械检验研究院•仿制药质量控制与评价重点实验室【正文语种】中文【中图分类】R917;R972【相关文献】1.不同厂家比沙可啶肠溶片仿制制剂与参比制剂溶出曲线的相似性评价2.相似因子法评价雷美替胺片自研制剂与原研制剂溶出曲线的相似性3.光纤药物溶出实时测定仪结合f2法评价阿折地平片自制制剂与原研制剂溶出曲线的相似性4.茶碱缓释片的制备及与参比制剂的体外溶出相似性评价5.市售盐酸二甲双胍片仿制制剂溶出曲线与溶出度研究因版权原因,仅展示原文概要,查看原文内容请购买。


仿制药一致性评价RevisedbyPetrelat2021仿制药致性评价一:背景信息1)2016年2月6日,国务院颁布了国办发【2016】8号文件“国务院办公厅关于开展仿制药质量和疗效一致性评价的意见”,随后国家又在短时间内连续颁布了一系列关于仿制药一致性评价工作的相关文件和指南,真正拉开了仿制药产品质量和疗效一致性评价工作的序幕。
2)根据国家仿制药一致性评价政策和时限要求,289种基本药物要求在2018年底完成评价,其他仿制药产品若能成为“首名”或“前三名”,对市场和经济意义重大。
否则,则存在较大风险失去市场甚至文号不保。
二:认识与建议虽然国家提出了明确的时间要求和评价原则,但当前的法规并不具体,企业左右为难。
公司基于长期从事药政法规研究的工作经验,我们对本轮要求的认识如下:1)不等不靠、主动研究。
2)要充分重视对产品的信息回顾与整理,重视仿制药评价的整体策划,为开展研究工作打基础。
3)重视药学研究。
仿制药研究的重点是药学部分,也是国家评价仿制药质量的重要内容,CMC是重中之重,必须先行。
4)时间紧迫仿制药一致性评价,尤其是基本药物的评价看起来还有2年多时间,实际分解后留给企业的时间已经非常紧张,即使一切顺利也需要20-25个月。
5)逐步开展,分段进行。
仿制药一致性评价是一个综合工程,链条长,费用高。
尤其是早年完成注册的产品,注册研究与申报资料更加薄弱,适合先初步摸索产品质量现状再进行系统深入的研究。
6)仿制药一致性评价应分为四个阶段开展审评跟踪、审评补充与批准三:优势与业绩仿制药一致性评价工作是一个系统工程,需要完整的服务链条和资源配置,包括CMC 研发实验室、动物GLP实验室、BE临床实验基地、生化分析实验室和注册服务团队,以及良好的公共资源,并且各个环节都要有经验丰富的技术团队与完善的质量管理体系,要经得起注册现场核查与临床现场核查。
1)咨询拥有的优势包括以下几方面:建立了高水平配置的国家级CMC研发实验室,拥有专业的研发人员和十多年药品研发经验,为中国三大医药技术研究成果转化平台之一,国家基本药物标准溶出度曲线制作承担单位之一;合作动物GLP实验室,比格犬等大动物试验不是问题;集团内拥有BE临床试验基地与通过FDA审计的生化分析实验室(海口、长春、南京3个BE基地),BE试验优先快速安排;18年丰富经验的注册团队,轻松化解技术难题。

溶出手把手教你做出仿制药四条溶出曲线文章来自丁香园insight数据库,已经取得授权2016 年 3 月 5 日,国务院办公厅印发了《关于开展仿制药质量和疗效一致性评价的意见》,仿制药一致性评价工作正式展开。
仿制药一致性评价工作中,首先需要评价的是仿制制剂与参比制剂在体外溶出曲线要一致。
然而,将仿制制剂与参比制剂做到体外四条溶出曲线一致,并不是一件容易的工作。
作者将平日的工作经验总结出来,欲与大家交流分享。
开始前的准备将 BCS 再次分类生物药剂学分类系统(BCS,biopharmaceutics classification system)是 1995 年由 Amidon 提出的基于药物溶解性质和渗透性差异的分类系统,分为四类。
对于体外四条溶出曲线而言,溶解性性质比渗透性更实用,因此根据溶解性质的差异将 BCS 再次分类,分为 A 类(Ⅰ 和Ⅲ)和 B 类(Ⅱ 和Ⅳ)。
之所以这样二次分类,是因为Ⅰ 和Ⅲ、Ⅱ 和Ⅳ 分别在体外呈现出相同的溶解度性质。
将化合物根据 pH-溶解度差异来分类《仿制药质量一致性评价·口服固体制剂溶出曲线测定与比较指导原则》中提出,在进行溶出度实验之前,建议绘制化合物pH-溶解度图。
那么根据 pH-溶解度的差异性,也可以将化合物分为两类:一类是溶解度不存在pH 依赖性或差异性。
暂且将饱和溶解度无pH 依赖性的原料药分为 a 类。
另一类是溶解度存在 pH 依赖性或差异性,其饱和溶解度随 pH 值增加而增加,或随pH 值增加而降低。
将这类化合物分为b 类,比如NAISD 类的布洛芬、双氯芬酸钠等。
这样分类如何应用呢?举个例子。
如表1 所示,双氯芬酸钠在不同介质中的饱和溶解度差异性较大,再结合根据上述BCS 的二次分类,那么可将双氯芬酸钠可定义为Bb 类化合物。
之所以这样区分,是为了建立自我工作模型,以后在工作遇到相同的化合物,直接进行套用,从而降低工作量。
如何快速有效地做出四条溶出曲线?根据化合物性质不同,其溶出曲线难易程度也是各有差别。

药物一致性评价中什么样的溶出曲线具有区分力溶出度试验的重要性已毋庸置疑,该实验在口服固体制剂品质评价和仿制药研发中愈来愈发挥出举足轻重的作用,而寻找到客观存在、并确定出最能体现原研制剂内在优良品质、具有区分力的溶出曲线才是本实验的重中之重,本文将就此展开论述。
溶出度试验在评价口服固体制剂内在品质和仿制药研发中愈来愈发挥出举足轻重的作用,尤“在多种pH值溶出介质中溶出曲线的测定和比对”更是成为“剖析”和“肢解”固体制剂内在品质一种擘肌分理、抽丝剥茧的重要手段;成为口服固体制剂内在品质呈现于外在的一种“表象”、“映射”和“载体”。
现今,人们愈发关注溶出度试验的区分力,即什么样的溶出曲线最能代表产品内在品质、最适合评估仿制制剂处方筛选与工艺开发、最能在仿制制剂与原研制剂体外溶出行为比较时体现出差异性,最具有体内外相关性,且最适合制订入质量标准等问题,本文将就此展开讨论。
既有质量标准的局限性国内质量标准由于之前的多年,国内对溶出度试验未给予高度重视和科学认知,无论是在新药审评还是在药检机构后期制订国家标准时,出发点皆是为了让既有产品合格来拟定试验参数,故目前很多国内质量标准均拟定得较为宽松(如高转速、加入高浓度表面活性剂甚至有机溶剂、采用溶解度大的溶出介质等),无区分力,借鉴性不强。
国外质量标准与进口质量标准由于溶出度试验的重要性,故在我们所能查阅获取的质量标准中,很多品种的溶出度试验均具有一定的“隐晦性”。
当我们获取数批原研制剂、按照质量标准试验条件检测时,结果往往都是较为快速释放情形(最多30min就已达85%)。
深度剖析测定原研制剂后发现,实验条件很多不具区分力,即便美国FDA公布的溶出度数据库中有的品种也如此。
在FDA/CDER属下的仿制药办公室于2012年下半年推出的、“质量源于设计”理念应用于仿制药研发——速释与缓控释制剂2个研发模板中就列举了以上条件不具备区分力的研发案例。
所以,若我们盲目迷信以上这些标准,秉承“照本宣科的拿来主义”,就会在仿制药开发和品质评价时误入歧途,甚至剑走偏锋,造成错误判断。

一致性评价重磅参考资料溶出度试验的开发和验证溶出度试验是一种重要的药物品质控制方法,用于评估药物的释放速度和特性。
为了确保一致性评价的准确性和一致性,美国药典(USP)发布了有关溶出度试验的通用章节USP1092、这个章节详细说明了溶出度试验的开发和验证,对于药物制剂的一致性评价提供了重要的参考资料。
首先,USP1092章节说明了溶出度试验的目的和应用范围。
该章节强调了溶出度试验的重要性,以及它在药物品质控制中的应用。
溶出度试验可以用于评估药物的释放速度、药物的溶解特性以及药物的可溶性。
它可以帮助药物生产企业确保产品的一致性和质量,并帮助监管机构进行监管和评估。
然后,USP1092章节详细介绍了溶出度试验的开发和验证过程。
该章节首先说明了试验设备和试验条件的选择。
试验设备的选择应基于产品的特性和试验的要求,而试验条件的选择则应符合美国药典的要求。
章节中列出了一些可用于溶出度试验的设备和条件,以提供参考。
接着,USP1092章节讨论了试验方法的开发和验证。
在试验方法的开发过程中,需要选择适当的介质和操作条件,并设定合适的试验时间。
章节中提供了一些建议用于试验方法的开发。
在试验方法的验证过程中,需要验证试验方法的准确性、精确性、线性范围、灵敏度和选择性。
章节中给出了验证试验方法时应进行的测试和要求。
USP1092章节还强调了溶出度试验的数据分析和结果解释。
对于试验结果的分析和解释,需要参考适当的统计学原理和方法。
章节中提供了一些常用的统计学方法和指导,以帮助药物生产企业正确分析和解释试验结果。
最后,USP1092章节还包括了对溶出度试验的质量控制的要求。
该章节指出了试验过程中的质量控制点和限度。
它强调了每个质量控制点的重要性,以确保试验的准确性和一致性。
总之,USP1092章节是一本关于溶出度试验开发和验证的重要参考资料。
它详细阐述了试验的目的和应用范围,详细描述了试验方法的开发和验证过程,并给出了一些统计学原理和方法的指导。

仿制药一致性评价一:背景信息1) 2016年2月6日,国务院颁布了国办发【2016】8号文件“国务院办公厅关于开展仿制药质量和疗效一致性评价的意见”,随后国家又在短时间内连续颁布了一系列关于仿制药一致性评价工作的相关文件和指南,真正拉开了仿制药产品质量和疗效一致性评价工作的序幕。
2) 根据国家仿制药一致性评价政策和时限要求,289种基本药物要求在2018年底完成评价,其他仿制药产品若能成为“首名”或“前三名”,对市场和经济意义重大。
否则,则存在较大风险失去市场甚至文号不保。
二:认识与建议虽然国家提出了明确的时间要求和评价原则,但当前的法规并不具体,企业左右为难。
公司基于长期从事药政法规研究的工作经验,我们对本轮要求的认识如下:1) 不等不靠、主动研究。
2) 要充分重视对产品的信息回顾与整理,重视仿制药评价的整体策划,为开展研究工作打基础。
3) 重视药学研究。
仿制药研究的重点是药学部分,也是国家评价仿制药质量的重要内容,CMC是重中之重,必须先行。
4) 时间紧迫仿制药一致性评价,尤其是基本药物的评价看起来还有2年多时间,实际分解后留给企业的时间已经非常紧张,即使一切顺利也需要20-25个月。
5) 逐步开展,分段进行。
仿制药一致性评价是一个综合工程,链条长,费用高。
尤其是早年完成注册的产品,注册研究与申报资料更加薄弱,适合先初步摸索产品质量现状再进行系统深入的研究。
6) 仿制药一致性评价应分为四个阶段开展三:优势与业绩仿制药一致性评价工作是一个系统工程,需要完整的服务链条和资源配置,包括CMC研发实验室、动物GLP实验室、BE临床实验基地、生化分析实验室和注册服务团队,以及良好的公共资源,并且各个环节都要有经验丰富的技术团队与完善的质量管理体系,要经得起注册现场核查与临床现场核查。
1) 咨询拥有的优势包括以下几方面:✔建立了高水平配置的国家级CMC研发实验室,拥有专业的研发人员和十多年药品研发经验,为中国三大医药技术研究成果转化平台之一,国家基本药物标准溶出度曲线制作承担单位之一;✔合作动物GLP实验室,比格犬等大动物试验不是问题;✔集团内拥有BE临床试验基地与通过FDA审计的生化分析实验室(海口、长春、南京3个BE基地),BE试验优先快速安排;✔18年丰富经验的注册团队,轻松化解技术难题。
一致性评价之体外溶出试验——溶出数据库溶出度试验是评价口服固体制剂内在质量的一种重要手段,旨在保证不同生产企业生产的同一药品的口服固体制剂具有相同的品质和疗效。
按照《总局关于发布化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)的通告》(2016年第120号),溶出曲线相似性评价研究资料是申报的必备材料之一。
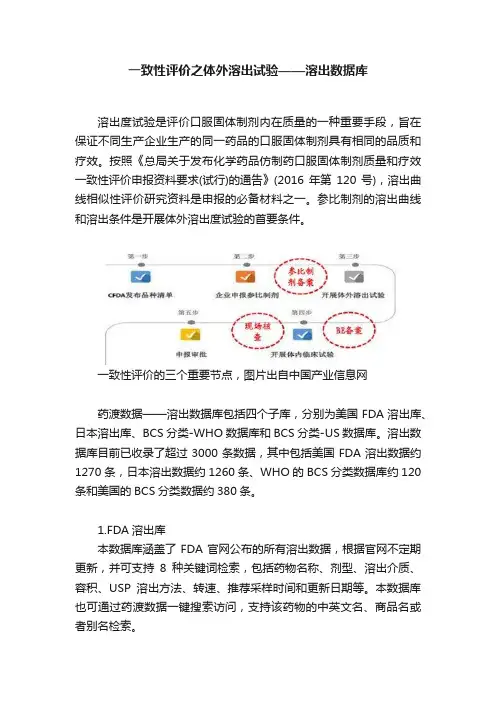
参比制剂的溶出曲线和溶出条件是开展体外溶出度试验的首要条件。
一致性评价的三个重要节点,图片出自中国产业信息网药渡数据——溶出数据库包括四个子库,分别为美国FDA溶出库、日本溶出库、BCS分类-WHO数据库和BCS分类-US数据库。
溶出数据库目前已收录了超过3000条数据,其中包括美国FDA溶出数据约1270条,日本溶出数据约1260条、WHO的BCS分类数据库约120条和美国的BCS分类数据约380条。
1.FDA溶出库本数据库涵盖了FDA官网公布的所有溶出数据,根据官网不定期更新,并可支持8种关键词检索,包括药物名称、剂型、溶出介质、容积、USP溶出方法、转速、推荐采样时间和更新日期等。
本数据库也可通过药渡数据一键搜索访问,支持该药物的中英文名、商品名或者别名检索。
本数据库检索结果包括药物名称、剂型、USP溶出方法、转速(RPMs)、溶出介质、容积(mL)、推荐采样时间(minutes)和更新日期,其中可根据需要按照药物名称首字母排序或更新日期早晚调整结果显示顺序。
图中字体为蓝色的药物名称,点击后可进一步查看完整的溶出信息。
本文以Calcifediol为例,点击黄色圆框内的“Calcifediol”,跳转到黄色箭头所示窗口,该窗口可以完整展示列表页未展示完全的内容。
2.日本溶出库本数据库涵盖了日本橙皮书官网所有药物的溶出数据,每月更新一次,并可提供活性成分(日文/英文)、参比制剂(全部/是/否)、溶出试验(全部/是/否)和药品品质情报(全部/是/否)等关键词检索。
本数据库可提供日英文活性成分名、参比制剂、溶出试验和药品品质情报集等信息,可根据需要按照日英文活性成分名调整显示顺序。


附件2通俗口服固体系体例剂溶出曲线测定与比较指点原则本指点原则实用于仿造药质量一致性评价中通俗口服固体系体例剂溶出曲线测定办法的树立和溶出曲线类似性的比较.一.布景固体系体例剂口服给药后,药物的接收取决于药物从制剂中的溶出或释放.药物在心理前提下的消融以及在胃肠道的渗入渗出等,是以,药物的体内溶出和消融对接收具有主要影响.体外溶出实验经常应用于指点药物制剂的研发.评价制剂批内批间质量的一致性.评价药品处方工艺变动前后质量和疗效的一致性等.通俗口服固体系体例剂,可采取比较仿造制剂与参比制剂体外多条溶出曲线类似性的办法,评价仿造制剂的质量.溶出曲线的类似其实不料味着两者必定具有生物等效,但该法可下降两者消失临床疗效差别的风险.二.溶出实验办法的树立溶出实验办法应能客不雅反应制剂特色.具有恰当的敏锐度和区分力.可参考有关文献,懂得药物的消融性.渗入渗出性.pKa常数等理化性质,考核溶出装配.介质.搅拌速度和取样距离期等实验前提,肯定合适的实验办法.(一)溶出仪溶出仪需知足相干的技巧请求,应可以或许经由过程机械验证及机能验证实验.须要时,可对溶出仪进行恰当改装,但需充分评价其须要性和可行性.溶出实验推举应用桨法.篮法,一般桨法选择50—75转/分钟,篮法选择50—100转/分钟.在溶出实验办法树立的进程中,转速的选择推举由低到高.若转速超出上述划定应供给充分辩明.(二)溶出介质溶出介质的研讨应根据药物的性质,充分斟酌药物在体内的情形,选择多种溶出介质进行,须要时可斟酌参加适量概况活性剂.酶等添加物.应考核药物在不合pH值溶出介质中的消融度,推举绘制药物的pH-消融度曲线.在肯定药物主成分稳固性知足测定办法请求的前提下,推举选择许多于3种pH值的溶出介质进行溶出曲线考核,如选择pH值1.2.4.5和6.8的溶出介质.对于消融度受pH值影响大的药物,可能需在更多种pH值的溶出介质中进行考核.推举应用的各类pH值溶出介质的制备办法见附件.当采取pH7.5以上溶出介质进行实验时,应供给充分的根据.水可作为溶出介质,但应用时应考核其pH值和概况张力等身分对药物及辅料的影响.推举选择500ml.900ml或1000ml.(三)溶出曲线的测定取样时光点可为5和/或10.15和/或20.30.45.60.90.120分钟,此后每隔1小时进行测定.以下任何一个前提均可作为考核截止时光点选择的根据.(1)持续两点溶出量均达85%以上,且差值在5%以内.—3.0)中考核时光不超出2小时.(2)在其他各pH值溶出介质中考核时光不超出6小时.(四)溶出前提的优化在截止时光内,药物在所有溶出介质中平均溶出量均达不到85%时,可优化溶出前提,直至消失一种溶出介质达到85%以上.优化次序为进步转速,参加适量的概况活性剂.酶等添加物.概况活性剂浓度推举在0.01%—1.0%(W/V)规模内依次递增,特别品种可适度增长浓度.某些特别药品的溶出介质可应用人工胃液和人工肠液.(五)溶出办法的验证办法树立后应进行须要的验证,如:精确度.周详度.专属性.线性.规模和耐用性等.三.溶出曲线类似性的比较溶出曲线类似性的比较,多采取非模子依附法中的类似因子(f2)法.该法溶出曲线类似性的比较是将受试样品的平均溶出量与参比样品的平均溶出量进行比较.平均溶出量应为12片(粒)的均值.盘算公式:R t为t时光参比样品平均溶出量;T t为t时光受试样品平均溶出量;n为取样时光点的个数.(一)采取类似因子(f2)法比较溶出曲线类似性的请求类似因子(f2)法最合适采取3—4个或更多取样点且应知足下列前提:1.应在完整雷同的前提下对受试样品和参比样品的溶出曲线进行测定.2.两条溶出曲线的取样点应雷同.时光点的拔取应尽可能以溶出量等分为原则,并统筹整数时光点,且溶出量超出85%的时光点不超出1个.3.第1个时光点溶出成果的相对尺度误差不得过20%,自第2个时光点至最后时光点溶出成果的相对尺度误差不得过10%.(二)溶出曲线类似性剖断尺度1.采取类似因子(f2)法比较溶出曲线类似性时,一般情形下,当两条溶出曲线类似因子(f2)数值不小于50时,可以为溶出曲线类似.2.当受试样品和参比样品在15分钟的平均溶出量均不低于85%时,可以为溶出曲线类似.四.其他(一)溶出曲线类似性的比较应采取同剂型.同规格的制剂.(二)当溶出曲线不克不及采取类似因子(f2)法比较时,可采取其他合适的比较法,但在应用时应赐与充分论证.附:溶出介质制备办法附溶出介质制备办法一.盐酸溶液取表1中划定量的盐酸,用水稀释至1000ml,摇匀,即得.表1 盐酸溶液的配制取表2中划定物资的取样量,用水消融并稀释至1000ml,摇匀,即得.表2 醋酸盐缓冲溶液的配制摇匀,即得.三.磷酸盐缓冲液取0.2mol/L磷酸二氢钾溶液250ml与表3中划定量的0.2mol/L 氢氧化钠溶液混杂,用水稀释至1000ml,摇匀,即得.表3 磷酸盐缓冲液稀释至1000ml.0.2mol/L氢氧化钠溶液:取氢氧化钠8.00g,加水消融并稀释至1000ml.以上为推举采取的溶出介质配制办法,若有须要,研讨者也可根据具体情形采取其他的溶出介质以及响应的配制办法.。
酒石酸美托洛尔片一致性评价参比制剂/溶出曲线测定
(草案) 【酒石酸美托洛尔】
中文名:酒石酸美托洛尔 英文名:Metoprolol Tartrate 分子式:(C 15H 25NO 3)2·C 4H 6O 4 分子量:684.82
结构式:
解离常数(25℃):
pKa ≈9.7
pH1.2 :1.0g/ml 以上 pH4.0:1.0g/ml 以上
在各溶出介质中的溶解度(37℃):pH6.8 :1.0g/ml 以上
pH 1.2:24小时内稳定 pH 4.0:24小时内稳定
在各溶出介质中的稳定性(37℃):pH 6.8:24小时内稳定
光:未测定
《标准溶出曲线》
<25mg 规格 片剂
>
<50mg规格片剂>
溶出曲线测定方法:
照高效液相色谱法(中国药典2010年版二部附录VD)测定。
色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂,以0.1mol/L高氯酸钠溶液(用0.85%高氯酸调节pH值至3.2)-乙腈(3:1)为流动相,检测波长为274nm,柱温为30℃,调整流速使美托洛尔主峰保留时间约为8分钟,理论板数按美托洛尔峰计算不低于2000,拖尾因子不大于1.5。
测定法取本品,照溶出度测定法(中国药典2010年版二部附录XC第二法),分别以规定的三种溶剂900ml为溶出介质,转速为每分钟50转,依法操作,经5、10、15、30、45、60分钟时,取溶液适量(5~10ml),滤过,并及时补充相同温度相同体积的溶出介质,取续滤液作为供试品溶液;另取酒石酸美托洛尔对照品适量,精密称定,加相应的溶出介质溶解并定量稀释制成每1ml 中约含28µg(25mg规格)、56µg(50mg规格)的溶液,作为对照品溶液。
精密量取供试品溶液和对照品溶液各50μl,分别注入液相色谱仪,记录色谱图,按外标法以峰面积计算每片在不同时间点的溶出量。
以12片的平均溶出量为纵坐标,时间为横坐标,绘制溶出曲线。
三种溶出介质配制方法:
(1) pH 1.2溶出介质的配制:取氯化钠2.0g和盐酸7.0ml,加水溶解并稀释至1000ml,即得。
(2) pH 4.0溶出介质的配制:取0.05mol/L醋酸溶液与0.05mol/L醋酸钠溶液按照16.4:3.6(V/V)比例混合,即得。
(3) pH 6.8溶出介质的配制:取磷酸盐缓冲液(pH 6.8)(取磷酸二氢钾3.40g 及无水磷酸氢二钠3.55g用水溶解并稀释至1000ml)适量,用水稀释1倍。
溶出曲线相似性比较与判定方法:
溶出曲线相似性的比较是将仿制制剂的平均溶出量与参比制剂的平均溶出量进行比较。
平均溶出量应为12片(粒)的均值。
(1) 25mg规格:在三种溶出介质中,均采用相似因子(f2)法比较溶出曲线相似性,比较的时间点为10、15和30分钟,两条溶出曲线相似因子(f2)数值不小于50,可认为溶出曲线具有相似性。
或在三种溶出介质中,当参比制剂在15分钟时,平均溶出量不低于85%,仿制制剂在15分钟时,平均溶出量不低于85%;或与参比制剂平均溶出量的差值均不大于10%,可认为溶出曲线相似。
(2) 50mg规格:在三种溶出介质中,均采用相似因子(f2)法比较溶出曲线相似性。
比较的时间点为10、15和30分钟,两条溶出曲线相似因子(f2)数值不小于50,可认为溶出曲线具有相似性。