多晶型及其检测
- 格式:pdf
- 大小:234.84 KB
- 文档页数:4

晶型药物常用的检测分析方法(2012-02-08 13:54:05)物质在结晶时由于受各种因素影响,使分子内或分子间键合方式发生改变,致使分子或原子在晶格空间排列不同,形成不同的晶体结构。
同一物质具有两种或两种以上的空间排列和晶胞参数,形成多种晶型的现象称为多晶现象(polymorphism)。
虽然在一定的温度和压力下,只有一种晶型在热力学上是稳定的,但由于从亚稳态转变为稳态的过程通常非常缓慢,因此许多结晶药物都存在多晶现象。
固体多晶型包括构象型多晶型、构型型多晶型、色多晶型和假多晶型。
药物分子通常有不同的固体形态,包括盐类,多晶,共晶,无定形,水合物和溶剂合物;同一药物分子的不同晶型,在晶体结构,稳定性,可生产性和生物利用度等性质方面可能会有显著差异,从而直接影响药物的疗效以及可开发性。
如果没有很好的评估并选择最佳的药物晶型进行研发,可能会在临床后期发生晶型的变化,从而导致药物延期上市而蒙受巨大的经济损失,如果上市后因为晶型变化而导致药物被迫撤市,损失就更为惨重。
因此,药物晶型研究和药物固态研发在制药业具有举足轻重的意义。
由于药物晶型的重要性,美国药监局(FDA)和中国药监局(SFDA)在药物申报中对此提出了明确规定,要求对药物多晶型现象进行研究并提供相应数据。
正因如此,任何一个新药的研发,都要进行全面系统的多晶型筛选,找到尽可能多的晶型,然后使用各种固态方法对这些晶型进行深入研究,从而找到最适合开发的晶型;选定最佳晶型后,下一步就是开发能始终如一生产该晶型的化学工艺;最后一步是根据制剂对原料药固态性质的要求,对结晶工艺进行优化和控制,确定生产具有这些固态性质的最佳工艺参数,从而保证生产得到的晶型具有理想的物理性质,比如晶体表象,粒径分布,比表面积等。
这种通过实验设计来保证质量的方法必须对药物晶型具有非常全面深刻的理解才能实现。
原研药公司对药物分子的晶型申请专利,可以延长药物的专利保护,从而使自己的产品具有更长时间的市场独享权。


医药制剂中的晶型分析目录前言 (1)1.XRD分析 (1)红外分析 (4)拉曼分析 (5)前言原料药(activepharmaceutica1ingredients,API)的晶型分析相信大家都比较熟悉,可以用XRD、IR、拉曼和DSC等。
但是当将原料药和大量辅料混合,做成制剂后,晶型的分析难度一下提高了很多,尤其是制剂中AP1含量很低的时候。
不少同行向我请教过这个问题,恰好上次有个公司委托我做了这些分析,今天我把这些结果给大家分析一下。
出于保密需要,具体药品名我就不透露了。
该片剂每片只含有5mgAPI,单片重量未知,一般20Omg比较常见。
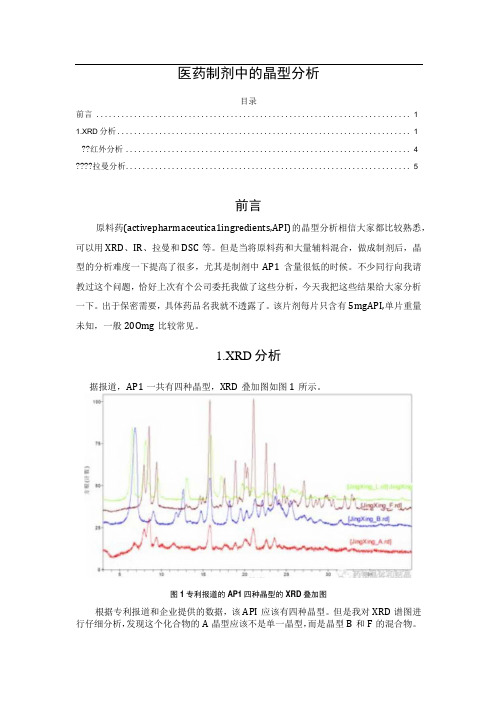
1.XRD分析据报道,AP1一共有四种晶型,XRD叠加图如图1所示。
图1专利报道的AP1四种晶型的XRD叠加图根据专利报道和企业提供的数据,该API应该有四种晶型。
但是我对XRD谱图进行仔细分析,发现这个化合物的A晶型应该不是单一晶型,而是晶型B和F的混合物。
A晶型的结晶度不高,所有的衍射峰在B和1都能找到对应的峰。
晶型B、F和1三种晶型重叠峰较少,应该是独立的晶型。
对制剂进行晶型的XRD分析时,需要先将每种辅料进行XRD分析,看看制剂的衍射峰里,哪些是辅料的,哪些是原料药的。
因为原料药含量很低,辅料的特征峰往往比原料药高。
参见图2,对比甲厂制剂与辅料的XRD图,发现制剂中的峰主要是乳糖的峰,其次含有少量Mg(OH)2,只有2。
二7.0和8.2处的小峰是辅料中没有的。
I _ ~~I"f1 - ------ ----------- ---⅜⅜⅜f,—- ----- ∣[]WΓθ∣5M>20101Sn111______ ______ _H卜. ___ __ 19∣RuTβra200Mu.101∙S2030V1I v O u it n»tr≡Mrτ2∙(t)图2甲厂制剂与每种固体辅料的XRD叠加图然后我们再将制剂的XRD谱图与原料药的对比进行分析(图3)o根据2θ=7.0和8.2处的峰,可以推测甲厂制剂中原料药的晶型应该主要是B,可能含有少量1。


多晶质量检验、抽样范围:生产中沉积的多晶硅、被测性能:外观及断面检查型号、电阻率、氧、碳含量,重金属杂质含量1〕:基磷电阻率和寿命:取300毫米长的多晶棒段H2气氛下区熔一次检测次数:¢≤40毫米硅棒:全检回收料不定期抽样、检验仪器:区熔炉,回探针电阻率测试仪,小数载流子寿命测试仪2〕基硼电阻率:从试验棒上取¢150×200 毫米,磷硼检验炉、次数:批量的20%检验仪器:区熔炉,回探针电阻率测试仪,3〕氧、碳含量—从多晶硅棒上取片检测、次数:按要求抽查、检验仪器:室温红外分光光度计4〕重金属杂质:硅芯表面金属杂质的含量,电感耦合等离子体质谱仪、次数:每年不小于45次5〕断面腐蚀:肉眼检测几何形状和表面状况、次数:100%〈二〉工艺技术规范 1:主要原辅材料1〕纯SIHCL3::Fe≤ 10ppb B≤ 0.03ppb p ≤ 0.1ppb Al≤ 10ppb 纯H2:露点≤ -50℃O2≤ 5ppm、2硅芯:直径¢7-8mm有效长度:2.800米、N型:电阻率50~250欧姆、弯曲度:≤3/10003〕:N2气:含N2 ≥ 99.99%,氧≤5PPm露点:≤-50℃、4〕:纯水:电阻率10≥欧姆, Al≤ 4ppm Cu≤ 0.8ppb Ca≤ 4ppb B≤ 0.3ppb P≤ 0.5ppb、5〕石墨:光谱纯,调密质,内部结构均匀,无空洞,灰分≤ 0.01%,比电阻≤20欧姆抗压强度≥900kg/cm2,假比重≥1.8g/Cm3、硬度45-80Kg/Cm2,加工成型后,经水浸泡,纯水煮至中性,干燥,高温煅烧后备用SIHCL3杂质分析中痕量硼的分析根据SIHCL3部分水解物能吸附杂质的实践经验,使SIHCL3中杂质硼被部分水解物吸附,而让基体SIHCL3自然挥发,用氢氟酸除去SIO2,加入三苯基-氯甲烷络合剂,使之生成稳定的络合物,它的分解温度为150℃,而SIHCL3的沸点为31.5℃,所以可以在较高的温度下挥发基体SIHCL3,残渣用ICP分析测定。
药物晶型常用的检测分析方法物质在结晶时由于受各种因素影响,使分子内或分子间键合方式发生改变,致使分子或原子在晶格空间排列不同,形成不同的晶体结构。
同一物质具有两种或两种以上的空间排列和晶胞参数,形成多种晶型的现象称为多晶现象(polymorphism)。
虽然在一定的温度和压力下,只有一种晶型在热力学上是稳定的,但由于从亚稳态转变为稳态的过程通常非常缓慢,因此许多结晶药物都存在多晶现象。
固体多晶型包括构象型多晶型、构型型多晶型、色多晶型和假多晶型。
物质在结晶时由于受各种因素影响,使分子内或分子间键合方式发生改变,致使分子或原子在晶格空间排列不同,形成不同的晶体结构。
同一物质具有两种或两种以上的空间排列和晶胞参数,形成多种晶型的现象称为多晶现象(polymorphism)。
虽然在一定的温度和压力下,只有一种晶型在热力学上是稳定的,但由于从亚稳态转变为稳态的过程通常非常缓慢,因此许多结晶药物都存在多晶现象。
固体多晶型包括构象型多晶型、构型型多晶型、色多晶型和假多晶型。
药物分子通常有不同的固体形态,包括盐类,多晶,共晶,无定形,水合物和溶剂合物;同一药物分子的不同晶型,在晶体结构,稳定性,可生产性和生物利用度等性质方面可能会有显著差异,从而直接影响药物的疗效以及可开发性。
如果没有很好的评估并选择最佳的药物晶型进行研发,可能会在临床后期发生晶型的变化,从而导致药物延期上市而蒙受巨大的经济损失,如果上市后因为晶型变化而导致药物被迫撤市,损失就更为惨重。
因此,药物晶型研究和药物固态研发在制药业具有举足轻重的意义。
由于药物晶型的重要性,美国药监局(FDA)和中国药监局(SFDA)在药物申报中对此提出了明确规定,要求对药物多晶型现象进行研究并提供相应数据。
正因如此,任何一个新药的研发,都要进行全面系统的多晶型筛选,找到尽可能多的晶型,然后使用各种固态方法对这些晶型进行深入研究,从而找到最适合开发的晶型;选定最佳晶型后,下一步就是开发能始终如一生产该晶型的化学工艺;最后一步是根据制剂对原料药固态性质的要求,对结晶工艺进行优化和控制,确定生产具有这些固态性质的最佳工艺参数,从而保证生产得到的晶型具有理想的物理性质,比如晶体表象,粒径分布,比表面积等。
药物制剂开发中对晶型的考虑随着药物开发研究的深入,难溶性药物在新药中的比例不断的增大,晶型问题越来越被重视。
不同晶型会影响药物在体内的溶出、吸收,进而影响药物的临床疗效和安全性,特别是一些难溶性口服固体和半固体制剂。
因此,对于多晶型药物,在研制成固体和半固体口服制剂时,对晶型进行研究有利于开发一种在临床治疗上有意义且稳定可控的晶型。
目前,国内对晶型问题也愈来愈重视,2015版药典附录新增“9105 多晶型药品的质量控制技术与方法指导原则”指出: 固体药物及其制剂中存在多晶型现象时,应使用“优势药物晶型物质状态”作为药物原料及其制剂晶型,以保证药品临床有效性、安全性与质量可控性。
下面就药物审评工作中遇到的晶型问题,以及研究开发这类多晶型药物所需注意的问题谈一些看法。
1、什么是药物的多晶型现象?固体物质根据其组成分子、原子、离子在三维空间的排列方式可以分为晶型(包括假晶型)和非晶体型。
晶体型的组成单元在三维空间排列固定有序,非晶型则相反。
所有晶型可以归纳为三斜晶系、单斜晶系、正交晶系、四方晶系、立方晶系、三方晶系、六角晶系共七个晶系14种晶格(如右图)。
有机药物的晶体大多为分子晶体,当药物分子中存在溶剂或分子时,因为药物分子易与溶剂或水分子形成氢键,药物分子与不同的溶剂分子结合,就会形成不同的晶型;不含溶剂的药物也可能由于分子的对称排列规律不同而存在的多晶现象,如药物结晶时的溶解、温度、湿度等,制剂过程中粉碎、混悬、压片等都会影响药物的晶型。
2、多晶现象会如何影响药物性质?众所周知,结构决定功能,同一药物的不同晶型在外观、溶解度、熔点、溶出度、生物有效性等方面都可能会有显著不同,从而影响了药物的稳定性、生物利用度及疗效,该种现象在口服固体制剂方面表现得尤为明显。
1)对药物理化性质及工业制剂的影响多晶型固体药物每个晶型有不同的表面自由能,而表面自由能大小是影响其溶出度的因素之一,像亚稳态的非极性表面自由能与稳态晶型基本相同,但极性表面自由能大于稳态的,因而总的表面自由能较大,更易被水润湿,在固体制剂崩解后形成的混悬液中,由于亚稳态粒子表面易水化,较厚的水化膜的反絮凝作用优于稳态晶型物,因而亚稳态的晶体粒子更易分散,顾有高的溶出度,如无味氯霉素共有A、B、C 3种晶型及无定型,我国1975年以前生产的无味氯霉素原料片剂、胶囊都为无效的A 型,后经改进生产工艺才生产出有生物活性的B型,并在质量标准中增加了非活性晶型的限度。
9种最常见的药物晶型检测方法药物分子通常有不同的固体形态,包括盐类,多晶,共晶,无定形,水合物和溶剂合物;同一药物分子的不同晶型,在晶体结构,稳定性,可生产性和生物利用度等性质方面可能会有显著差异,从而直接影响药物的疗效以及可开发性。
如果没有很好的评估并选择最佳的药物晶型进行研发,可能会在临床后期发生晶型的变化,从而导致药物延期上市而蒙受经济损失,如果上市后因为晶型变化而导致药物被迫撤市,损失就很严重。
因此,药物晶型研究和药物固态研发在制药业具有举足轻重的意义。
一、X-射线衍射法(X-ray diffraction)X-射线衍射是研究药物晶型的主要手段,该方法可用于区别晶态和非晶态,鉴别晶体的品种,区别混合物和化合物,测定药物晶型结构,测定晶胞参数(如原子间的距离、环平面的距离、双面夹角等),还可用于不同晶型的比较。
X-射线衍射法又分为粉末衍射和单晶衍射两种,前者主要用于结晶物质的鉴别及纯度检查,后者主要用于分子量和晶体结构的测定。
1、单晶衍射单晶衍射是国际上公认的确证多晶型的最可靠方法,利用该方法可获得对晶体的各晶胞参数,进而确定结晶构型和分子排列,达到对晶型的深度认知。
而且该方法还可用于结晶水/溶剂的测定,以及对成盐药物碱基、酸根间成键关系的确认。
然而,由于较难得到足够大小和纯度的单晶,因此该方法在实际操作中存在一定困难。
2、粉末衍射粉末衍射是研究药物多晶型的最常用的方法。
粉末法研究的对象不是单晶体,而是众多取向随机的小晶体的总和。
每一种晶体的粉末X-射线衍射图谱就如同人的指纹,利用该方法所测得的每一种晶体的衍射线强度和分布都有着特殊的规律,以此利用所测得的图谱,可获得出晶型变化、结晶度、晶构状态、是否有混晶等信息。
该方法不必制备单晶,使得实验过程更为简便,但在应用该方法时,应注意粉末的细度,而且在制备样品时需特别注意研磨过筛时不可发生晶型的转变。
二、红外吸收光谱法不同晶型药物分子中的某些化学键键长、键角会有所不同,致使其振动-转动跃迁能级不同,与其相应的红外光谱的某些主要特征如吸收带频率、峰形、峰位、峰强度等也会出现差异,因此红外光谱可用于药物多晶型研究。
多晶型及其检测
多晶型是某些化合物所具有的性能,能够以两种或多种结构不同的形式结晶。
能以不同晶形存在的物质称为多晶体。
不同晶形的物质具有非常不同的物理、化学或生物性能,有时在药物至食品的一系列行业中具有重要的价值。
如上所述,物质的各个多晶体形态在主要的物理性能方面各不相同,例如熔融温度、颜色、溶解性、折光指数、硬度或导电导热性。
然而,不同的晶形在熔融时得到同一个液相。
多晶体为同素异形现象,例如表现为硫、碳(石墨、钻石)、磷和大量矿物和有机化合物。
聚合物也会以多晶体形式出现,例如全同立构聚丙烯或聚四氟乙烯。
药物多晶体[1]具有极大的实际重要性。
由于各个多晶体的溶解性和分解性能常常非常不同,所以在人体中的再吸收和生物药效率[2]也不同。
因此,治疗功效取决于存在的晶形,例如亚稳晶形的活性可能是稳定晶形的两倍。
多晶型不仅对药理功效很重要,而且甚至在多晶型物质的生产(结晶和干燥条件)、加工和成型(黏性、流动性外观)的开始阶段也很重要。
各个晶形在特定温度范围内是稳定的,可用希腊字母(α、β)或罗马数字(I、II、III)表示。
此外,亚稳态晶形也能存在,为了有所区别,例如表示为α'。
它们向稳定形的逐渐转变需要若干个小时,有时甚至几年。
实践中,将多晶体类型区分为互变形和单变形两类:
y可逆的固-固转变称为互变形,见图13.1左。
由低温α形态向高温β形态的α→β转变是吸热的,因而α形态的熔融焓高于β形态的熔融焓[3]。
当然,α形态的熔点只有当α→β转变很慢时才能观察到;
y放热的亚稳态变为稳定态的固 固转变称为单变形,因为转变只向单方向进行并不可逆转,见图13.1右。
较低温度熔融的β'晶形的熔融焓通常小于较高温度熔融的稳定态β的熔融焓
[1]。
熔融热之差等于单向转变焓。
根据Ostwald规则,较不稳定的晶形常常在由熔体冷却时先
结晶,然后逐步转变到更稳定的形态。
该过程称为Ostwald熟化。
在DSC中,通过慢慢加热无定形物质(由熔体骤冷得到)至玻璃化转变温度以上,常常可得到亚稳态晶形[4]。
也可从特定溶剂的溶液结晶得到亚稳态晶形。
许多化合物在由溶液结晶析出时形成具有不同物理性能的溶剂化物。
如果溶剂是水,则称为水合物。
如果将这种溶剂化物在气密耐压的容器内加热,则在比非溶剂化的化合物低得多的温度下熔融。
不同的熔融温度与多晶型化合物类似,因而其行为常称为假多晶型[2]。
有些由拉伸的具有偶极性能的分子组成的有机化合物,在单次转变中不能达到各向同性的液体聚集态。
在熔融过程以上温度出现称为中间相的特殊液相。
该相为存在于晶相和各向同性液相间的液晶相。
继续加热时形成各向同性的液相。
还有加热时首先形成塑性晶态的类似球体的分子,继续加热时得到各向同性的熔体。
trs fus II
fus II fus I
Enantiotropic Solid-Solid Transition
Monotropic Solid-Solid Transition 图13.1. 互变相图(左)和单变相图
(右)的示意图。
由不同相的蒸气压与温度的关系表示。
亚稳态晶型β’的熔点常常仅在快速加热时达到,否则由单向转变形成稳定晶型β。
多晶体的检测
可对一个化合物的各个多晶型的不同物理性能进行表征。
以下为检测多晶体的常用方法: y DSC :所有的一级相变均涉及焓变。
自由焓函数可由cp 温度函数计算得到;
y 热学-光学分析[1]:试样置于交叉的偏振滤光片之间进行观察。
试样在温度程序控制下,通过双折射的变化来观察各个相转变,见图13.2。
参阅第十七章“热光分析法”;
图13.2. 磺胺嘧啶的两个多晶型在偏振光中呈现不同的双折射。
y 溶解性研究;
y X-光方法,例如Debye-Scherrer 粉末衍射测量;
y 红外光谱法。
磺胺嘧啶多晶体的DSC检测
先将试样加热至熔融(图13.3中的第一次升温),然后用自动进样器将熔融试样热坩埚放到室温25°C的金属转盘上使试样骤冷,形成无定形玻璃态。
第二次升温测量(图13.3中的红色曲线)表明,玻璃化转变后跟随着放热的冷结晶。
所形成的亚稳相经过放热的单向固-固转变变为更稳定的晶形,而后该晶形于179°C熔融。
最后,由熔体结晶为稳定相并于190°C熔融。
图13.3. 磺胺嘧啶的DSC测量曲线。
以5K/min升温速率进行的第一次测量(黑色曲线),稳定晶形的熔融温度约为190°C。
下面的红色曲线为第二次升温测量,单向固-固转变发生在约125°C。
参考文献
[1] METTLER TOLEDO Data Sheet: “The DSC-Microscopy System”.
[2] J. L. Ford and P. Timmins, Pharmaceutical Thermal Analysis, Ellis Horwood, 1989.
[3] D. Giron, J. Pharmaceutical & Biomedical Analysis, Vol. 4, No. 6, 755–770, 1986.
[4] R. Hilfiker, Polymorphism in the Pharmaceutical Industry, Wiley-VCH, 2006.
[5] G. Widmann, Thermochimica Acta, 112 (1987), 137–140.
[6]《热分析应用手册系列丛书》: 《药物》, (东华大学出版社), 计划中.。