眼科遗传病基因检测简介
- 格式:pptx
- 大小:3.31 MB
- 文档页数:20





基因检测案例14|Leber先天性黑朦疾病简介Leber先天性黑朦(Leber congenital amaurosis LCA)是一种导致儿童双眼先天盲的遗传性视网膜疾病,占儿童先天性双眼盲的10-20%,占遗传性视网膜变性类疾病的5%。
该病呈常染色体隐性遗传,目前已知有20多个致病基因,临床上以眼球震颤、固视障碍、畏光、指压眼球为特征。
眼底检查早期多为正常,随着病变进行性进展,数年后可见眼底椒盐样色素沉着、骨细胞样色素、视网膜血管狭窄、广泛视网膜色素上皮和脉络膜萎缩。
视网膜电图表现为a、b波平坦,甚至消失。
眼底检查眼底表现多样,不同基因引起的表现也不尽相同。
但以视网膜污秽、色素增生和脱失、黄斑萎缩及缺损多见。
大致可分为:①黄斑区正常,根据是否伴有视网膜色素沉着,又可分为两类:(a)视网膜无色素沉着;(b)视网膜散在色素沉着,视网膜色素可表现为骨细胞样、团块状、点状及椒盐样,颜色可为黑褐色、白色及黄色等,可伴有视网膜色泽灰暗、血管变细。
②黄斑萎缩伴有视网膜色素沉着:黄斑萎缩可呈金箔样反光、色素变动、沉着,视网膜色素可以表现为骨细胞样、团块状、点状及椒盐样,颜色可为黑褐色、白色及黄色等。
③黄斑缺损样改变:黄斑区边界清楚的视网膜、脉络膜组织萎缩缺损,透见脉络膜大血管甚至巩膜,可伴有不同类型的色素沉着、血管变细等改变。
基因治疗目前针对LCA2 RPE65 突变已经有美国FDA批准上市的视网膜下腔注射Luxturna (载体为腺相关病毒)。
针对其他基因突变所致的Leber先天性黑朦的临床试验也有一些处于开展中。
以下是关于Leber先天性黑朦的相关基因治疗的临床研究进展。
案例分享临床症状先证者,男,32岁,临床诊断为双眼LCA,先证者亲属状况不明。
检测项目眼科遗传病基因检测(441个基因)。
检测方法从受检者外周血中提取基因组DNA,构建基因组文库,通过探针杂交捕获相关的目的基因外显子及相邻部分内含子区域,进行富集。

眼科遗传疾病的分子遗传学分析眼科遗传病是指由基因突变引起的眼部疾病,具有遗传性和多样性。
随着分子遗传学研究的深入,越来越多的眼科遗传疾病的病因和发病机制被揭示。
本文将通过病例分析、分子遗传学进展、诊断与治疗等方面,深入探讨眼科遗传疾病的分子遗传学分析的重要性和应用价值。
以先天性视网膜劈裂症为例,该病是一种少见的眼科遗传疾病,主要表现为视网膜劈裂、囊泡形成和视力严重受损。
通过分子遗传学研究,发现该病是由MYO7A基因突变引起的一种常染色体隐性遗传病。
MYO7A基因编码肌球蛋白7A,在视网膜色素上皮细胞中表达,参与视细胞和视网膜色素上皮细胞之间的信号传递。
基因突变会导致肌球蛋白7A功能障碍,进而引发视网膜劈裂症。
随着分子遗传学的发展,基因组学、蛋白质组学、代谢组学等技术广泛应用于眼科遗传疾病的研究。
基因组学研究可以帮助我们发现新的致病基因及其作用机制;蛋白质组学研究可以揭示蛋白质表达谱的变化和相互作用网络;代谢组学研究则可以揭示疾病发生过程中代谢产物的变化。
这些研究方法不仅为我们提供了更多的疾病诊疗靶点,也为眼科遗传疾病的精准治疗提供了可能。
分子遗传学的发展为眼科遗传疾病的诊断和治疗提供了新的思路和方法。
基因检测可以用于疾病的诊断和分类,帮助医生制定个性化的治疗方案。
针对基因突变的治疗方法,如基因替代疗法、基因矫正疗法等,已经在部分眼科遗传疾病中取得了显著的效果。
蛋白质组学和代谢组学的研究也为药物研发提供了新的靶点,有助于开发更有效的治疗策略。
在物理治疗方面,如针对先天性视网膜劈裂症,可以采用光凝术、玻璃体切除术等方法来减轻症状。
心理干预在眼科遗传疾病的治疗中也起着重要的作用,可以帮助患者及其家庭成员应对疾病带来的心理压力。
分子遗传学在眼科遗传疾病的诊断和治疗中发挥着越来越重要的作用。
通过对基因突变、基因表达和调控、蛋白质组学和代谢组学等方面的研究,我们可以更深入地了解眼科遗传疾病的发病机制,为患者提供更为精准的治疗方案。

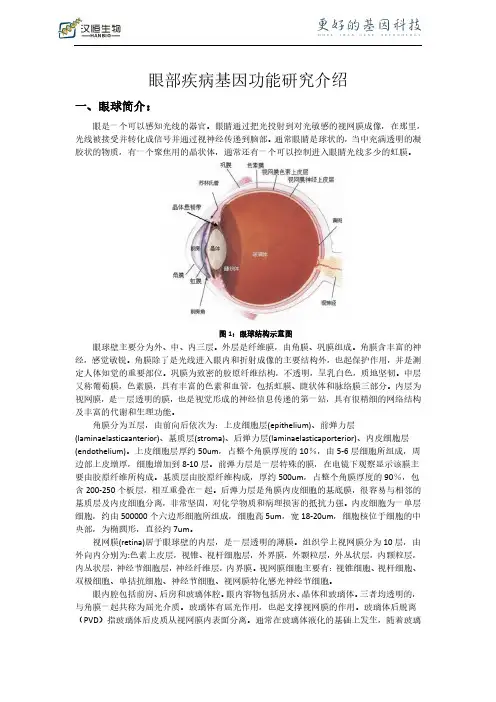
眼部疾病基因功能研究介绍一、眼球简介:眼是一个可以感知光线的器官。
眼睛通过把光投射到对光敏感的视网膜成像,在那里,光线被接受并转化成信号并通过视神经传递到脑部。
通常眼睛是球状的,当中充满透明的凝胶状的物质,有一个聚焦用的晶状体,通常还有一个可以控制进入眼睛光线多少的虹膜。
图1:眼球结构示意图眼球壁主要分为外、中、内三层。
外层是纤维膜,由角膜、巩膜组成。
角膜含丰富的神经,感觉敏锐。
角膜除了是光线进入眼内和折射成像的主要结构外,也起保护作用,并是测定人体知觉的重要部位。
巩膜为致密的胶原纤维结构,不透明,呈乳白色,质地坚韧。
中层又称葡萄膜,色素膜,具有丰富的色素和血管,包括虹膜、睫状体和脉络膜三部分。
内层为视网膜,是一层透明的膜,也是视觉形成的神经信息传递的第一站,具有很精细的网络结构及丰富的代谢和生理功能。
角膜分为五层,由前向后依次为:上皮细胞层(epithelium)、前弹力层(laminaelasticaanterior)、基质层(stroma)、后弹力层(laminaelasticaporterior)、内皮细胞层(endothelium)。
上皮细胞层厚约50um,占整个角膜厚度的10%,由5-6层细胞所组成,周边部上皮增厚,细胞增加到8-10层。
前弹力层是一层特殊的膜,在电镜下观察显示该膜主要由胶原纤维所构成。
基质层由胶原纤维构成,厚约500um,占整个角膜厚度的90%,包含200-250个板层,相互重叠在一起。
后弹力层是角膜内皮细胞的基底膜,很容易与相邻的基质层及内皮细胞分离,非常坚固,对化学物质和病理损害的抵抗力强。
内皮细胞为一单层细胞,约由500000个六边形细胞所组成,细胞高5um,宽18-20um,细胞核位于细胞的中央部,为椭圆形,直径约7um。
视网膜(retina)居于眼球壁的内层,是一层透明的薄膜。
组织学上视网膜分为10层,由外向内分别为:色素上皮层,视锥、视杆细胞层,外界膜,外颗粒层,外丛状层,内颗粒层,内丛状层,神经节细胞层,神经纤维层,内界膜。

案例分享基因辅助诊断眼科罕见病——Leber遗传性视神经病变Leber遗传性视神经病变Leber遗传性视神经病变(Leber hereditary optic neuropathy,LHON)是⼀种由线粒体DNA突变所引起的母系遗传性视神经萎缩。
好发于青年男性。
主要临床表现为双眼先后发⽣的⽆痛性视⼒突然急剧下降,疾病早期视盘充⾎、⽑细⾎管扩张,进展⾄后期视神经萎缩。
病因和流⾏病学LHON是线粒体DNA突变所导致的疾病,位于线粒体基因组上的第11778、第14484和第3460位点为该病的3个原发位点,90%以上的LHON病⼈中能够发现G11778A、G3460A、T14484C三个突变中的⼀个,尤以G11778A最多,占50%~80%。
上述突变可影响线粒体呼吸链复合体Ⅰ的正常功能,导致线粒体ATP合成减少及活性氧(reactive oxygen species,ROS)产⽣增多,引起神经节细胞的凋亡。
除上述原发位点,⾄今已发现50多个线粒体DNA上的继发突变位点,与疾病的外显率、病情严重程度等相关。
除线粒体DNA突变外,核基因突变、环境因素和线粒体单倍型均可能参与疾病的发⽣发展。
LHON是最常见的线粒体病,东北英格兰地区的患病率为1/27 000,⽽在⼀个以欧洲⼈群为基础的荟萃分析中,该病的患病率为1/45 000。
在我国邢台地区的⼀项以医院为基础的流⾏病学研究中,粗略估计邢台地区LHON患病率下限为1.092/100 000。
该病存在外显不全的特点,线粒体基因突变携带者可能不发病。
LHON男⼥患者⽐例⼤致为5:1,发病年龄通常为15~35岁。
临床表现主要症状为双眼先后发⽣的⽆痛性视⼒突然急剧下降,双眼发作间隔⼀般为数周到数⽉。
眼底检查在疾病早期表现为视盘充⾎⽔肿、视盘旁⽑细⾎管迂曲扩张,疾病后期视盘⽔肿和⽑细⾎管扩张消退,最终视盘颞侧或全部呈萎缩性改变。
多数LHON病⼈只存在眼部表现,少数可合并全⾝其他系统症状如智⼒障碍、癫痫、听⼒障碍、肌张⼒障碍等。

遗传性眼底病的基因治疗遗传性眼底病的基因治疗在美国开展的比较早,庞继景教授在美期间是基因治疗实验的全程参与者。
如今,庞继景教授回国加盟厦门眼科中心,开设遗传性眼病咨询门诊,为国内遗传性眼病患者提供专业基因诊疗服务。
因遗传因素而罹患的眼病称为遗传性眼病,在庞继景教授看来,遗传性眼病有四大难点,一是种类繁多,目前已知的遗传性眼病有700种以上,二是有失明危险,遗传性眼病对眼的结构和功能危害严重,有些会造成先天失明,或随着病情发展,视力逐渐遭到破坏,最终导致失明。
三是目前绝大多数通过传统的方法很难治愈。
四是,还会遗传给下一代。
庞继景教授开着遗传性眼病咨询门诊的初衷在于提供临床及遗传咨询服务,以患者的需求为出发点,融遗传检测、诊断、咨询及疾病管理、预防于一体。
为将来国内外合作开展基因治疗临床试验或商业治疗尽快做好准备。
基因治疗的前提是基因检测,通过基因检测查出致病基因。
基因疗法原理通过一个载体把正常的相应基因转入到病变细胞内,弥补遗传病的基因缺陷,或者纠正/修复相应的突变基因,从而达到减轻或治愈疾病的目的。
基因疗法优势基因疗法在视网膜遗传病治疗上独具优势,目前大多数具有隐性遗传的视网膜变性类疾病,特别是涉及基因较小的疾病,基本可以通过基因替代疗法得到治疗,基因疗法为低视力患者提供一个新的治疗手段。
基因诊疗越早越好基因诊疗强调一个早字,某些视网膜遗传病如果没有进行早期治疗,可以造成视细胞的死亡,对基因治疗的预后产生不良影响。
除了基因检测以外,另一个备受瞩目的是遗传咨询。
“我有视网膜色素变性,我的孩子会得这种病吗?”,庞继景教授称,通过对病人家庭调查、实验室检查及结合临床特点,对疾病做出正确诊断,判断其是否为遗传病,判明是新的突变还是遗传。
其次确定遗传方式,是患者进行孕前专业遗传咨询的前提和基础。
通过孕前/产前相应专业遗传咨询/检测,可以最大程度地避免某些遗传病的发生,也是优生优育的重要环节。
2017年,已有700多个导致眼病的突变基因以及近300个导致眼底病的突变基因被发现。

基因检测案例15|结晶样视网膜色素变性疾病简介结晶样视网膜变性(Bietti crystalline corneoretinal dystrophy,BCD)是一种相对罕见的视网膜变性,1937年由Bietti首次报道。
典型改变为黄白色闪光结晶样物质沉积于视网膜,伴有视网膜色素上皮和脉络膜萎缩,部分患者近角膜缘部角膜基质浅层也可见到沉积的结晶。
世界范围内BCD并非常见,患者以中国和日本比较多见。
遗传方式常染色体隐性遗传临床表现症状:多数患者20-40岁发病,双眼受累。
临床表现为进行性视力下降,或夜盲,或两者兼有。
亦有无自觉症状,因眼底检查才被发现者。
色觉早期正常,晚期可有红绿色盲或全色盲。
临床上主要分为局限性或弥漫性两种类型:局限性:视网膜病变主要局限于后极部,病人常表现为旁中心暗点;弥漫性:表现为广泛性RPE病变和色素沉积,病人常表现为周边视野缺损、中心视力下降和ERG改变。
裂隙灯显微镜检查:少数(约三分之一)病例在角膜缘浅层基质内可见到结晶样、黄白色、细小点状沉着物。
眼底检查:早期:视网膜后极部散布有黄色、结晶样、圆形细小或不规则形较大的闪辉亮点。
多数结晶样病变位于视网膜RPE细胞层,部分病变位于神经视网膜内。
进展期:随着病情进展,视网膜色素上皮和脉络膜毛细血管逐渐萎缩,此期萎缩区外可见骨细胞样或不规则形色素沉着,萎缩区内结晶样物质很少存在,可见暴露的脉络膜大血管。
晚期:广泛的视网膜色素上皮和脉络膜毛细血管萎缩,偶见结晶样沉积。
视乳头和视网膜血管早期正常,晚期出现视神经萎缩和视网膜动脉变细。
Yuzawa et al.将BCD临床分为3期:第一期:后极部RPE细胞萎缩,伴细小、均匀、白色的结晶沉积;第二期:RPE萎缩区扩大,后极部RPE萎缩区内可见脉络膜毛细血管萎缩;第三期:RPE 和脉络膜毛细血管广泛萎缩。
辅助检查色觉:病变累及黄斑区,可伴有色觉异常。
视野:萎缩区范围内视野缺损。
局灶性萎缩多表现为中心暗点,弥漫性萎缩以周边视野缺损为主。
中华实验眼科杂志2018 年 7 月第36 卷第7 期Chi: JExp Ophthalmo&July 2018,V〇1.36,N〇.7! 481 !•专家建议与推荐•眼遗传病基因诊断方法专家共识中国眼遗传病诊疗小组中国眼科遗传联盟通信作者:睢瑞芳,Email:hrfsui@163. comDO I: 10. 3760/cm a. j. issn. 2095-0160. 2018. 07. 001基金项目:中国医学科学院医学与健康科技创新工程经费资助项目(2016-I2M-1-002 );国家重点研发计划项目(2016YFC0901500)眼遗传病是一组由于基因缺陷导致的眼部疾病。
2018年4月27日在人类孟德尔遗传在线数据库 (Online Mendelian Inheritance in Man,OMIM) (http:// W W /)中以“eye”作为关键词搜索疾病表型可得到440种疾病条目。
临床常见的眼遗传病有视网 膜变性、先天性青光眼、先天性白内障、遗传性视神经 病变、先天性眼外肌异常及累及眼部的一些综合征等。
遗传方式包括常染色体显性遗传、常染色体隐性遗传、X性连锁遗传、双基因遗传及线粒体遗传等。
此外,眼遗传病同样存在着等位基因异质性和基因座异质性。
同时,不同地域、不同民族之间的基因变异和表型均存 在着较大的差异,这些因素为眼遗传病的临床诊断和 分子检测带来了巨大的挑战。
随着人类基因组参考序 列的完成、基因芯片和高通量测序等技术的问世以及 生物信息技术对海量生物数据高效分析和处理技术的 发展,近几年来单基因遗传病的分子诊断效率迅速提 升,技术方法取得了很大的突破,为患者和临床医师的遗 传咨询提供了技术保障,更为将来的基因治疗奠定了基 础。
目前,在众多基因组技术中二代测序(next-/eneration sequencin/NGS)技术以其特有的优势在包括眼遗传病 在内的单基因遗传病的研究中发挥重要作用,并越来 越多地用于眼遗传病的分子检测。
视网膜色素变性RPGR基因ORF15检测基因检测过程中,数据解读是很重要的一个环节。
如果对疾病了解不清楚,或者对数据不敏感,很有可能会漏检。
后续我们会为大家介绍解读过程中遇到的各种坑儿。
今天为大家带来的是一个视网膜色素变性RPGR基因ORF15的检测案例。
1.视网膜色素变性简介视网膜色素变性(retinitis pigmentosa,RP)是一组以感光细胞和色素上皮细胞进行性选择性丧失为特点的遗传致盲眼病,世界范围内发病率为1/3000,是致盲的主要病因之一,目前无有效疗法。
该病早期症状为夜盲,随病程发展,中心视力逐渐减退,最终可完全失明。
RP可以在眼部单独存在,也可以是全身多个系统疾病或综合征的眼部组成部分。
RP可呈常染色体显性遗传、常染色体隐性遗传、X连锁遗传、线粒体遗传和双基因遗传。
在所有RP患者中,常染色体显性遗传占15-25%,常染色体隐性遗传占5-22%,X连锁遗传占5-15%。
2.RPGR是X连锁视网膜色素变性的主要致病基因,外显子ORF15具有复杂结构在各类遗传型的RP患者中,X连锁视网膜色素变性( X-linked retinitis pigmentosa,XLRP)临床表现最为严重,患者发病早,通常在20岁之前出现夜盲等视觉障碍,在40岁之前可进展为部分或完全失明。
RPGR是XLRP最重要的的致病基因,能解释XLRP 70%-75%的病因[参考文献1]。
RPGR又称为视网膜色素变性GTP酶调控因子(retinitis pigmentosa GTPase regulator),位于Xp21.1。
该基因有多种剪切体,最初发现的剪切体A含有19个外显子,编码815个氨基酸的蛋白。
检测该转录本上的变异,能够找到10-20%的XLRP 患者的致病变异。
后来剪切体C的发现是RPGR研究的一个重要的里程碑,该转录本含有15个外显子,编码1152个氨基酸。
该转录本含有一个新的外显子ORF15,它是由原来的Exon15及一部分Intron15序列组合构成。
基因检测助力遗传病诊断——Leber先天性黑矇Leber先天性黑矇(leber congenital amaurosis, LCA; OMIM 204000)是一种严重的遗传性致盲性视网膜营养不良,其特征为一出生或1岁以内即出现严重的视力受损、眼球震颤和严重的视网膜功能障碍。
LCA的患病率为2-3/100000活产婴儿,占遗传性视网膜变性疾病的5%和学龄儿童先天性双眼盲的20%。
1869年德国眼科医生Theodor Leber首次提出了LCA,并指出该疾病的特点为先天性盲合并眼球震颤及复杂多变的眼底表现。
该病的早期诊断存在一定困难, 容易漏诊误诊。
鉴别诊断包括色素性视网膜炎,Alström综合征,Joubert综合征,Stargardt病,Senior-Loken综合征,Conorenal综合征和婴儿神经元蜡样脂褐质沉着症(NCL)。
如NCL的ERG可以显示一个负波形,与神经认知衰退和癫痫有关。
Senior-Loken综合征即眼-肾综合征眼底及视功能损害与本病相似。
但前者有肾功能不全,不同于本病。
临床表现LCA的特点是出生后或出生后几个月出现严重的视力障碍,眼球运动或眼球震颤,瞳孔光反应差,眼指征(戳、揉和/或按压眼睛),全视野视网膜电图(ERG)检测不到或严重异常。
致病机制和原因LCA为常染色体隐性遗传,也有少数显性遗传的报道。
迄今为止,已报道编码视网膜特异性蛋白质的基因突变导致LCA,包括AIPL1,CRB1,CRX,GUCY2D,LRAT,RPE65,RPGRIP1,CEP290,RDH12,LCA5,TULP1,RD3,IMPDH1,SPATA7,NMNAT1,KCNJ13,PRPH2,IQCB1,OTX2和MERTK 。
这20个基因约占患病人群的70%,另外还有20%~30%的患者致病基因尚不清楚[1]。
这些基因功能涉及视网膜光信号向电信号的传导过程(GUCY2D)、视网膜内维生素A在光信号中的代谢循环(RPE65、LRAT、RDH12)、鸟嘌呤的合成(IMPDH1)、光感受器纤毛转运过程(CEP290、LCA5、RPGRIP1、TULP1)、锥细胞外节的吞噬作用(MERTK)、视网膜光感受器细胞的分化和发育(CRX、CRB1)、感光细胞内蛋白转运和正常分布(AIPL1、RPGRIP1)和鸟嘌呤合成受阻(IMPDH1)等[2]。
可编辑修改精选全文完整版眼科主要致盲、致畸疾病的分子遗传学研究进展一、Leber’s先天性黑矇的基因研究进展Leber’s先天性黑矇(Leber congenital amaurosis, LCA),是发生最早、最严重的遗传性视网膜病变, 出生时或出生后一年内双眼锥杆细胞功能完全丧失,导致婴幼儿先天性盲。
LCA占遗传性视网膜病变的5%以上,是导致儿童先天性盲的主要疾病(占10%-20%)。
多呈常染色体隐性遗传,临床上以眼球震颤、固视障碍、畏光、指压眼球为特征。
眼底检查早期多为正常,随着病变进行性进展,数年后可见眼底椒盐样色素沉着、骨细胞样色素、视网膜血管狭窄、广泛视网膜色素上皮和脉络膜萎缩。
视网膜电图表现为a、b波平坦,甚至消失。
可伴有圆锥角膜、远视、发育迟缓和神经系统异常等。
本病原因不明,亦无有效治疗手段。
近年来发现数种与LCA相关的致病基因,主要包括RPE65,GUCY2D, CRX, RPGRIP1,CRBI和AIPL1等。
表1 与Leber’s先天性黑矇相关的致病基因及位点位点基因编码蛋白1p31 RPE65 RPE651q31-q32.1 CRB1 CRB16q21.3 TULP1 Tubby-like protein 114q11 RPGRIP1 Retinitis pigmentosa GTPase regulatorinteracting protein17P13 RetGC1(GUCY2D) Photoreceptor specific uanylate cyclase17p13.1 AIPL1 Arylhydrocarbon-interacting protein-like-119q13.3 CRX Transcription factor二、视网膜色素变性的基因研究进展视网膜色素变性(retinitis pigmentosa,RP)是视网膜感光细胞和色素上皮细胞功能受到严重损害的一种视网膜变性疾病,其发病率约为1/3500-4000,全球至少有150万人患有此病,且有逐年上升趋势,是目前主要的致盲性眼病之一。
如何解读眼科遗传病基因检测报告遗传性眼病不同于普通眼病,普通眼病可以通过药物或者手术控制甚至治愈,但是大部分的遗传性眼病目前没有有效的治疗药物,加之遗传性眼病危害的不是一个人,而是有血缘关系的整个家族,因此,对于遗传性眼病患者来说,遗传性眼病基因检测就显得非常重要。
通过基因检测,可以帮助遗传性眼病患者明确致病基因,通过遗传阻断从家族中排除致病基因,让家族摆脱遗传性眼病的困扰。
针对很多患者反映,拿到检测报告看不懂的情况,本期给大家做一期详细的报告解读。
遗传性眼病基因检测报告分为基因检测报告和遗传学分析报告两大部分,其中第一部分基因检测报告包括样本信息、检测信息、检测结果、家系图及家族验证;遗传学分析报告包括基因检测结果及相应的遗传解析,另附有疾病说明、参考文献、方法说明等。
倘若检测到临床意义不明确但是有可能致病的变异就会在临床意义未名的变异附表呈现,遗传解析同样紧跟其后。
最后是本项目检测的基因列表。
跟大家一起解读这份报告样本信息:即先证者信息(先证者即第一个来就诊的患者),家族史是鉴别诊断遗传性眼病患者重要指标,通过家族史问诊帮助医生初步判断。
从检者一般是患者的直系亲属,患者检测结果结合从检者检测结果可以判断突变基因的父母来源,同时可以验证检测结果的准确性,提高检出率。
检测信息:包含检测项目和检测方法。
检测项目根据患者的临床症状选择相应的检测项目。
检测方法通常采用是靶向捕获。
检测结果:是报告的核心部分,包含检测到基因名称、参考序列、核苷酸改变、氨基酸改变、纯合还是杂合、致病性分析、遗传方式、变异来源。
其中遗传方式主要包括常染色显性遗传(AD)、常染色体隐性遗传(AR)、和X连锁的隐性遗传(XLAR)等。
找到突变基因(即核苷酸和氨基酸发生改变),分析该基因是否是患者的病因,还需要考虑突变基因的遗传方式和杂合纯合,比如,如果突变基因遗传方式是AD,那么杂合就能致病。
反之,如果突变基因遗传方式是AR,那么纯合才能致病。
遗传性视神经病变检测方案遗传性视神经病变是由核基因组或线粒体DNA突变导致视神经损伤的一大类疾病,可以独立存在,也可以是全身综合征的一部分,是儿童和青少年盲的一个重要原因。
按照遗传方式不同,遗传性视神经病变可分为四种:Leber遗传性视神经病(Leber’s hereditary optic neuropathy,LHON)、常染色体显性遗传视神经萎缩(Autosomal dominant optic atrophy,ADOA)、常染色体隐性遗传视神经萎缩(Autosomal recessive optic atrophy,AROA)和X连锁隐性遗传视神经萎缩(X-linked recessive optic atrophy,XROA),临床中以前两种最为常见,其中,LHON为线粒体遗传。
1.常染色体显性遗传性视神经萎缩(ADOA)特定的核基因突变可以导致遗传性视神经病变,称为常染色体遗传性视神经萎缩。
该病患者可仅表现为视神经萎缩,也可以伴发白内障、眼外肌麻痹、耳聋,周围神经病变、心脏病等,称为遗传性视神经萎缩附加症状。
通过连锁分析,目前已确定了8个视神经萎缩致病基因的染色体候选位点,其中OPA1是最早克隆的基因(表1)。
另外两个已克隆的基因为OPA3和TMEM126A。
多项研究表明,75%以上的常染色体显性遗传性视神经萎缩(ADOA)是由OPA1基因突变所致。
OPA1基因在人体各组织广泛表达,在人视网膜、脑组织、心脏、前列腺、肌肉组织表达最高。
OPA1基因编码蛋白是一种发动蛋白相关GTP酶,含有5个功能域:N端线粒体导向序列,N端盘曲螺旋样结构域,GTP酶结构域,中心发动蛋白结构域和C端盘曲螺旋结构域。
OPA1定位于线粒体内膜,主要参与调节线粒体内膜融合、维持线粒体嵴网状结构。
表1 常染色体遗传性视神经萎缩致病基因染色体位点和基因及其突变导致的临床表现围神经病变、多发性硬化OPA2 chrXp11.4-p11.21 致病基因未确定X-Linked -OPA3 chr19q13.2-q13.3 OPA3AD或AR 白内障、周围神经病变OPA4 chr18q12.2-q12.3 致病基因未确定AD -OPA5 chr22q12.1-q13.1 致病基因未确定AD -OPA6 chr8q21-q22 致病基因未确定AR - OPA7 chr11q14.1-q21TMEM126A AR 耳聋、心脏病OPA8 chr16q21-q22 致病基因未确定AD 耳聋、心脏病注:AD:常染色体显性遗传;AR:常染色体隐性遗传;X-linked:X 连锁遗传。