Pichia pastoris
- 格式:pdf
- 大小:361.22 KB
- 文档页数:7



井冈山大学学报(自然科学版)34文章编号:1674-8085(2020)03-0034-05菊粉内切酶在毕赤酵母中的表达及酶学性质测定王淮1,罗文浩2,普元倩1,徐春萍1,*杨健康1(1.大理大学基础医学院,云南,大理671000;2. 中山大学光华口腔医学院,广东,广州510080)摘要:为了实现菊粉内切酶在毕赤酵母(Pichia pastoris)中的高活性表达并对表达的菊粉内切酶酶学性质、水解菊粉的产物进行分析。
通过下载无花果曲霉菊粉内切酶基因的编码序列,做了密码子优化后进行全基因合成,然后在毕赤酵母GS115中表达。
对表达的菊粉内切酶的酶活、最适反应温度、最适pH值、热稳定性进行了分析,最后对酶水解菊粉的产物进行了高效液相色谱(HPLC)分析。
摇瓶表达的酶活力为420 U/mL,酶的最适反应温度为55 ℃,最适反应pH为6.0。
HPLC分析表明:表达的菊粉内切酶酶解底物菊粉的主要产物为低聚果糖,聚合度为3~5之间。
构建的工程酵母可以高效表达菊粉内切酶,可以用于生产低聚果糖。
关键词:毕赤酵母;菊粉内切酶;表达;酶学性质;水解产物中图分类号:Q786 文献标识码:A DOI:10.3969/j.issn.1674-8085.2020.03.007 EXPRESSION OF ENDO INULINASE IN PICHIA PASTORIS AND DETERMINATION OF ITS ENZYMATIC PROPERTIESWANG Huai1, LUO Wen-hao2, PU Yuan-qian1, XU Chun-ping1, *YANG Jian-kang1(1.School of Basic Medical Sciences, Dali University, Dali, Y unnan 671000, China;2. Guanghua School of Stomatology, Sun Yat-sen University, Guangzhou, Guangdong 510080, China)Abstract: To achieve high activity expression of endo inulinase in Pichia pastoris, and to analyze the enzymatic properties and the products of hydrolyzed inulin, we downloaded the coding sequence of the endo inulinase gene of Aspergillus ficuum, performed the whole gene synthesis after codon optimization, and expressed in P. pastoris GS115. The enzymatic activity, optimum reaction temperature, optimum pH value and thermal stability were analyzed. Finally, the products of inulin hydrolysis were analyzed by high performance liquid chromatography (HPLC). The enzymatic activity expressed in shake flask was 420 U/ml. The optimum reaction temperature of the enzyme was 55 ℃, and the optimum pH was 6.0. HPLC analysis showed that the main products of inulin hydrolysis by endo inulinase were oligofructoses with a degree of polymerization of 2~5. The constructed genetically engineered yeast can efficiently express endo inulinase and be used to produce oligofructose.Key words:Pichia pastoris; endo inulinase; expression; enzymatic properties; hydrolysate1998年Uhm等人从无花果曲霉(Aspergillus ficuum)中分离到了一种新的菊粉内切酶基因,命名为inu2[1]。



.2 巴斯德毕赤酵母表达外源蛋白的降解机理及其控制策略1.2.1 巴斯德毕赤酵母表达外源蛋白的降解机理在外源蛋白的表达过程中,宿主菌毕赤酵母的胞内和胞外均有一定量的蛋白酶的表达,因此,不论是胞内表达亦或是分泌表达,大多数外源蛋白均面临着被降解的问题,这也是影响表达量的一个重要因素,同时,还增加了纯化目的蛋白的难度。
近年来,蛋白酶的研究是P.pastoris表达系统一个重点和热点。
越来越多的蛋白酶的遗传背景和生理生化性质得到深入的研究[52; 53]。
P.pastoris能根据细胞生长环境(碳源的改变以及细胞或细胞器的胁迫)来调整自身酶系,以合成与降解不同的蛋白和细胞器,液泡是蛋白质降解最主要的场所[54],另一降解场所是细胞基质蛋白酶体中。
但是,对于外源蛋白来说,其降解常在表达和分离纯化的第一步,主要是由培养基中胞外蛋白酶,细胞外膜结合蛋白酶(cell-bound proteases)[55]和细胞自噬或裂解释放的胞内蛋白酶降解的[5]。
胞内蛋白酶主要涉及降解蛋白质前体产生活性蛋白;切除转运出膜后的蛋白质信号肽;使调控蛋白失活;降解变异或不需要的蛋白质;提供营养,前体和能量。
胞外蛋白酶分泌较少,主要降解部分蛋白质提供氨基酸和多肽等营养[22]。
根据蛋白酶的分泌和作用地点,P.pastoris的胞内蛋白酶可以分为三种类别,即液泡蛋白酶(vacuolar proteases),细胞基质蛋白酶体(the cytosolic proteosome)以及分泌途径的蛋白酶(proteases located along the secretory pathway)[52; 56]。
表1.2 毕赤酵母液泡蛋白酶和分泌途径蛋白酶[57]Table 1.2 Proteases of Pichia pastoris vacuole and secretory pathway[57]Enzyme Type Gene Zymogen formVacuole PrA Aspartic PEP4 YesPrB Serine PRB1 YesCpY Serine PRC1 YesCpS Metallo-( Zn2+) CPS1 UnknownApI Metallo-( Zn2+) LAP4 YesApCo Metallo-(Co2+) DAP2 NoDPAP-B Serine SEC11 UnknownSecretory pathway signal peptidase Kex2protease Serine KEX2 YesKex1 carboxypeptidase Serine KEX1 NoDPAP-A Serine STE13 Unknown,predict noYeast aspartyl protease ш Aspartic YAP3 Unknown,predict yes酵母液泡位于基质中,一方面是维持胞内pH和盐离子平衡,储藏盐离子的功能;另一方面,由于液泡中含有大量的非特异性的水解酶,较宽底物范围的内生和异源蛋白酶,液泡是降解蛋白甚至细胞器的一个重要场所,大约80%的蛋白在液泡中降解[57]。


毕赤酵母热致死动力学模型的建立与检验田康明;段威;王正祥;路福平【摘要】毕赤酵母作为最成熟的蛋白质过量表达平台微生物之一,广泛应用于酶制剂、功能性大分子等大宗产品的发酵制造.建立毕赤酵母常规温度下(50~60,℃)的热致死动力学模型,有助于指导毕赤酵母发酵制造体系的菌体后处理工艺,增加菌体利用价值.为了建立毕赤酵母在50~60,℃条件下热致死动力学模型,将不同菌体量的毕赤酵母菌液在50、55、60,℃条件下进行不同时间热灭活处理,通过平板培养活细胞计数的方法评价不同处理方式下毕赤酵母的热致死效果,得到的致死曲线用Weibull模型和多元回归方程进行拟合构建热致死动力学模型,并对建立的模型进行数学检验和实用性检验.结果表明:建立的毕赤酵母热致死动力学模型可靠,具有实际使用价值.本研究所建立的模型能较好地模拟不同温度、不同处理时间对不同菌体量的毕赤酵母热致死效果的影响.并可以为毕赤酵母热灭活处理条件的确定提供计算依据.%Pichia pastoris,as one of the most mature protein overexpression platforms in the study of microorganisms,was widely used for bulk product manufacturing such as enzyme and functional macromolecule.Establishing a lethal thermal kinetics model of Pichiapastorisunder various heat treatment conditions from 50,℃ to 60,℃ will supply a guide to Pichia pastoris cell manufacturing system and increase the utilization value of cellmass.The lethal thermal kinetics of Pichiapas-torisunder various heat treatment conditions from 50,℃ to 60,℃ was investigated,and the availability and feasibility of the model was verified by live cell lethal tests.The cell culture of Pichiapastoris GS115 with different optical density was incu-bated at 50,℃,55,℃ and 60,℃ for variousheating time respectively,then the obtained lethal curves were fitted usingthe Weibull model and calculated using multiple regression equation to establish the lethal thermal kinetics model.Subsequently the feasibility and pratical lethality were confirmed by live cell lethal tests.APichiapastoris lethal thermal kinetics model was thus established,and its availability and feasibility were confirmed.The established model in this study can be used to describe the lethal thermal kinetics of Pichiapastorisunder various heat treatment conditions.The methods constructed here can be used for getting the suitable heat treatment process for heat inactivation of Pichiapastoris.【期刊名称】《天津科技大学学报》【年(卷),期】2017(032)002【总页数】6页(P24-29)【关键词】毕赤酵母;致死动力学;预测微生物学;Weibull模型【作者】田康明;段威;王正祥;路福平【作者单位】天津科技大学化工与材料学院,天津 300457;工业发酵微生物教育部重点实验室,天津科技大学生物工程学院,天津 300457;天津科技大学化工与材料学院,天津 300457;工业发酵微生物教育部重点实验室,天津科技大学生物工程学院,天津 300457【正文语种】中文【中图分类】Q939.97毕赤酵母(Pichia pastoris)表达体系具有表达率高、遗传稳定、产物可分泌、发酵工艺成熟等许多优点,这使得其成为应用最广泛的真核表达体系[1].从20世纪80年代初开始,人们已经用毕赤酵母表达体系成功表达疫苗、激素、抗菌肽、干扰素、工业酶、膜受体蛋白、细胞毒素及其衍生物等上千种蛋白,毕赤酵母生产体系在工业上已是仅次于大肠杆菌(E.coli)的最常用蛋白表达系统[2–3],所涉及到的领域有水产业[4]、饲料加工业[5]以及动物传染病的防治[6]等.以毕赤酵母表达生产工业酶为例,发酵生产过程完成后,需要对发酵液中的酶进行板框过滤除去菌体,陶瓷膜过滤除去细胞碎片和大分子蛋白等,并通过添加相应的辅助材料获得工业酶制剂产品.分离提取过程形成的菌体残渣通常通过烘干的方式制备成副产品用于饲料蛋白的添加.发酵结束后产品的分离提取过程及菌体残渣的烘干过程属于典型的开放式操作,很容易造成拥有自主知识产权的重组菌种流失.基因重组的毕赤酵母如果进入到环境中,可能污染人类赖以生存的自然环境,影响到人体健康和生态环境,还可能打破原有的生态平衡[7].因此,如何采用简单易行且安全可靠的方式在充分保证产品活力前提下彻底灭活提取液中的活细胞是工业酶生产中的关键环节,对于表达产物在胞内的菌液显得尤为重要.工业上对酵母发酵液灭活处理有酶法、超声波、机械研磨、高压匀浆等多种方法,但是热处理灭活却是最有效、最经济、最简单的方法.酵母对高温敏感[8],可以在对发酵产品影响较小的情况达到全部灭活的目的.通过热处理也有利于酵母营养物质的提取[9]和目标产物的分离.本文主要考察毕赤酵母在50~60,℃的致死规律,并建立相关模型,为毕赤酵母生产体系中建立有效的菌种热灭活方法提供技术参考.1.1 菌种与培养基毕赤酵母(Pichia pastoris)GS115购自Invitrogen公司,现保藏于本研究室.YPD培养基(g/L):酵母提取物 10,蛋白粉 20,葡萄糖20.1.2 样品处理取培养 24,h的毕赤酵母菌液,放入灭菌的离心管中,8,000,r/min离心5,min,弃上清液.再用YPD培养基将菌体重悬,配制成不同浓度的毕赤酵母菌液.1.3 热处理取10,mL不同浓度的毕赤酵母菌液,放入50,mL离心管中.将离心管于30,℃条件下维持5,min,然后分别放入50、55和60,℃的水浴摇床,转速为100,r/min,放入后开始计时,处理一定时间.处理结束后将处理液放入冰水中.1.4 微生物计数及致死效果的检测根据 GB/4789.15—2010方法,对热处理前后的菌液进行菌落计数[10].吸取1,mL加热处理前后的稀释菌液到灭菌的平皿中,再进行倾注,混匀后放置30,℃培养,48,h后统计单菌落数.致死效果用致死率(R)和残存对数(lg,S)表示.式中:N为热处理一段时间后剩余的活细胞数量;N0为初始的活细胞数量.1.5 热致死动力学模型1.5.1 一级模型将毕赤酵母残存对数为纵坐标,热处理时间为横坐标,绘制致死曲线,用 Weibull 模型进行拟合,Weibull模型为式中:t为热处理时间(min);参数 b为模型尺度因子;参数n为曲线形状因子.当n=l时,曲线呈线性趋势;n>l时,曲线呈上凸状,表示死亡速率持续增加;n<l时,曲线呈凹状,表示细菌死亡速率逐渐减小,存活的细菌对环境压力产生一定抗性[11].1.5.2 二级模型建立不同变量(温度和菌体量)的与一级失活模型参数(Weibull模型中的b和n)之间的多元回归方程,将响应值即参数b和n进行适当的对数或平方根变换.多元回归模型为式中:y为b或n的响应值;b0、bj、bjj、bjl为不同的常量系数;xj、xl为编码的变量温度和菌体量.1.6 实验数据分析和模型验证应用Sigmaplot 12.5软件对毕赤酵母在不同处理条件下的致死曲线数据进行分析和模型拟合.采用参数 R2、Af、Bf、RMSE、SSE评价模型拟合度[12].当R2越接近1时,表示相关的预测模型参考价值越高;Af为准确因子,反映了预测值和实际值的偏离程度,Af值越小,表明模型预测值与真实值越接近,模型的精确程度越高;Bf为偏差因子,Bf越接近 1,模型拟合度越高;均方根误差(RMSE)可作为衡量预测准确度指标,可以反映模型预测的离散程度;误差平方和SSE值越小,模型精确度越高.式中:SSE为误差平方和;SST为总离差平方和;µ0为实验观测值;µ为预测值;n为观测值个数.2.1 温度、作用时间及不同菌体量下的毕赤酵母致死规律毕赤酵母对高温敏感,在32,℃以上生长则不利于蛋白的表达,一般在45,℃时就停止生命活动,致死温度一般在 50~60,℃[13].且温度越高致死速率越快.菌体量对致死速率同样有着影响,单位体积的细胞越多,致死所需要的热量就越多,且菌液的热传递效果越差,这样同样条件下的致死率越低.以热处理时间为横坐标,毕赤酵母残存对数为纵坐标,绘制特定温度和特定菌体量(吸光度)的致死曲线,如图1 所示.毕赤酵母残存的细胞量的对数随着热处理时间的延长而降低,同时,随着温度的升高毕赤酵母的死亡速率也随之加快.相同温度处理下的致死曲线趋势大致相似,菌液的浓度越大,致死曲线的位置就更加靠后,达到相同灭菌效率的时间越长.随着处理温度的升高,致死曲线的形状趋于直线.温度较低的50,℃下处理的致死曲线是“S”形的,在热处理开始和结束时的曲线平缓.而在60,℃下处理的致死曲线的形状类似一条直线,没有明显的滞后和拖尾的现象.2.2 毕赤酵母的一级热致死动力学模型一级模型是一个数学方程或数学函数,用来表示微生物数量与时间的关系.在微生物的动力学研究中,失活规律通常是非线性的,在早期的研究中曾经提出对数残留定律,既微生物的热致死的对数曲线为直线.但是,在很多情况下灭活曲线并不遵循这种关系,而是存在“肩部”或“拖尾”现象[14].因为微生物菌群里含有很多独立的群体,而残存曲线是菌群各自动力学模式的综合体现[15].因此有研究将失活曲线看作概率模型[16].例如有研究发现,单增李斯特菌在高压协同温度处理时的失活模型可以用 loglogistic模型来拟合[17],改进的Gompertz模型能很好地预测细菌的热灭活趋势[18],Albert等[19]利用设计的Weibull模型成功拟合了预测细菌的灭活规律.但这些热灭活模型研究的微生物主要是食源性的致病细菌,对于广泛使用的温敏型工程菌毕赤酵母这一真菌研究较少.其中 Weibull分布函数被广泛应用于微生物存活数据的分析,近年来,Weibull模型已经成功拟合了很多微生物的非线性热失活模型曲线[14,,20].将热处理后的毕赤酵母残存数据,采用 Weibull模型进行曲线拟合,得到的相关数据见表1.拟合方程中 n>1,即所有毕赤酵母的致死曲线呈上凸的形状,这表明在初始阶段随着加热时间的延长,致死速率也随之加快,出现“肩部”情况;在热处理后期,致死速率却变慢.这种情况在较低温度处理下更加明显.这是由于在初始阶段,菌液处于升温阶段,热量没有传递均匀,此时的热处理对部分细胞没有很大影响.待温度平衡,致死速率也趋于稳定,随着时间的延长,毕赤酵母对高温出现了一定的耐受性,体现在致死曲线上就是“拖尾”.模型的决定系数 R2为0.960,9~0.999,9,同时SSE较小,P<0.001,表明Weibull模型表现出较好的拟合度,拟合所得到的结果接近于实验真实值.2.3 基于Weibull模型参数的二级模型的建立由以上分析可知,温度对致死率的影响和菌体量对致死率的影响可以用 Weibull模型来描述(拟合效果理想).但是 Weibull模型的参数却随着温度和菌体量变化,因此需要建立一个温度和菌体量对一级模型参数影响的二级模型.采用多元回归方程拟合出模型为式中:x为热处理的温度,y为菌体量(吸光度);b和n为 Weibull模型的参数.两个模型的相关系数(R2)分别为0.974,1和0.946,6,说明该模型所描述的温度和菌液吸光度与一级模型中的参数之间的关系是可信的.从拟合方程的系数上看,温度对一级模型参数的影响有较大的影响,其次是菌体量(吸光度).2.4 毕赤酵母热致死模型的验证对于验证一个模型是否有效,偏差因子和准确因子被认为作为首要的标准,且被证明是验证模型预测模型性能的非常有价值的工具.如果 Bf值在 0.9~1.05的范围之内,认为该模型能够很好地预测微生物生长速度和生长状况;如果 Bf值在0.7~0.9或者1.06~1.15范围之内,则该模型是可以被接受的;如果 Bf值大于1.15或者小于 0.7,则说明该模型是失败的[21].偏差因子不能表示参数估计的平均准确性,因此常结合准确性因子(Af)对模型进行数学检测,Af值越大表示平均准确性越低,而 Af值等于 1表示预测值与观测值之间完全吻合.用53、57,℃处理A600分别为20、45、120的毕赤酵母菌液,验证热致死模型的可靠性.由表 2可知,RMES、Bf、Af的值均在可靠范围,模型的准确性较高.并通过代入温度和菌体量数据到模型中,得到达到特定致死率的预测时间,表3列举的实验组和预测组所达到50%,、90%,、99%,、99.9%,致死率的时间相近,这亦说明本研究建立的毕赤酵母热致死模型是成功的.实验中A600为169,处理温度60,℃,代入二级模型中可得到一级模型的参数 b =0.363,6,n=1.032,1.一级模型为 lg(N/N0)=–0.363,6t1.032,1,代入 99.99%,致死率即 lg(N/N0)=–4,获得达到 99.99%,致死率的时间为 14.690,3,min,实际实验中60,℃处理 15,min的菌液,热处理完成后直接倾注的平板在30,℃下培养96,h后平板上未见菌落形成.本研究采用 Weibull模型对毕赤酵母的热致死曲线进行拟合,并且建立了温度和菌体量(吸光度)对一级模型参数影响的二级模型,结果表明此模型能够较准确地模拟毕赤酵母的热致死规律.这为实际生产中热处理毕赤酵母菌液提供了一定的理论依据.【相关文献】[1]Ahmad M,Hirz M,Pichler H,et al. Protein expression in Pichia pastoris:Recent achievements and perspectives for heterologous protein production[J]. Applied Microbiology Biotechnology,2014,98(12):5301–5317.[2]朱泰承,李寅. 毕赤酵母表达系统发展概况及趋势[J]. 生物工程学报,2015,31(6):929–938.[3]Macauley-Patrick S,Fazenda M L,McNeil B,et al.Heterologous protein production using the Pichia pastoris expression system[J]. Yeast,2005,22(4):249–270.[4]陈瑞东. N–酰基高丝氨酸内酯酶基因克隆、表达、性质研究与水产养殖应用[D]. 北京:中国农业科学院,2010.[5]许英蕾,孙建义,刘明启. 巴斯德毕赤酵母表达系统及其在饲料添加剂开发上的应用前景[J]. 饲料工业,2005,26(2):40–42.[6]丁嘉烽. 巴斯德毕赤酵母表达系统在动物传染病防制中应用的研究进展[J]. 当代畜牧,2013,38(14):89–90.[7]杨楠桢. 生物性污染废水处理技术研究[D]. 上海:复旦大学,2010.[8]Sree N K,Sridhar M,Suresh K,et al. Isolation of thermotolerant,osmotolerant,flocculating Saccharomyces cerevisiae for ethanol production[J]. Bioresource Technology,2000,72(1):43–46.[9]路福平,杨华,王玉,等. 高温和H2O2诱导酵母细胞产生活性衍生物的研究[J]. 微生物学通报,2004,31(5):28–32.[10]中华人民共和国卫生部. GB 4789.15—2010食品安全国家标准·食品微生物学检验·霉菌和酵母计数[S].北京:中国标准出版社,2010.[11]Chen H,Hoover D G. Use of Weibull model to describe and predict pressure inactivation of Listeria monocytogenes Scott A in whole milk[J]. Innovative Food Science and Emerging Technologies,2004,5(3):269–276.[12]王军,董庆利,丁甜. 预测微生物模型的评价方法[J]. 食品科学,2011,32(21):268–272.[13]Li P,Anumanthan A,Gao X,et al. Expression of recombinant proteins in Pichiapastoris[J]. Applied Biochemistry and Biotechnology,2007,142(2):105–124.[14]Boekel M A J S. On the use of the Weibull model to describe thermal inactivation of microbial vegetative cells[J]. International Journal of Food Microbiology,2002,74(1/2):139–159.[15]Doyle M E,Mazzotta A S,Wang T,et al. Heat resistance of Listeria monocytogenes[J]. Journal Food Protection,2001,64(3):410–429.[16]董庆利. Weibull模型拟合细菌非线性失活曲线的应用研究[J]. 上海理工大学学报,2009,31(4):387–391.[17]Chen H,Hoover D G. Modeling the combined effect of high hydrostatic pressure and mild heat on the inactiva-tion kinetics of Listeria monocytogenes Scott A in whole milk[J]. Innovative Food Science and Emerging Technologies,2003,4(1):25–34.[18]Gil M M,Brandão T R S,Silva C L M. A modified Gompertz model to predict microbial inactivation under time-varying temperature conditions[J]. Journal of Food Engineering,2006,76(1):89–94.[19]Albert I,Mafart P. A modified Weibull model for bacterial inactivation[J]. International Journal of Food Microbiology,2005,100(1/2/3):197–211.[20]Buzrul S,Alpas H,Bozoglu F. Use of Weibull frequency distribution model to describe the inactivation of Alicyclobacillus acidoterrestris by high pressure at different temperatures[J]. Food Research International,2005,38(2):151–157.[21]Ross T,McMeekin T A,Baranyi J. Predictive microbiology and foodsafety[M]//Tortorello C A B L. Encyclopedia of Food Microbiology.2nd ed. Oxford:Academic Press,2014:59–68.。

LiCl法转化毕赤酵母LiCl法转化毕赤酵母感受态毕赤酵母的制备1. 接种Pichia pastoris到50 mL YPD培养基中,30 ℃摇菌过夜(约24~28h)培养到OD值为0.8~1.0(约108 Cells/ml);培养基里有流沙样的菌体在流动2. 收获细胞,用25 mL无菌水洗涤一次,室温下5000 rpm离心10 min;3. 重悬细胞于1 mL 100 mM LiCl溶液中,将悬液转入1.5 mL离心管;4. 12000 rpm离心15 s沉淀菌体,重悬菌体于400 μL 100 mM LiCl溶液中;5. 按50 μL /管分装,立即进行转化;注:不要将感受态酵母菌冰浴;毕氏酵母的转化1.煮沸1ml鲑鱼精DNA 5min,迅速冰浴以制备单链担体DNA;鲑鱼精DNA主要是防止目的基因的降解,协助目的基因的转化2. 将感受态酵母菌离心,以Tips去除残余的LiCl溶液;3. 对于每一个转化,按以下顺序加入:50% PEG3350 240 μL1M LiCl 36 μL2mg/ml 单链Salmon sperm DNA 25 μL5~10 μg/50 μL H2O 质粒DNA 50 μL4. 剧烈旋涡混匀直至沉淀菌体完全分布均匀(约1min);5. 30 ℃水浴孵育30 min;6. 42 ℃水浴热休克20~25 min;7. 8000rpm离心10 min,收集酵母菌体;8. 重悬酵母于500 μL YPD培养基,30℃摇床孵育;9. 1~4 h后,取25~100 μL菌液涂布于选择性培养基平板(YPD+Zeocin),于30 ℃培养2-5天鉴定;试剂1M LiCl <用去离子蒸馏水配制,0.22um滤膜过滤除菌;必要时用消毒去离子水稀释>50% PEG3350 < 用去离子蒸馏水配制,0.45um滤膜过滤除菌,用较紧的盖子的瓶子分装>(氯化锂转化法只能PEG3350,不能用PEG8000)2 mg/ml salmon sperm DNA / TE(10mM Tris-Cl, pH8.0, 1.0mM EDTA)-20℃保存注:醋酸锂对毕氏酵母无效,对酿酒酵母有效,仅氯化锂有效;PEG3350可屏蔽高浓度LiCl的毒害作用。


毕赤酵母表达(pichia pastoris expression )实验手册2010-07-15 10:54:56| 分类:毕赤酵母| 标签:|字号大中小订阅一.毕赤酵母表达常用溶液及缓冲液的配制二.毕赤酵母表达的培养基配制三.主要试验环节的操作 3.1 酵母菌株的分离纯化 3.2 pPICZαA原核宿主菌TOP10F’的活化培养 3.3毕赤酵母表达的试验方法 3.4 毕赤酵母电转化方法 3.5 Pichia酵母表达直接PCR鉴定重组子的方法 3.6 毕赤酵母基因组提取方法 3.7 Mut+表型重组酵母的诱导表达实验关键词:酵母实验毕赤酵母表达 pichia pastoris expression 毕赤酵母酵母菌株大肠杆菌表达系统最突出的优点是工艺简单、产量高、周期短、生产成本低。
然而,许多蛋白质在翻译后,需经过翻译后的修饰加工,如磷酸化、糖基化、酰胺化及蛋白酶水解等过程才能转化成活性形式。
大肠杆菌缺少上述加工机制,不适合用于表达结构复杂的蛋白质。
另外,蛋白质的活性还依赖于形成正确的二硫键并折叠成高级结构,在大肠杆菌中表达的蛋白质往往不能进行正确的折叠,是以包含体状态存在。
包含体的形成虽然简化了产物的纯化,但不利于产物的活性,为了得到有活性的蛋白,就需要进行变性溶解及复性等操作,这一过程比较繁琐,同时增加了成本。
大肠杆菌是用得最多、研究最成熟的基因工程表达系统,当前已商业化的基因工程产品大多是通过大肠杆菌表达的,其主要优点是成本低、产量高、易于操作。
但大肠杆菌是原核生物,不具有真核生物的基因表达调控机制和蛋白质的加工修饰能力,其产物往住形成没有活性的包涵体,需要经过变性、复性等处理,才能应用。
近年来,以酵母作为工程菌表达外源蛋白日益引起重视,原因是与大肠杆菌相比,酵母是低等真核生物,除了具有细胞生长快,易于培养,遗传操作简单等原核生物的特点外,又具有真核生物时表达的蛋白质进行正确加工,修饰,合理的空间折叠等功能,非常有利于真核基因的表达,能有效克服大肠杆菌系统缺乏蛋白翻译后加工、修饰的不足。

利用重组酵母菌生产人胰岛素摘要:利用重组酵母菌生成人胰岛素为以下流程:获得目的基因→构建重组质粒→构建基因工程菌→工程菌发酵→产物分离纯化→产品检验。
其中前三个流程为产人胰岛素酵母工程菌的构建,根据人胰岛素的氨基酸序列和酵母偏爱的密码子,采用基因半合成技术合成人胰岛素基因。
将合成的胰岛素基因克隆到pPIC9质粒,构建了毕赤(Pichia)酵母重组表达载体pPINS319,用BglII线性化后用电转化法导入毕赤酵母GSll5菌株,经过营养筛选和PCR 复筛,得到含人胰岛素基因的毕赤酵母工程菌株。
后三个流程为胰岛素的大规模发酵生产,通过补料-分批发酵获得发酵液,分离纯化后获得人胰岛素。
纯化后的重组人胰岛素产品经SDS-PAGE检测,氨基酸组成分析及小鼠惊厥实验证明制得产品为有生物活性的人胰岛素。
关键词:重组酵母;人胰岛素;发酵;纯化;鉴定Using recombinant yeast to produce human insulinHuSheng 1142043040Abstract:The proceedings of produce human insulin by recombinant yeast show as follow:get purpose gene→c onstruct the recombinant plasmid→construct the genetic engineering bacteria→fermentation of e ngineering bacterium→separation and purification of the product→identification of the product. The front three processes is building engineering bacteria of human insulin yeasts . Insulin gene was synthesized according to the amino acid sequence of human insulin and yeast preferential codons. The insulin gene was cloned in to pPIC9 vector and expression vector pPINS319 was constructed. The expression vector was digested with BgllI and then used to transform Pichia Pastoris GSll5 by electroporation. The expression analysis showed that the insulin gene w s able to expressed efficiently in Pichia Pastoris. The posterior three processes is large-scale production by fermentation.Through fed - batch fermentation getting fermentation liquor, separation and purification the fermentation liquor getting human insulin.The final products purified by supedrex 75showed one band by SDS-PAGE analysis and were identical with the native human insulin by all critetia employed.(SDS-PAGE analysis,amin acid composition analysis and bioidentity assay) Keywords:recombinant yeast;human insulin; fermentation;purification;identification 胰岛素是FDA批准的第一个用于人类的基因重组药物。



甲醇营养型毕赤酵母表达系统介绍巴斯德毕赤酵母(Pichia pastoris)是近几年发展起来的较为完善的、
被广泛用来表达外源蛋白的甲醇营养型酵母表达系统。
目前通过毕赤
酵母表达了很多种性质不同的蛋白,越来越多的实验室及公司开始搭
建毕赤酵母表达系统的平台,相信随着对毕赤酵母表达系统的研究越
来越深入,会有更多成功表达并进行商业化应用的案例出现。
1.其优点主要为:
1) 具有强有力的乙醇氧化酶(Alochol Oxidase,AOX1)基因启动子,可严格调控外源蛋白的表达;
2) 作为真核表达系统,可对表达的蛋白进行翻译后的加工与修饰,
从而使表达出的蛋白具有生物活性;
3) 营养要求低、生长快、培养基廉价,便于工业化生产; 4) 可高密度发酵培养,在发酵罐中细胞干重甚至可达120g/L以上; 5) 表达量高,许多蛋白可达到g/L以上水平;
6) 在P. pastoris中表达的蛋白既可存在于细胞内,又可分泌到胞外,
自身分泌的蛋白非常少,十分有利于纯化;
7) 糖基化程度低,与S. cerevisiae相比,P. pastoris不产生过度的糖基化。
P. pastoris表达的糖蛋白的糖链长度为8-14个甘露糖,而S. cerevisiae 糖链中甘露糖多达50-150个;S. cerevisiae分泌的糖蛋白的核心寡聚糖具有终端α-1,3糖苷连接,使分泌的糖蛋白的抗原性明
显增强,而P.pastoris的糖基化位点与哺乳类细胞的相同,其所分泌
的糖蛋白的免疫原性较低,更利于临床应用。
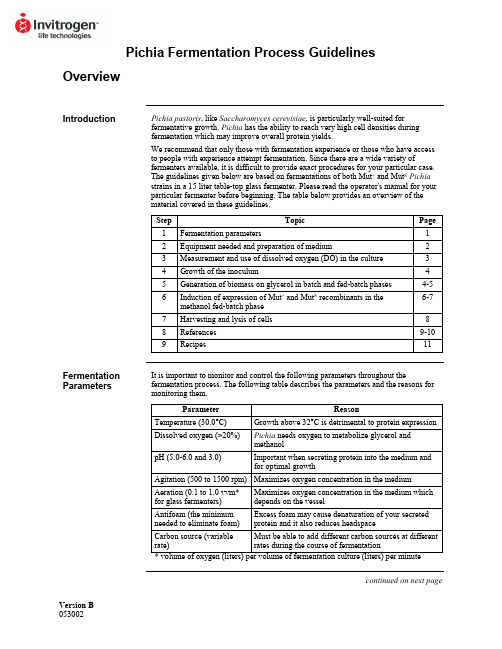
Version B Pichia Fermentation Process GuidelinesOverviewIntroduction Pichia pastoris, like Saccharomyces cerevisiae, is particularly well-suited forfermentative growth. Pichia has the ability to reach very high cell densities duringfermentation which may improve overall protein yields.We recommend that only those with fermentation experience or those who have accessto people with experience attempt fermentation. Since there are a wide variety offermenters available, it is difficult to provide exact procedures for your particular case.The guidelines given below are based on fermentations of both Mut+ and Mut S Pichiastrains in a 15 liter table-top glass fermenter. Please read the operator's manual for yourparticular fermenter before beginning. The table below provides an overview of thematerial covered in these guidelines.Step Topic Page1 Fermentationparameters 12 Equipment needed and preparation of medium 23 Measurement and use of dissolved oxygen (DO) in the culture 34 Growth of the inoculum 45 Generation of biomass on glycerol in batch and fed-batch phases 4-56 Induction of expression of Mut+ and Mut S recombinants in themethanol fed-batch phase6-77 Harvesting and lysis of cells 88 References 9-109 Recipes 11Fermentation Parameters It is important to monitor and control the following parameters throughout thefermentation process. The following table describes the parameters and the reasons for monitoring them.Parameter Reason Temperature (30.0°C) Growth above 32°C is detrimental to protein expression Dissolved oxygen (>20%) Pichia needs oxygen to metabolize glycerol andmethanolpH (5.0-6.0 and 3.0) Important when secreting protein into the medium andfor optimal growthAgitation (500 to 1500 rpm) Maximizes oxygen concentration in the mediumAeration (0.1 to 1.0 vvm*for glass fermenters)Maximizes oxygen concentration in the medium whichdepends on the vesselAntifoam (the minimumneeded to eliminate foam)Excess foam may cause denaturation of your secretedprotein and it also reduces headspaceCarbon source (variablerate)Must be able to add different carbon sources at differentrates during the course of fermentationcontinued on next pageOverview, continuedRecommended Equipment Below is a checklist for equipment recommendations.• A jacketed vessel is needed for cooling the yeast during fermentation, especially during methanol induction. You will need a constant source of cold water (5-10°C). This requirement may mean that you need a refrigeration unit to keep the water cold. • A foam probe is highly recommended as antifoam is required.• A source of O2--either air (stainless steel fermenters at 1-2 vvm) or pure O2(0.1-0.3 vvm for glass fermenters).• Calibrated peristaltic pumps to feed the glycerol and methanol.• Automatic control of pH.Medium Preparation You will need to prepare the appropriate amount of following solutions:• Fermentation Basal Salts (page 11)• PTM1Trace Salts (page 11)• ~75 ml per liter initial fermentation volume of 50% glycerol containing 12 ml PTM1 Trace Salts per liter of glycerol.• ~740 ml per liter initial fermentation volume of 100% methanol containing 12 mlPTM1Trace Salts per liter of methanol.Monitoring the Growth of Pichia pastoris Cell growth is monitored at various time points by using the absorbance at 600 nm (OD600) and the wet cell weight. The metabolic rate of the culture is monitored by observing changes in the concentration of dissolved oxygen in response to carbon availability (see next page).Dissolved Oxygen (DO) MeasurementIntroduction The dissolved oxygen concentration is the relative percent of oxygen in the mediumwhere 100% is O2-saturated medium. Pichia will consume oxygen as it grows, reducing the dissolved oxygen content. However, because oxygen is required for the first step ofmethanol catabolism, it is important to maintain the dissolved oxygen (DO) concentra-tion at a certain level (>20%) to ensure growth of Pichia on methanol. Accuratemeasurement and observation of the dissolved oxygen concentration of a culture willgive you important information about the state and health of the culture. Therefore, it isimportant to accurately calibrate your equipment. Please refer to your operator's manual.Maintaining the Dissolved Oxygen Concentration (DO) 1. Maintaining the dissolved oxygen above 20% may be difficult depending on theoxygen transfer rates (OTR) of the fermenter, especially in small-scale glassvessels. In a glass vessel, oxygen is needed to keep the DO above 20%, usually~0.1-0.3 vvm (liters of O2per liter of fermentation culture per minute). Oxygen consumption varies and depends on the amount of methanol added and the protein being expressed.2. Oxygen can be used at 0.1 to 0.3 vvm to achieve adequate levels. This can beaccomplished by mixing with the air feed and can be done in any glass fermenter.For stainless steel vessels, pressure can be used to increase the OTR. Be sure toread the operator's manual for your particular fermenter.3. If a fermenter cannot supply the necessary levels of oxygen, then the methanol feedshould be scaled back accordingly. Note that decreasing the amount of methanol may reduce the level of protein expression.4. To reach maximum expression levels, the fermentation time can be increased todeliver similar levels of methanol at the lower feed rate. For many recombinantproteins, a direct correlation between amount of methanol consumed and theamount of protein produced has been observed.Use of DO Measurements During growth, the culture consumes oxygen, keeping the DO concentration low. Note that oxygen is consumed whether the culture is grown on glycerol or methanol. The DO concentration can be manipulated to evaluate the metabolic rate of the culture and whether the carbon source is limiting. The metabolic rate indicates how healthy the culture is. Determining whether the carbon source is limiting is important if you wish to fully induce the AOX1 promoter. For example, changes in the DO concentrations (DO spikes) allow you to determine whether all the glycerol is consumed from the culture before adding methanol. Secondly, it ensures that your methanol feed does not exceed the rate of consumption. Excess methanol (> 1-2% v/v) may be toxic.Manipulation of DO If carbon is limiting, shutting off the carbon source should cause the culture to decrease its metabolic rate, and the DO to rise (spike). Terminate the carbon feed and time how long it takes for the DO to rise 10%, after which the carbon feed is turned back on. If the lag time is short (< 1 minute), the carbon source is limiting.Fermenter Preparation and Glycerol Batch PhaseInoculum Seed Flask Preparation Remember not to put too much medium in the baffled flasks. Volume should be 10-30% of the total flask volume.1. Baffled flasks containing a total of 5-10% of the initial fermentation volume ofMGY or BMGY are inoculated with a colony from a MD or MGY plate or from a frozen glycerol stock.2. Flasks are grown at 30°C, 250-300 rpm, 16-24 hours until OD600= 2-6. Toaccurately measure OD600> 1.0, dilute a sample of your culture 10-fold before reading.Glycerol Batch Phase 1. Sterilize the fermenter with the Fermentation Basal Salts medium containing 4%glycerol (see page 11).2. After sterilization and cooling, set temperature to 30°C, agitation and aeration tooperating conditions (usually maximum rpm and 0.1-1.0 vvm air), and adjust the pH of the Fermentation Basal Salts medium to 5.0 with 28% ammonium hydroxide(undiluted ammonium hydroxide). Add aseptically 4.35 ml PTM1trace salts/liter of Fermentation Basal Salts medium.3. Inoculate fermenter with approximately 5-10% initial fermentation volume from theculture generated in the inoculum shake flasks. Note that the DO will be close to 100% before the culture starts to grow. As the culture grows, it will consumeoxygen, causing the DO to decrease. Be sure to keep the DO above 20% by adding oxygen as needed.4. Grow the batch culture until the glycerol is completely consumed (18 to 24 hours).This is indicated by an increase in the DO to 100%. Note that the length of timeneeded to consume all the glycerol will vary with the density of the initial inoculum.5. Sampling is performed at the end of each fermentation stage and at least twice daily.We take 10 ml samples for each time point, then take 1 ml aliquots from this 10 mlsample. Samples are analyzed for cell growth (OD600and wet cell weight), pH, microscopic purity, and protein concentrations or activity. Freeze the cell pellets and supernatants at -80°C for later analysis. Proceed to Glycerol Fed-Batch Phase,page 5.Yield A cellular yield of 90 to 150 g/liter wet cells is expected for this stage. Recombinant protein will not yet be produced due to the absence of methanol.Introduction Once all the glycerol is consumed from the batch growth phase, a glycerol feed isinitiated to increase the cell biomass under limiting conditions. When you are ready toinduce with methanol, you can use DO spikes to make sure the glycerol is limited.Glycerol Fed-Batch Phase 1. Initiate a 50% w/v glycerol feed containing 12 ml PTM1trace salts per liter of glycerol feed. Set the feed rate to 18.15 ml/hr /liter initial fermentation volume.2. Glycerol feeding is carried out for about four hours or longer (see below). A cellularyield of 180 to 220 g/liter wet cells should be achieved at the end of this stage while no appreciable recombinant protein is produced.Note The level of expressed protein depends on the cell mass generated during the glycerolfed-batch phase. The length of this feed can be varied to optimize protein yield. A rangeof 50 to 300 g/liter wet cells is recommended for study. A maximum level of 4%glycerol is recommended in the batch phase due to toxicity problems with higher levelsof glycerol.Important If dissolved oxygen falls below 20%, the glycerol or methanol feed should bestopped and nothing should be done to increase oxygen rates until the dissolvedoxygen spikes. At this point, adjustments can be made to agitation, aeration, pressure oroxygen feeding.Proteases In the literature, it has been reported that if the pH of the fermentation medium islowered to 3.0, neutral proteases are inhibited. If you think neutral proteases aredecreasing your protein yield, change the pH control set point to 3.0 during the glycerolfed-batch phase (above) or at the beginning of the methanol induction (next page) andallow the metabolic activity of the culture to slowly lower the pH to 3.0 over 4 to 5 hours(Brierley, et al., 1994; Siegel, et al., 1990).Alternatively, if your protein is sensitive to low pH, it has been reported that inclusion ofcasamino acids also decreases protease activity (Clare, et al., 1991).Introduction All of the glycerol needs to be consumed before starting the methanol feed to fullyinduce the AOX1 promoter on methanol. However, it has been reported that a "mixedfeed" of glycerol and methanol has been successful to express recombinant proteins(Brierley, et al., 1990; Sreekrishna, et al., 1989). It is important to introduce methanolslowly to adapt the culture to growth on methanol. If methanol is added too fast, it willkill the cells. Once the culture is adapted to methanol, it is very important to use DOspikes to analyze the state of the culture and to take time points over the course ofmethanol induction to optimize protein expression. Growth on methanol also generates alot of heat, so temperature control at this stage is very important.Mut+ Methanol Fed-Batch Phase 1. Terminate glycerol feed and initiate induction by starting a 100% methanol feedcontaining 12 ml PTM1trace salts per liter of methanol. Set the feed rate to 3.6 ml/hr per liter initial fermentation volume.2. During the first 2-3 hours, methanol will accumulate in the fermenter and thedissolved oxygen values will be erratic while the culture adapts to methanol.Eventually the DO reading will stabilize and remain constant.3. If the DO cannot be maintained above 20%, stop the methanol feed, wait for theDO to spike and continue on with the current methanol feed rate. Increaseagitation, aeration, pressure or oxygen feeding to maintain the DO above 20%. 4. When the culture is fully adapted to methanol utilization (2-4 hours), and is limitedon methanol, it will have a steady DO reading and a fast DO spike time (generally under 1 minute). Maintain the lower methanol feed rate under limited conditions for at least 1 hour after adaptation before doubling the feed. The feed rate is then doubled to ~7.3 ml/hr/liter initial fermentation volume.5 After 2 hours at the 7.3 ml/hr/liter feed rate, increase the methanol feed rate to~10.9 ml/hr per liter initial fermentation volume. This feed rate is maintainedthroughout the remainder of the fermentation.6. The entire methanol fed-batch phase lasts approximately 70 hours with a total ofapproximately 740 ml methanol fed per liter of initial volume. However, this may vary for different proteins.Note: The supernatant may appear greenish. This is normal.Yield The cell density can increase during the methanol fed-batch phase to a final level of 350 to 450 g/liter wet cells. Remember that because most of the fermentation is carried out ina fed-batch mode, the final fermentation volume will be approximately double the initialfermentation volume.Fermentation of Mut S Pichia Strains Since Mut S cultures metabolize methanol poorly, their oxygen consumption is very low. Therefore, you cannot use DO spikes to evaluate the culture. In standard fermentations of a Mut S strain, the methanol feed rate is adjusted to maintain an excess of methanol in the medium which does not exceed 0.3% (may be determined by gas chromatography). While analysis by gas chromatography will insure that nontoxic levels of methanol are maintained, we have used the empirical guidelines below to express protein in Mut S strains. A gas chromatograph is useful for analyzing and optimizing growth of Mut S recombinants.continued on next pageMethanol Fed-Batch Phase, continuedMut S Methanol Fed- Batch Phase The first two phases of the glycerol batch and fed-batch fermentations of the Mut S strains are conducted as described for the Mut+ strain fermentations. The methanol induction phases of the Mut+ and Mut S differ in terms of the manner and amount in which the methanol feed is added to the cultures.1. The methanol feed containing 12 ml PTM1trace salts per liter of methanol is initiated at 1 ml/hr/liter initial fermentation volume for the first two hours. It is then increased in 10% increments every 30 minutes to a rate of 3 ml/hr which ismaintained for the duration of the fermentation.2.. The vessel is then harvested after ~100 hours on methanol. This time may be variedto optimize protein expression.Harvesting and Lysis of CellsIntroduction The methods and equipment listed below are by no means complete. The amount of cells or the volume of supernatant will determine what sort of equipment you need.Harvesting Cells and Supernatant For small fermentations (1-10 liters), the culture can be collected into centrifuge bottles (500-1000 ml) and centrifuged to separate the cells from the supernatant.For large fermentations, large membrane filtration units (Millipore) or a Sharples centrifuge can be used to separate cells from the supernatant. The optimal method will depend on whether you need the cells or the supernatant as the source of your protein and what you have available.Supernatants can be loaded directly onto certain purification columns or concentrated using ultrafiltration.Cell Lysis We recommend cell disruption using glass beads as described in Current Protocols inMolecular Biology, page 13.13.4. (Ausubel, et al., 1990) or Guide to ProteinPurification (Deutscher, 1990). This method may be tedious for large amounts of cells.For larger amounts, we have found that a microfluidizer works very well. Frenchpressing the cells does not seem to work as well as the glass beads or the microfluidizer.ReferencesIntroduction Most of the references below refer to papers where fermentation of Pichia wasperformed. Note that some of these are patent papers. You can obtain copies of patentsusing any of the following methods.• Patent Depository Libraries. U. S. patents and international patents granted underthe Patent Cooperation Treaty (PCT) are available on microfilm. These can be copiedand mailed or faxed depending on length. There is a fee for this service. The referencelibrarian at your local library can tell you the location of the nearest Patent DepositoryLibrary.• Interlibrary Loan. If you are not near a Patent Depository Library, you may request acopy of the patent through interlibrary loan. There will be a fee for this service.• U. S. Patent Office. Requests may be made directly to the Patent Office, Arlington,VA. Please call 703-557-4636 for more information on cost and delivery.• Private Library Services. There are private companies who will retrieve and sendyou patents for a fee. Two are listed below:Library Connection: 804-758-3311Rapid Patent Services: 800-336-5010Citations Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A.,Struhl, K., eds (1990) Current Protocols in Molecular Biology. GreenePublishing Associates and Wiley-Interscience, New York.Brierley, R. A., Siegel, R. S., Bussineau, C. M. Craig, W. S., Holtz, G. C., Davis, G. R.,Buckholz, R. G., Thill, G. P., Wondrack, L. M., Digan, M. E., Harpold, M. M.,Lair, S. V., Ellis, S. B., and William, M. E. (1989) Mixed Feed RecombinantYeast Fermentation. International Patent (PCT) Application. Publication No.WO 90/03431.Brierley, R. A., Bussineau, C., Kosson, R., Melton, A., and Siegel, R. S. (1990)Fermentation Development of Recombinant Pichia pastoris Expressing theHeterologous Gene: Bovine Lysozyme. Ann. New York Acad. Sci.589: 350-362.Brierley, R. A., Davis, G. R. and Holtz, G. C. (1994) Production of Insulin-Like GrowthFactor-1 in Methylotrophic Yeast Cells. United States Patent5,324,639.Clare, J. J., Romanos, M. A., Rayment, F. B., Rowedder, J. E., Smith, M. A., Payne, M.M., Sreekrishna, K. and Henwood, C. A. (1991) Production of EpidermalGrowth Factor in Yeast: High-level Secretion Using Pichia pastoris StrainsContaining Multiple Gene Copies. Gene105: 205-212.Cregg, J. M., Tschopp, J. F., Stillman, C., Siegel, R., Akong, M., Craig, W. S.,Buckholz, R. G., Madden, K. R., Kellaris, P. A., Davis, G. R., Smiley, B. L.,Cruze, J., Torregrossa, R., Veliçelebi, G. and Thill, G. P. (1987) High-levelExpression and Efficient Assembly of Hepatitis B Surface Antigen in theMethylotrophic Yeast Pichia pastoris. Bio/Technology5: 479-485.Cregg, J. M., Vedvick, T. S. and Raschke, W. C. (1993) Recent Advances in theExpression of Foreign Genes in Pichia pastoris. Bio/Technology11: 905-910.Deutscher, M. P. (1990) Guide to Protein Purification. In: Methods in Enzymology (J.N. Abelson and M. I. Simon, eds.) Academic Press, San Diego, CA.continued on next pageReferences, continuedCitations, continuedDigan, M. E., Lair, S. V., Brierley, R. A., Siegel, R. S., Williams, M. E., Ellis, S. B., Kellaris, P. A., Provow, S. A., Craig, W. S., Veliçelebi, G., Harpold, M. M. andThill, G. P. (1989) Continuous Production of a Novel Lysozyme via Secretionfrom the Yeast Pichia pastoris. Bio/Technology7: 160-164.Hagenson, M. J., Holden, K. A., Parker, K. A., Wood, P. J., Cruze, J. A., Fuke, M., Hopkins, T. R. and Stroman, D. W. (1989) Expression of Streptokinase inPichia pastoris Yeast. Enzyme Microbiol. Technol.11: 650-656.Laroche, Y., Storme, V., Meutter, J. D., Messens, J. and Lauwereys, M. (1994) High-Level Secretion and Very Efficient Isotopic Labeling of Tick AnticoagulantPeptide (TAP) Expressed in the Methylotrophic Yeast, Pichia pastoris.Bio/Technology12: 1119-1124.Romanos, M. A., Clare, J. J., Beesley, K. M., Rayment, F. B., Ballantine, S. P., Makoff,A. J., Dougan, G., Fairweather, N. F. and Charles, I. G. (1991) RecombinantBordetella pertussis Pertactin p69 from the Yeast Pichia pastoris High LevelProduction and Immunological Properties. Vaccine9: 901-906.Siegel, R. S. and Brierley, R. A. (1989) Methylotrophic Yeast Pichia pastoris Produced in High-cell-density Fermentations With High Cell Yields as Vehicle forRecombinant Protein Production. Biotechnol. Bioeng.34: 403-404.Siegel, R. S., Buckholz, R. G., Thill, G. P., and Wondrack, L. M. (1990) Production of Epidermal Growth Factor in Methylotrophic Yeast Cells. International Patent(PCT) Application. Publication No. WO 90/10697.Sreekrishna, K., Nelles, L., Potenz, R., Cruse, J., Mazzaferro, P., Fish, W., Fuke, M., Holden, K., Phelps, D., Wood, P. and Parker, K. (1989) High LevelExpression, Purification, and Characterization of Recombinant Human TumorNecrosis Factor Synthesized in the Methylotrophic Yeast Pichia pastoris.Biochemistry28(9): 4117-4125.©2002 Invitrogen Corporation. All rights reservedRecipesFermentation Basal Salts Medium For 1 liter, mix together the following ingredients:Phosphoric acid, 85% (26.7 ml)Calcium sulfate 0.93 gPotassium sulfate 18.2 gMagnesium sulfate-7H2O 14.9gPotassium hydroxide 4.13 gGlycerol 40.0g Water to 1 literAdd to fermenter with water to the appropriate volume and sterilize.PTM1 Trace Salts Mix together the following ingredients:Cupric sulfate-5H2O 6.0gSodium iodide 0.08 gManganese sulfate-H2O 3.0gSodium molybdate-2H2O 0.2gBoric Acid 0.02 g Cobalt chloride 0.5 g Zinc chloride 20.0 gFerrous sulfate-7H2O 65.0gBiotin 0.2gSulfuric Acid 5.0 mlWater to a final volume of 1 literFilter sterilize and store at room temperature.Note: There may be a cloudy precipitate upon mixing of these ingredients. Filter-sterilize as above and use.11。
ORIGINAL PAPERImproved glutathione production by gene expression in Pichia pastorisLiwen Fei ÆYan Wang ÆShaoxin ChenReceived:19October 2008/Accepted:1January 2009/Published online:20January 2009ÓSpringer-Verlag 2009Abstract To utilize Pichia pastoris to produce glutathi-one,an intracellular expression vector harboring two genes (gsh1and gsh2)from Saccharomyces cerevisiae encoding enzymes involved in glutathione synthesis and regulated by the glyceraldehyde-3-phosphate dehydrogenase (GAP)promoter was transformed into P.pastoris GS115.Through Zeocin resistance and expression screening,a transformant that had higher glutathione yield (217mg/L)in flask cul-ture than the host strain was obtained.In fed-batch culture process,this recombinant strain displayed high activity for converting precursor amino acids into glutathione.The glutathione yield and biomass achieved 4.15g/L and 98.15g (dry cell weight,DCW)/L,respectively,after 50h fermentation combined with addition of three amino acids (15mmol/L glutamic acid,15mmol/L cysteine,and 15mmol/L glycine).Keywords Glutathione production ÁRecombinant Pichia pastoris ÁFed-batch culture IntroductionGlutathione (GSH)is the most abundant non-protein thiol found in all living organisms [1].With its important physiological functions such as antioxidant,detoxification of xenobiotics and immune booster [2–8],GSH has been widely used in medical treatment,food additive and cos-metic industry [9,10].In recent years,the demand for GSH is tending to increase.However,the high price of GSH limits its applications.Therefore,it is of great importance to develop an efficient way to produce pared with chemical and enzymatic method,fermentative production of GSH is an efficient approach in practical process [11].To date,Saccharomyces cerevisiae is the most commonly described microorganism in literatures for GSH production.Many attempts have focused on screening GSH over-producers by mutagenesis and process optimization to achieve high content of GSH [12–16].Recently,Wang et al.reported a strategy to increase the yield of GSH by amino acid modulation and high-cell-density culture of S.cerevisiae [12].Although these processes have improved the pro-duction of GSH,the volumetric yield of GSH was still low because most of the strains belonging to S.cerevisiae were hard to attain a very high-cell-density using minimal media due to Crabtree effect [17].Pichia pastoris is being widely used as a host organism for the production of heterologous proteins [18].The most advantageous characteristic of this microorganism is that it is able to grow to high biomass in simple minimal defined media [above 130g/L dry cell weight (DCW)][19].Since P.pastoris has the ability to reach higher cell density than that of S.cerevisiae during fermentation,the recombinant P.pastoris has the potential to produce higher volumetric yield of GSH.In this work,a recombinant P.pastoris expressing the genes of GSH synthesis enzymes,gsh1and gsh2,was constructed to increase GSH production.To our knowl-edge,this is the first example of GSH production by recombinant P.pastoris .This paper demonstrated that the GSH production could be efficiently enhanced in P.pastoris in fed-batch culture combined with addition of precursor amino acids.L.Fei ÁY.Wang ÁS.Chen (&)Department of Biochemistry,Shanghai Institute of Pharmaceutical Industry,Beijing Road West 1320,200040Shanghai,People’s Republic of China e-mail:sxzlb@ Bioprocess Biosyst Eng (2009)32:729–735DOI 10.1007/s00449-009-0297-xMaterials and methodsPlasmids,strains,enzyme,and chemicalsAll plasmids and strains used in this study are listed in Table1.Restriction endonuclease,T4DNA ligase,and DNA polymerase were obtained from TaKaRa(Dalian, China).All other chemicals were of analysis grade and commercially available.Construction of integrative vectorsGeneral recombinant DNA procedures were carried out using standard procedure[20].The constructionflow chart of integrative plasmid pGAPZHGSH is described in Fig.1.gsh1(Gene ID.853344)and gsh2(Gene ID.854108) were amplified by PCR from genomic of S.cerevisiae.The forward primer and reverse primer used for amplification of gsh1gene were50-CCATCGATACCATGGGACTCT TAGCTTTG-30(underlined sequence was Cla I site)and 50-GCGGCCGCTTAACATTTGCTTTCTAT-30(Not I site was underlined),respectively.The obtained2056-bp product was inserted into pMD-18T to generate plasmid pMDGSH1.The forward primer and reverse primer used for ampli-fication of gsh2gene were50-CCATCGATACCATGGCA CACTATCCA-30(underlined was Cla I site)and50-GCG GCCGCCCTAGTAAAGAATAATACTG-30(underlined was Not I site),respectively.The obtained1496-bp PCR product was inserted into pMD-18T,deriving plasmid pMDGSH2.his4DNA fragment was amplified from plasmid pAO815with a forward primer50-GCGGATCCTAATGC GGTAGTTTATCACAGTTA-30(underlined was Bam HI site)and an antisense primer50-GAAGATCTTCATGACA TTTCCCTTGCTACCTG-30(underlined was Bgl II site). The obtained PCR product was inserted into pMD-18T to generate plasmid pMDHis4.The2921-bp Bam HI-to-Bgl II fragment from plasmid pMDHis4was cloned into the Bam HI site of pGAPZ a A to generate pGAPZ a AH.Then,the2034-bp Cla I-to-Not I fragment containing gsh1from pMDGSH1was inserted into the Not I-to-Bst bI site of plasmid pGAPZ a AH,obtaining pGAPZHG1.Similarly,pGAPZG2was constructed by inserting Cla I-to-Not I fragment from pMDGSH2into Bstb I-to-Not I site of pGAPZ a A.Finally,the Bgl II-to-Bam HI fragment of pGAPZG2containing GAP promoter,gsh2 and AOX1terminator was subcloned into the Bgl II site of pGAPZHG1,obtaining pGAPZHGSH.Transformation and strain selectionTransformation of P.pastoris was achieved by electro-poration according to the protocol of manufacture (Invitrogen).The plasmid pGAPZHGSH linearized by Bsp EI in the his4DNA fragment was transformed into P.pastoris GS115by electroporation,and resulted trans-formants were selected on yeast extract peptone dextrose medium(YPD)plates containing10g/L yeast extract, 20g/L peptone,20g/L glucose with addition of1mol/L sorbitol and l00l g/mL Zeocin.Table1Plasmids and strains used in this studyPlasmids Relevant characteristics Source or referencepMD-18T Amp resistant TaKaRapGAPZ a A Zeocin resistance,pGAP,a-factor,AOX1terminator InvitrogenpAO815G418resistant,pAOX1,his4Invitrogen pMDGSH1gsh1in pMD-18T,Amp resistant This work pMDGSH2gsh2in pMD-18T,Amp resistant This work pMDHis4his4in pMD-18T,Amp resistant This work pGAPZ a AH his4in pGAPZ a A,Zeocin resistance,pGAP,a-factor,AOX1terminator This work pGAPZHG1gsh1in pGAPZ a AH,Zeocin resistance,his4,pGAP,AOX1terminator This work pGAPZG2gsh2in pGAPZ a A,Zeocin resistance,pGAP,AOX1terminator This work pGAPZHGSH gsh1and gsh2in pGAPZ a A,Zeocin resistance,his4,pGAP,AOX1terminator This work StrainsE.coli DH5a F-,SupE44,la cU169,a80/lac Z D M15,deoR,hsdR17,phoA,rec A1,end A1,gyrA96,k-,thi-1,rel A1AmershamP.pastoris GS115his4-,Mut?InvitrogenP.pastoris D18gsh1and gsh2integrated into P.pastoris GS115This workS.cerevisiae Wild type strain containing the gsh1and gsh2gene Our laboratoryEach colony growing on the plate was picked into a 250mL shakeflask containing15mL YPD medium and cultured at30°C and250rpm for48h.1mL of each culture was centrifuged at4,0009g for5min and the collected cells were washed twice with deionized water before resuspended in1mL deionized water.Intracellular GSH was extracted from the cells by freezing the resus-pended cells at-20°C overnight,and then incubating at 83°C for15min.After centrifugation,the resultant supernatant was used for assay of GSH content by colori-metric method described previously[21,22].The posi-tive colonies were subjected to PCR for confirmation of the presence of gsh1and gsh2.The sense primer was 50-GGTCTGACGCTCAGTGGAACGAAAC-30,and the antisence primer was50-GATTCGATAGATCCACAGGA CGGGTG-30.To examine the genetic stability of the recombinant strain,the colony on YPD agar plate was picked to a YPD agar slant and cultivated at30°C for48h.Biomass formed on this slant was called generation F0.Generation F1–F5 slant were produced by inoculating the biomass from their parent slant followed with48h cultivation at30°C.Cells on each generation of slant were separately inoculated to a 250mL shakeflask containing15mL YPD medium andcultured at30°C for18h.Ten percent(v/v)of the culture was transferred into15mL of fresh YPD medium for cultivation of48h,and the GSH content was analyzed as described above.Medium optimizationVarious carbon and nitrogen sources were tested to deter-mine the best components for GSH production.P.pastoris D18growing on YPD agar slant was cultured in liquid YPD medium at30°C and250rpm for18h as seed cul-ture.For all experiments,10%inoculum(v/v)of seed culture was used in a500mL shakeflask containing 50mL culture medium and cultivated at30°C and 250rpm for48h.The GSH content was analyzed as described above.Fed-batch fermentationFed-batch fermentation was carried out in a5L fermentor (B.Braun,Biostat B).Strain P.pastoris D18grown on YPD agar plate was inoculated into liquid YPD medium and cultured at30°C until OD600value reached5.0,and then was used as seed culture.Ten percent inoculum(v/v) was transferred into the bioreactor containing50g/L glucose,5g/L yeast extract,11g/L(NH4)2SO4,7g/L K2HPO4Á3H2O,5g/L MgSO4Á7H2O,0.5g/L CaCl2Á2H2O, 0.5g/L KCl with pH5.0–5.5.Initial agitation was250rpm and the temperature was30°C.Dilute oxygen(DO)was maintained above25%through adjustment of agitation speed.Air was used and the aeration rate in the fed-batch culture was1vvm.The pH of the medium was adjusted to 5.0with10%ammonium hydroxide.Feeding of80%(w/v) glucose was carried out after16h of culture at a rate of 2.9mL/L h by peristaltic pump.Dry cell weight was determined after yeast cells were centrifuged at4,0009g for10min and washed twice with deionized water before drying at105°C to constant weight,and the GSH con-centration was analyzed by HPLC.Analytical methodGlutathione was extracted from the fermentation broth by adjusting the pH of samples to1.5with2.0%(v/v)H2SO4 and incubated at85°C for5min.And then GSH con-taining supernatant was centrifuged at4,0009g for5min andfiltered through0.45l m membrane before subjected to HPLC analysis.The column used was C18(Hypersil BDS, 15094.6mm,5l m).The mobile phase consisted of methanol and0.11%(w/v)1-heptanesulfonate(sodium salt)in potassium phosphate buffer(50mmol/L,pH3.0)in the ratio of5:95(v/v).Theflow rate was1.0mL/min,and detection wavelength was set at210nm.GSH from Koway (Japan)was used as calibration standard.Results and discussionConstruction and selection of recombinant P.pastoris The biosynthesis pathway of GSH has been elucidated, which includes a two-step enzymatic catalyzed process [23–25].First,gamma-glutamylcysteine is synthesized from L-glutamic acid and L-cysteine via gamma-glutamyl-cysteine synthetase(GSH1);second,glycine is added to the C-terminal of gamma-glutamylcysteine by the GSH syn-thetase(GSH2).And the DNA sequences of the two enzymes have been identified in S.cerevisiae[26,27]. Therefore,an integrative plasmid pGAPZHGSH was con-structed in order to obtain a recombinant P.pastoris for GSH synthesis(Fig.1).The plasmid contains a strong constitutive GAP promoter,the his4DNA fragment for homologous recombination into the his4locus on the chromosome of P.pastoris,the gsh1and gsh2,the AOX1 terminator,and the Zeocin-resistant gene.In addition,the a-signal factor was deleted from the pGAPZ a A,so that the enzymes could be expressed intracellularly in P.pastoris.pGAPZHGSH was linearized by Bsp EI and transformed into P.pastoris GS115.Transformants that appeared on the YPD plate containing1mol/L sorbitol and100l g/mL Zeocin after48–72h revealed that plasmid had been integrated into the yeast chromosome through homologous recombination.And the presence of the gsh1and gsh2in recombinant P.pastoris was also confirmed by PCR using the primers described in the‘‘Materials and methods’’.The copy number of the integrated plasmid may vary in dif-ferent transformants resulting in diverse expression levels of cloned gene.Three hundred colonies were picked to assess the GSH-producing capability.As expected,the GSH levels varied from different colonies transformed by the same linearized plasmid.As shown in Fig.2,the GSH content ranged from35to230mg/L,and approximately 10%of the strains produced GSH higher than200mg/L. By isolation and purification for several times,the one colony originally designated as P.pastoris D18,which produced217mg/L GSH in theflask culture,4.7times higher than that of host strain(38mg/L),was selected for further study.The genetic stability of the recombinant strain was also taken into consideration by analyzing the GSH-production level.It was observed that the GSH-pro-duction capability showed no obvious change after serial cultivation forfive generations,suggesting that the plasmid pGAPZHGSH was stably integrated into the chromosome of P.pastoris.Fed-batch cultureIn order to enhance the synthesis level of GSH by P.pas-toris D18,the composition of the production medium was examined before batch culture.Several different carbon sources and nitrogen sources were investigated(Table2), and the glucose and yeast extract were found to be the best carbon source and nitrogen source for GSH synthesis, respectively.Finally,a medium described in the part of materials and methods was applied for fed-batch culture.To scale up the fermentation of P.pastoris D18,the batch fermentation for GSH production was carried out in a5L fermentor.The time course of fed-batch culture of P.pastoris D18was shown in Fig.3.During the process of fermentation,the cell density rose continually,and grew up to94.98g(DCW)/L at45h.The GSH content reached 0.92g/L at28h and tended to remain constant during further culture.Unexpectedly,as a biomass related product, the result showed that the GSH content did not increase with the accumulation of biomass after28h.It has been reported that GSH yield could be enhanced effectively by addition of precursor amino acids mixture [28–30].Therefore,the effect of concentrations of pre-cursor amino acids on GSH production was investigated in fed-batch culture of strain P.pastoris D18.As shown in Table3,the addition of amino acids significantly stimu-lated the GSH synthesis in the yeast cells,and the GSH yield and productivity were increased with increasing the concentration of each amino acid at0–15mmol/L.How-ever,in the case of20mmol/L amino acid,there was little increase of yield and productivity.In addition,we have observed that GSH yield was not only influenced by the concentration of amino acids but also related to the time that amino acids were added.When the amino acids were added at culture time earlier than30h,the effect of amino acids on GSH production was inconspicuous.The optimal feeding time was found to be40–45h when P.pastoris D18biomass reached above90g(DCW)/L.The time course of fed-batch cultivation of P.pastoris D18with amino acids addition was shown in Fig.4.When 15mmol/L of each amino acid was added at44h,the GSH content was increased rapidly from1.03to4.15g/L at 50h,which was351%higher than that without any amino acid addition.The biomass reached98.15g(DCW)/L at15%16% 59%10%0-50(mg/L)51-100(mg/L)101-200(mg/L)Table2Effects of carbon sources and nitrogen sources on GSH productionCarbon source a GSH yield(mg/L)Nitrogen source b GSH yield(mg/L)Glucose227.9Yeast extract285.4 Glycerol217.8Peptone265.9 Sucrose159.2Corn steep liquor powder208.9Corn gluten meal152.5P.pastoris D18cultured in the YPD medium in which GSH yield was 227.9mg/L was used as the control.Each trial was conducted in triplicatea2%(w/v)of various carbon sources were instead of glucose in the YPD mediumb3%(w/v)nitrogen sources were instead of yeast extract and peptone in the YPD mediumthe end of fermentation.The result indicated that the recombinant yeast displayed high GSH synthesis activity for converting the three precursor amino acids into GSH in high-cell-density culture process.During the fermentation process,the glucose was fed to obtain a high-density of biomass.The total consumption of glucose(including the glucose used in fermentation medium)was130g/L of fermentation broth,and the GSH yield on glucose was 32mg(GSH)/g(glucose).Recently,production of GSH by S.cerevisiae or Can-dida utilis were reported by several research groups,the reported yield of GSH in batch culture ranged from800to 2,000mg/L[12–14,16].Among them,Wen et al.obtained the maximal GSH yield(2.19g/L)by high-cell-density fed-batch culture of S.cerevisiae and amino acid modu-lation[14].In our study,the recombinant P.pastoris D18 showed higher biomass and GSH synthesis capability during fed-batch culture.Moreover,glucose was used as carbon source instead of glycerol,which is a common carbon source for P.pastoris fermentation,and the time course was controlled within50h.All the factors con-tribute to low production cost and therefore make it possible to produce GSH in industrial scale. ConclusionThe present studies show that the recombinant P.pastoris expressing heterologous GSH synthesis genes is an alter-native microorganism for GSH production.The high-cell-density culture and amino acid addition were successfully combined together to maximize GSH production by cul-turing the recombinant P.pastoris,which allows to obtain GSH with high yield and relatively low cost. Acknowledgments This work was supported by Creative Foundation Grant of Shanghai Institute of Pharmaceutical Industry.References1.Meister A,Andersen ME(1983)Glutathione.Annu Rev Biochem52:711–7602.Flohe L(1985)The glutathione peroxidase reaction:molecularbasis of the antioxidant function of selenium in mammals.Curr Top Cell Regul27:473–4783.Vartanyan LS,Gurevich SM,Kozachenko AI,Nagler LG,Lozovskaya EL,Burlakova EB(2000)Changes in superoxide production rate and in superoxide dismutase and glutathione peroxidase activities in subcellular organelles in mouse liver under exposure to low doses of low-intensity radiation.Biochem (Mosc)65:442–4464.Ray S,Watkins DN,Misso NL,Thompson PJ(2002)Oxidantstress induces gamma-glutamylcysteine synthetase and glutathi-one synthesis in human bronchial epithelial NCI-H292cells.Clin Exp Allergy32:571–5775.Singh RJ(2002)Glutathione:a marker and antioxidant for aging.J Lab Clin Med140:380–3816.Rolseth V,Djurhuus R,Svardal AM(2002)Additive toxicity oflimonene and50%oxygen and the role of glutathione in detox-ification in human lung cells.Toxicology170:75–887.Penninckx M(2000)A short review on the role of glutathione inthe response of yeasts to nutritional,environmental,and oxidative stresses.Enzyme Microb Technol26:737–7428.Droge W,Breitkreutz R(2000)Glutathione and immune func-tion.Proc Nutr Soc59:595–600Table3Effect of precursor amino acids on synthesis of glutathione in fed-batch culture of P.pastoris D18Amino acids mixture GSH yield(g/L)GSH productivity(mg/LÁh)Without addition of amino acid0.9232.85mmol/L glutamic acid,5mmol/L cysteine,5mmol/L glycine 2.0340.610mmol/L glutamic acid,10mmol/L cysteine,10mmol/L glycine 2.7254.415mmol/L glutamic acid,15mmol/L cysteine,15mmol/L glycine 4.1583.020mmol/L glutamic acid,20mmol/L cysteine,20mmol/L glycine 4.2683.5Fed-batch culture was carried out in a5L fermentor at30°C and pH5.0.The fermentation time was50h,and amino acids were added at44h9.Wei G,Li Y,Du G,Chen J(2003)Effect of surfactants onextracellular accumulation of glutathione by Saccharomyces cerevisiae.Process Biochem38:1133–113810.Yoshida K,Hariki T,Inoue H,Nakamura T(2002)External skinpreparation for whitening.JP Patent2,002,284,66411.Sakato K,Tanaka H(1992)Advanced control of glutathionefermentation process.Biotechnol Bioeng40:904–91212.Wang Z,Tan T,Song J(2007)Effect of amino acids addition andfeedback control strategies on the high-cell-density cultivation of Saccharomyces cerevisiae for glutathione production.Process Biochem42:108–11113.Zhang T,Wen S,Tan T(2007)Optimization of the medium forglutathione production in Saccharomyces cerevisiae.Process Biochem42:454–45814.Wen S,Zhang T,Tan T(2006)Maximizing production ofglutathione by amino acid modulation and high-cell-density fed-batch culture of Saccharomyces cerevisiae.Process Biochem 41:2424–242815.Kazue Y,Kaori Y(2006)Mutant yeast,method of producingglutathione-rich yeast,culture thereof,fraction thereof,yeast extract and glutathione-containing foods and drinks.WO Patent 2,006,013,73616.Santos LO,Gonzales TA,Ubeda BT,Alegre RM(2007)Influ-ence of culture conditions on glutathione production by Saccharomyces cerevisiae.Appl Microbiol Biotechnol77:763–76917.Leuenberger HG(1972)Cultivation of Saccharomyces cerevisiaein continuous culture II.Influence of the crabtree effect on the growth characteristics of Saccharomyces cerevisiae grown in a glucose limited chemostat.Arch Mikrobiol83:347–35818.Cereghino JL,Cregg JM(2000)Heterologous protein expressionin the methylotrophic yeast Pichia pastoris.FEMS Microbiol Rev24:45–6619.Shay LK,Hunt HR,Wegner GH(1987)High productivity fer-mentation process for cultivating industrial microorganisms.J Ind Microbiol2:79–8520.Sambrook J,Russell DW(2001)Molecular cloning:a laboratorymanual,3rd edn edn.Cold Spring Harbor Laboratory Press,New York21.Ellman GL(1959)Tissue sulfhydryl groups.Arch BiochemBiophys82:70–7722.Liu J,Wang Y-q,Liu G,Zhang B-r(2004)Comparison of threemethods for determination of glutathione(Chinese).J Beijing Univ Chem Technol31:35–3823.Johnston RB,Bloch K(1951)Enzymatic synthesis of glutathione.J Biol Chem188:221–24024.Snoke JE,Bloch K(1952)Formation and utilization of gamma-glutamylcysteine in glutathione synthesis.J Biol Chem199:407–41425.Snoke JE,Yanari S,Bloch K(1953)Synthesis of glutathionefrom gamma-glutamylcysteine.J Biol Chem201:573–58626.Ohtake Y,Yabuuchi S(1991)Molecular cloning of the gamma-glutamylcysteine synthetase gene of Saccharomyces cerevisiae.Yeast7:953–96127.Inoue Y,Sugiyama K,Izawa S,Kimura A(1998)Molecularidentification of glutathione synthetase(GSH2)gene from Sac-charomyces cerevisiae.Biochim Biophys Acta1395:315–320 28.Alfafara C,Kanda A,Shioi T,Shimizu H,Shioya S,Suga K(1992)Effect of amino acids on glutathione production by Sac-charomyces cerevisiae.Appl Microbiol Biotechnol36:538–540 29.Cha JY,Park JC,Jeon BS,Lee YC,Cho YS(2004)Optimalfermentation conditions for enhanced glutathione production by Saccharomyces cerevisiae FF-8.J Microbiol42:51–5530.Wen S,Zhang T,Tan T(2005)Optimization of the amino acidcomposition in glutathione fermentation.Process Biochem 40:3474–3479。