化学药物质量控制分析方法验证指导原则
- 格式:ppt
- 大小:2.22 MB
- 文档页数:23
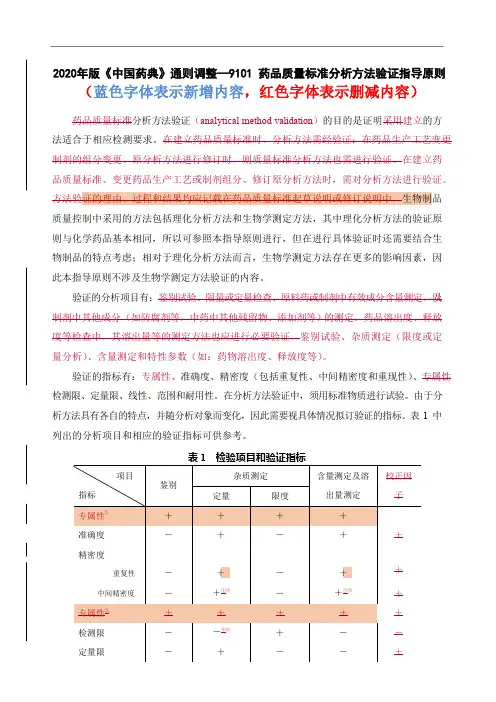
2020年版《中国药典》通则调整—9101 药品质量标准分析方法验证指导原则(蓝色字体表示新增内容,红色字体表示删减内容)药品质量标准分析方法验证(analytical method validation)的目的是证明采用建立的方法适合于相应检测要求。
在建立药品质量标准时,分析方法需经验证;在药品生产工艺变更、制剂的组分变更、原分析方法进行修订时,则质量标准分析方法也需进行验证。
在建立药品质量标准、变更药品生产工艺或制剂组分、修订原分析方法时,需对分析方法进行验证。
质量控制中采用的方法包括理化分析方法和生物学测定方法,其中理化分析方法的验证原则与化学药品基本相同,所以可参照本指导原则进行,但在进行具体验证时还需要结合生物制品的特点考虑;相对于理化分析方法而言,生物学测定方法存在更多的影响因素,因此本指导原则不涉及生物学测定方法验证的内容。
验证的分析项目有:鉴别试验、限量或定量检查、原料药或制剂中有效成分含量测定,以及制剂中其他成分(如防腐剂等,中药中其他残留物、添加剂等)的测定。
药品溶出度、释放度等检查中,其溶出量等的测定方法也应进行必要验证。
鉴别试验、杂质测定(限度或定量分析)、含量测定和特性参数(如:药物溶出度、释放度等)。
验证的指标有:专属性、准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐用性。
在分析方法验证中,须用标准物质进行试验。
由于分析方法具有各自的特点,并随分析对象而变化,因此需要视具体情况拟订验证的指标。
表1 中列出的分析项目和相应的验证指标可供参考。
方法验证内容如下。
三一、专属性专属性系指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的分析方法能正确测定出被测物的能力。
鉴别反应、杂质检査和含量测定方法,均应考察其专属性。
如方法专属性不强,应采用多种不同原理的方法予以补充。
1.鉴别反应应能区分可能共存的物质或结构相似的化合物。
不含被测成分的供试品,以及结构相似或组分中的有关化合物,应均呈阴性反应。

一、准确度准确度系指采用该方法测定的结果与真实值或参考值接近的程度,一般用回收率(%)表示。
准确度应在规定的范围内测定。
1.化学药含量测定方法的准确度原料药采用对照品进行测定,或用本法所得结果与已知准确度的另一个方法测定的结果进行比较。
制剂可在处方量空白辅料中,加入已知量被测物对照品进行测定。
如不能得到制剂辅料的全部组分,可向待测制剂中加人已知量的被测物对照品进行测定,或用所建立方法的测定结果与已知准确度的另一种方法测定结果进行比较。
准确度也可由所测定的精密度、线性和专属性推算出来。
2.化学药杂质定量测定的准确度可向原料药或制剂处方量空白辅料中加人已知量杂质进行测定。
如不能得到杂质或降解产物对照品,可用所建立方法测定的结果与另一成熟的方法进行比较,如药典标准方法或经过验证的方法。
在不能测得杂质或降解产物的校正因子或不能测得对主成分的相对校正因子的情况下,可用不加校正因子的主成分自身对照法计算杂质含量。
应明确表明单个杂质和杂质总量相当于主成分的重量比(%) 或面积比(% )。
3.中药化学成分测定方法的准确度可用对照品进行加样回收率测定,即向已知被测成分含量的供试品中再精密加人一定量的被测成分对照品,依法测定。
用实测值与供试品中含有量之差,除以加入对照品量计算回收率。
在加样回收试验中须注意对照品的加人量与供试品中被测成分含有量之和必须在标准曲线线性范围之内;加入对照品的量要适当,过小则引起较大的相对误差,过大则干扰成分相对减少,真实性差。
回收率:%= (C - A ) /S X 100%式中:A为供试品所含被测成分量;B 为加入对照品量;C 为实测值。
4.校正因子的准确度对色谱方法而言,绝对(或定量)校正因子是指单位面积的色谱峰代表的待测物质的量。
待测定物质与所选定的参照物质的绝对校正因子之比,即为相对校正因子。
相对校正因子计算法常应用于化学药有关物质的测定、中药材及其复方制剂中多指标成分的测定。
校正因子的表示方法很多,本指导原则中的校正因子是指气相色谱法和髙效液相色谱法中的相对重量校正因子。

国家食品药品监督管理局关于发布化学药物稳定性研究等16个技术指导原则的通知文章属性•【制定机关】国家食品药品监督管理局(已撤销)•【公布日期】2005.03.18•【文号】国食药监注[2005]106号•【施行日期】2005.03.18•【效力等级】部门规范性文件•【时效性】现行有效•【主题分类】药政管理正文国家食品药品监督管理局关于发布化学药物稳定性研究等16个技术指导原则的通知(国食药监注[2005]106号)各省、自治区、直辖市食品药品监督管理局(药品监督管理局):为促进我国药品的研究开发,指导药物研究单位用科学规范的方法开展药品研究工作,我局自2003年5月开始正式启动了化学药物研究技术指导原则的起草和修订工作。
指导原则的修订是以《中华人民共和国药品管理法》和《药品注册管理办法》为依据,以科学性、前瞻性、可操作性为指导思想,充分借鉴了ICH等技术指导原则。
目前我局已制定了化学药物稳定性等16个研究技术指导原则(见附件),现予发布,请参照执行。
附件:1.化学药物稳定性研究技术指导原则(略)2.化学药物原料药制备和结构确证研究的技术指导原则(略)3.化学药物杂质研究的技术指导原则(略)4.化学药物制剂研究基本技术指导原则(略)5.化学药物质量控制分析方法验证技术指导原则(略)6.化学药物残留溶剂研究的技术指导原则(略)7.化学药物临床药代动力学研究技术指导原则(略)8.化学药物制剂人体生物利用度和生物等效性研究技术指导原则(略)9.化学药物临床试验报告的结构与内容技术指导原则(略)10.化学药物和生物制品临床试验的生物统计学技术指导原则(略)11.化学药物急性毒性试验技术指导原则(略)12.化学药物长期毒性试验技术指导原则(略)13.化学药物一般药理学研究技术指导原则(略)14.化学药物刺激性、过敏性和溶血性研究技术指导原则(略)15.化学药物非临床药代动力学研究技术指导原则(略)16.化学药物质量标准建立的规范化过程技术指导原则(略)国家食品药品监督管理局二○○五年三月十八日。
9101 药品质量标准分析方法验证指导原则药品质量标准分析方法验证(analytical method validation)的目的是证明采用建立的方法适合于相应检测要求。
在建立药品质量标准时,分析方法需经验证;在药品生产工艺变更、制剂的组分变更、原分析方法进行修订时,则质量标准分析方法也需进行验证。
在建立药品质量标准、变更药品生产工艺或制剂组分、修订原分析方法时,需对分析方法进行①已有重现性验证,不需验证中间精密度。
②如一种方法不够专属,可用其他分析方法予以补充。
③视具体情况予以验证。
一二、准确度准确度系指采用该所建立方法测定的结果与真实值或参比值接近的程度,一般用回收率(%)表示。
准确度应在规定的线性范围内测定试验。
准确度也可由所测定的精密度、线性和专属性推算出来。
1.化学药含量测定方法的准确度原料药采用对照品可用已知纯度的对照品或供试品进行测定,或用本法所得所测定结果与已知准确度的另一个方法测定的结果进行比较。
制剂可在处方量空白辅料中,加入已知量被测物对照品进行测定。
如不能得到制剂辅料的全部组分,可向待测制剂中加入已知量的被测物进行测定,或用所建立方法的测定结果与已知准确度的另一种个方法测定结果进行比较。
准确度也可由所测定的精密度、线性和专属性推算出来。
对色谱方法而言,绝对(或定量)校正因子是指单位面积的色谱峰代表的待测物质的量。
待测定物质与所选定的参照物质的绝对校正因子之比,即为相对校正因子。
相对校正因子计算法常应用于化学药有关物质的测定、中药材及其复方制剂中多指标成分的测定。
校正因子的表示方法很多,本指导原则中的校正因子是指气相色谱法和髙效液相色谱法中的相对重量校正因子。
相对校正因子可采用替代物(对照品)和被替代物(待测物)标准曲线斜率比值进行比较获得;采用紫外吸收检测器时,可将替代物(对照品)和被替代物(待测物)在规定波长和溶剂条件下的吸收系数比值进行比较,计算获得。
54. 数据要求在规定范围内,取同一浓度(相当于100%浓度水平)的供试品,用至少测定6份样品的结果进行评价;或设计*此表源自AOAC《Guidelines for Single Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals》二三、精密度精密度系指在规定的测定条件下,同一份均匀供试品,经多次取样测定所得结果之间的接近程度。

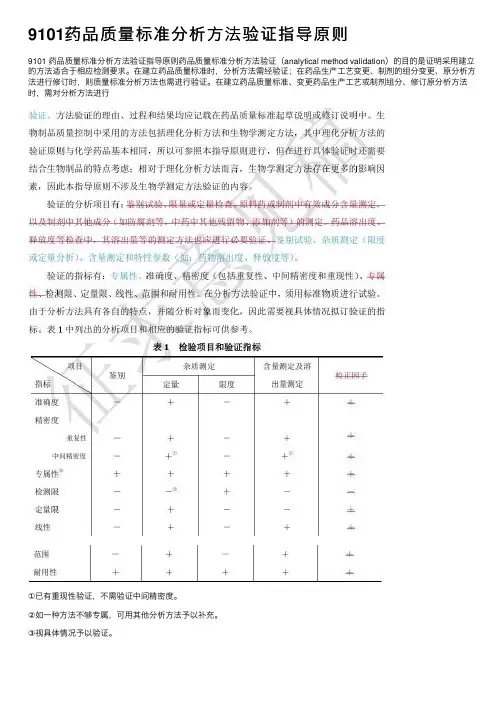
《中国药典》2015版四部9101药品质量标准分析⽅法验证指导原则《中国药典》2015版四部9101药品质量标准分析⽅法验证指导原则药品质量标准分析⽅法验证的⽬的是证明采⽤的⽅法适合于相应检测要求。
在建⽴药品质量标准时,分析⽅法需经验证;在药品⽣产⼯艺变更、制剂的组分变更、原分析⽅法进⾏修订时,则质量标准分析⽅法也需进⾏验证。
⽅法验证理由、过程和结果均应记载在药品质量标准起草说明或修订说明中。
⽣物制品质量控制中采⽤的⽅法包括理化分析⽅法和⽣物学测定⽅法,其中理化分析⽅法的验证原则与化学药品基本相同,所以可参照本指导原则进⾏,但在进⾏具体验证时还需要结合⽣物制品的特点考虑;相对于理化分析⽅法⽽⾔,⽣物学测定⽅法存在更多的影响因素,因此本指导原则不涉及⽣物学测定⽅法验证的内容。
验证的分析项⽬有:鉴别试验、限量或定量检查、原料药或制剂中有效成分含量测定,以及制剂中其他成分(如防腐剂等,中药中其他残留物、添加剂等)的测定。
药品溶出度、释放度等检查中,其溶出量等的测定⽅法也应进⾏必要验证。
验证指标有:准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐⽤性。
在分析⽅法验证中,须采⽤标准物质进⾏试验。
由于分析⽅法具有各⾃的特点,并随分析对象⽽变化,因此需要视具体⽅法拟订验证的指标。
表1中列出的分析项⽬和相应的验证指标可供参考。
表1检验项⽬和验证指标项⽬鉴别杂质测定含量测定及校正因⼦内容定量限度溶出量测定准确度-+-++精密度重复性-+-++中间精密度-+①-+①+专属性②+++++检测限--③+--定量限-+--+线性-+-++范围-+-++耐⽤性+++++①巳有重现性验证,不需验证中间精密度。
②如⼀种⽅法不够专属,可⽤其他分析⽅法予以补充。
③视具体情况予以验证。
⼀、准确度准确度系指采⽤该⽅法测定的结果与真实值或参考值接近的程度,⼀般⽤回收率(%)表⽰。
准确度应在规定的范围内测定。
9101药品质量标准分析⽅法验证指导原则9101 药品质量标准分析⽅法验证指导原则药品质量标准分析⽅法验证(analytical method validation)的⽬的是证明采⽤建⽴的⽅法适合于相应检测要求。
在建⽴药品质量标准时,分析⽅法需经验证;在药品⽣产⼯艺变更、制剂的组分变更、原分析⽅法进⾏修订时,则质量标准分析⽅法也需进⾏验证。
在建⽴药品质量标准、变更药品⽣产⼯艺或制剂组分、修订原分析⽅法时,需对分析⽅法进⾏①已有重现性验证,不需验证中间精密度。
②如⼀种⽅法不够专属,可⽤其他分析⽅法予以补充。
③视具体情况予以验证。
⼀⼆、准确度准确度系指采⽤该所建⽴⽅法测定的结果与真实值或参⽐值接近的程度,⼀般⽤回收率(%)表⽰。
准确度应在规定的线性范围内测定试验。
准确度也可由所测定的精密度、线性和专属性推算出来。
1.化学药含量测定⽅法的准确度原料药采⽤对照品可⽤已知纯度的对照品或供试品进⾏测定,或⽤本法所得所测定结果与已知准确度的另⼀个⽅法测定的结果进⾏⽐较。
制剂可在处⽅量空⽩辅料中,加⼊已知量被测物对照品进⾏测定。
如不能得到制剂辅料的全部组分,可向待测制剂中加⼊已知量的被测物进⾏测定,或⽤所建⽴⽅法的测定结果与已知准确度的另⼀种个⽅法测定结果进⾏⽐较。
准确度也可由所测定的精密度、线性和专属性推算出来。
对⾊谱⽅法⽽⾔,绝对(或定量)校正因⼦是指单位⾯积的⾊谱峰代表的待测物质的量。
待测定物质与所选定的参照物质的绝对校正因⼦之⽐,即为相对校正因⼦。
相对校正因⼦计算法常应⽤于化学药有关物质的测定、中药材及其复⽅制剂中多指标成分的测定。
校正因⼦的表⽰⽅法很多,本指导原则中的校正因⼦是指⽓相⾊谱法和髙效液相⾊谱法中的相对重量校正因⼦。
相对校正因⼦可采⽤替代物(对照品)和被替代物(待测物)标准曲线斜率⽐值进⾏⽐较获得;采⽤紫外吸收检测器时,可将替代物(对照品)和被替代物(待测物)在规定波长和溶剂条件下的吸收系数⽐值进⾏⽐较,计算获得。



药品质量标准分析方法验证指导原则《中国药典》2015年版药品质量标准分析方法验证的目的是证明采用的方法适合于相应检测要求。
在建立药品质量标准时,分析方法需经验证;在药品生产工艺变更、制剂的组分变更、原分析方法进行修订时,则质量标准分析方法也需进行验证。
方法验证理由、过程和结果均应记载在药品质量标准起草说明或修订说明中。
生物制品质量控制中采用的方法包括理化分析方法和生物学测定方法,其中理化分析方法的验证原则与化学药品基本相同,所以可参照本指导原则进行,但在进行具体验证时还需要结合生物制品的特点考虑;相对于理化分析方法而言,生物学测定方法存在更多的影响因素,因此本指导原则不涉及生物学测定方法验证的内容。
验证的分析项目有:鉴别试验、限量或定量检查、原料药或制剂中有效成分含量测定,以及制剂中其他成分(如防腐剂等,中药中其他残留物、添加剂等)的测定。
药品溶出度、释放度等检查中,其溶出量等的测定方法也应进行必要验证。
验证指标有:准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐用性。
在分析方法验证中,须采用标准物质进行试验。
由于分析方法具有各自的特点,并随分析对象而变化,因此需要视具体方法拟订验证的指标.表1中列出的分析项目和相应的验证指标可供参考。
一、准确度准确度系指采用该方法测定的结果与真实值或参考值接近的程度,一般用回收率(%)表示.准确度应在规定的范围内测定。
1。
化学药含量测定方法的准确度原料药采用对照品进行测定,或用本法所得结果与已知准确度的另一个方法测定的结果进行比较。
制剂可在处方量空白辅料中,加入已知量被测物对照品进行测定。
如不能得到制剂辅料的全部组分,可向待测制剂中加入已知量的被测物对照品进行测定,或用所建立方法的测定结果与已知准确度的另一种方法测定结果进行比较。
准确度也可由所测定的精密度、线性和专属性推算出来。
2。
化学药杂质定量测定的准确度可向原料药或制剂处方量空白辅料中加入已知量杂质进行测定。
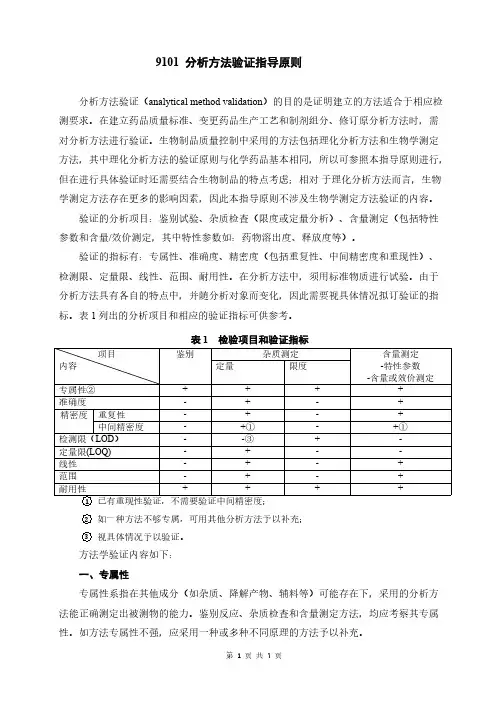
9101分析方法验证指导原则分析方法验证(analytical method validation)的目的是证明建立的方法适合于相应检测要求。
在建立药品质量标准、变更药品生产工艺和制剂组分、修订原分析方法时,需对分析方法进行验证。
生物制品质量控制中采用的方法包括理化分析方法和生物学测定方法,其中理化分析方法的验证原则与化学药品基本相同,所以可参照本指导原则进行,但在进行具体验证时还需要结合生物制品的特点考虑;相对于理化分析方法而言,生物学测定方法存在更多的影响因素,因此本指导原则不涉及生物学测定方法验证的内容。
验证的分析项目:鉴别试验、杂质检查(限度或定量分析)、含量测定(包括特性参数和含量/效价测定,其中特性参数如:药物溶出度、释放度等)。
验证的指标有:专属性、准确度、精密度(包括重复性、中间精密度和重现性)、检测限、定量限、线性、范围、耐用性。
在分析方法中,须用标准物质进行试验。
由于分析方法具有各自的特点中,并随分析对象而变化,因此需要视具体情况拟订验证的指标。
表1列出的分析项目和相应的验证指标可供参考。
表1检验项目和验证指标项目内容鉴别杂质测定含量测定-特性参数-含量或效价测定定量限度专属性②++++准确度-+-+精密度重复性-+-+中间精密度-+①-+①检测限(LOD)--③+-定量限(LOQ)-+--线性-+-+范围-+-+耐用性++++ 1已有重现性验证,不需要验证中间精密度;2如一种方法不够专属,可用其他分析方法予以补充;3视具体情况予以验证。
方法学验证内容如下:一、专属性专属性系指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的分析方法能正确测定出被测物的能力。
鉴别反应、杂质检査和含量测定方法,均应考察其专属性。
如方法专属性不强,应采用一种或多种不同原理的方法予以补充。
1.鉴别反应应能区分可能共存的物质或结构相似的化合物。
不含被测成分的供试品,以及结构相似或组分中的有关化合物,应均呈阴性反应。

分析方法验证一.方法验证概述1方法验证概念解析2各方指导原则3方法验证的常见困难4风险管理在分析方法中的应用5限度与定量验证的区别与适用情况6方法验证的前期准备7方法验证的方案和报告首先申明是方法验证:ICHQ2,分析方法的验证是为了证明该方法与预期的目的相一致。
中国药典,药品质量标准分析方法验证的目的是证明采用的方法适合于相应的检测要求。
USP1225,通过实验室研究确认方法的性能参数可以满足预期的检测要求。
什么是检测要求:结实(耐用性好)、准确、稳定是否需要验证?需要经过验证的:含量测定、有关物质、残留溶剂、基因毒杂质、其他特定杂质、鉴别(仪器)、溶出度、释放度、粒径(激光衍射法)、其他采用液相色谱及光谱法等检测项目。
不需要经过验证的:炽灼残渣、干燥失重、鉴别(化学法)、性状、水分、酸碱度、PH、其他理化检测。
注意事项:方法是否需要验证,应当基于风险评估的判断,惯例只是对于风险的一致认知,并不适用于所有情况。
注册用方法验证的一般要求代表性的样品:验证用到的样品应当能够代表上市批的质量,工艺一致,批量相当。
规范的过程:验证过程应当符合GMP或GMP 类似要求方法确认与转移相关指导原则12015年版中国药典四部91012.化学药物质量控制分析方法验证技术指导原则P12254.ICHQ25.FDA的Analytical procedures and method validation方法验证的常见困难:1 已经验证的方法转移失败。
2 已经验证的方法在日常检测过程中不适用。
3方法验证过程中发现方法不适用4 某些验证项目需要多次验证才能获得满意的结果。
分析方法风险应对前置于方法开发阶段1在方法设计阶段应当预测全生命周期内可能存在的风险,如方法转移、可能的OOT/OOS、系统适用性试验失败原因、监管法规的变化、技术进步等。
2 收集相关方的需求,包括QC实验室、合成部门、制剂部门等。
风险管理在方法验证中的应用1什么是风险?不确定性对目标的影响2什么是风险管理?在风险方面,指导和控制组织的协调活动3 方法验证中应当结合分析方法的生命周期进行系统的风险评估,识别可能存在的风险,进行与等级相应的应对。
化学药物分析方法验证的内容和评价质量可控是活性化合物成药及进行药品安全有效性评价的前提。
对药品的质量进行控制应该是多方面的,其中包括生产环境的控制、原辅料来源和质量的控制、生产工艺过程的控制,以及按照质量标准进行成品检验控制等。
但是,在药品研发过程中,揭示药品的品质、控制药品的质量,通常是要针对研制药品的特性 (主药的理化性质及制剂的质量要求等 )确定相应的研究测试项目,再根据测试项目的需要,建立适宜的分析方法 (包括方法的选择和方法的验证 ),最后是根据药品质量的要求及安全性研究结果确定各测试项目的限度,起草制订检验药品质量的质量标准。
分析方法是揭示药品品质的工具和手段,方法验证是判断采用的分析方法是否科学、可行的过程。
实际上,方法验证就是根据确定的检测项目的要求,预先设置一定的验证内容,并通过设计合理的试验来验证所采用的分析方法能否符合检测项目的要求。
下面就根据《化学药物质量控制分析方法验证技术指导原则》解读分析方法验证的内容和评价要点,在对目前申报资料中分析方法验证工作中存在的问题进行分析的基础上,以高效液相色谱法为例,阐述方法验证的思路及验证要点,希望能对药物研发者有所帮助。
一、方法验证的内容首先应该明确的是:方法验证的内容应根据检测项目的要求,同时结合所采用的分析方法的特点确定。
相同的分析方法用于不同的检测项目时,其验证要求是不同的。
例如,采用高效液相色谱法用于制剂的鉴别和杂质定量检查时的验证要求是不同的;前者重点要验证方法的专属性,而后者重点要验证方法的专属性、准确度和定量限。
通常需要验证的检测项目:鉴别、杂质检查 (限度试验、定量试验 )、定量测定 (含量测定、溶出度、释放度等 ),还有其他特定的检测项目 (粒径分布、分子量分布)等。
方法验证的内容包括专属性、线性、范围、准确度、精密度、检测限、定量限、耐用性 (粗放度 )和系统适用性等。
1、专属性专属性系指在其他成分 (如杂质、降解物、辅料等 )存在下,采用的分析方法能够正确鉴定、检出被测物的特性。
方法验证指导原则-《中国药典》2015年版第四部9101药品质量标准分析方法验证指导原则量。
③应注意固体制剂的晶型原料药含量应在标准曲线的线性范围内。
④应使用外标标准物质ai2o3对仪器及数据进行校正。
方法3差示扫描量热法(DSC)定置分析方法,获得供试品晶型含量数据。
采用DSC定量分析的晶型物质一般应具有不同的熔融吸热峰值,且晶型样品质量与吸热量呈正比关系。
(a)晶型原料药分析:精密称量不同质量晶型样品,建立质量与热量的热焓值的线性关系,绘制标准曲线,定量测定样品的晶型含量。
(b)混晶原料药分析:当不同晶型含量与热焓呈正比关系,采用精密称量配制不同晶型含量的混晶样品,建立晶型含量与热焓值的线性关系,绘制标准曲线,定量测定混晶样品中的晶型含量。
(c)方法说明:①仅适用于晶型原料药定量分析。
②对熔融吸热峰值相差大的混晶原料供试品,建立标准曲线时线性范围较宽;熔融吸热峰值相差小的混晶样品,建立标准曲线时线性范围较窄。
③有时DSC法仅能作为限量检测方法。
方法4红外光谱(IR)定量分析方法,获得供试品晶型含量数据。
采用IR法可以对晶型原料药或固体制剂进行定量分析,常用的方法为相对峰强度法。
晶型特征峰选取原则:①分别选取2种晶型特有的红外光谱吸收峰作为特征峰。
②2种晶型的特征峰应独立而不受对方干扰。
③特征峰强度应与晶型成分含量呈对应线性关系。
对压力可致晶型状态发生转变的晶型原料供试品,制样时应避免压片法。
(a)晶型原料药分析:采用相对峰强度法时分别选择2 种晶型成分的特征吸收峰位置匕与b2,在同一红外光谱图上读取2种晶型成分的特征吸收峰的吸光度值V1与42,计算二者特征吸收峰的吸光度比值r。
通过配制一系列不同晶型比例的混晶样品,建立特征吸收峰的吸光度比值的对数值与晶型含量间的线性关系,绘制标准曲线,实现对混晶样品的晶型含量进行定量分析。
(b)制剂中晶型原料药成分分析:采用相对峰强度法时分别选择晶型原料药特征吸收峰位置h与空白辅料的特征吸收峰位置b2,在同一红外光谱图上读取2种晶型成分的特征吸收峰的吸光度值八1与A2,计算二者特征吸收峰的吸光度比值~通过配制一系列含有不同质量晶型原料与空白辅料比例混合样品,建立特征吸收峰的吸光度比值的对数值与晶型原料药含量间的线性关系,绘制标准曲线,实现对固体制剂中晶型原料药含量进行定量分析。
(完整版)方法学验证指导原则一、准确度准确度系指采用该方法测定的结果与真实值或参考值接近的程度,一般用回收率(%)表示。
准确度应在规定的范围内测定。
1.化学药含量测定方法的准确度原料药采用对照品进行测定,或用本法所得结果与已知准确度的另一个方法测定的结果进行比较。
制剂可在处方量空白辅料中,加入已知量被测物对照品进行测定。
如不能得到制剂辅料的全部组分,可向待测制剂中加人已知量的被测物对照品进行测定,或用所建立方法的测定结果与已知准确度的另一种方法测定结果进行比较。
准确度也可由所测定的精密度、线性和专属性推算出来。
2.化学药杂质定量测定的准确度可向原料药或制剂处方量空白辅料中加人已知量杂质进行测定。
如不能得到杂质或降解产物对照品,可用所建立方法测定的结果与另一成熟的方法进行比较,如药典标准方法或经过验证的方法。
在不能测得杂质或降解产物的校正因子或不能测得对主成分的相对校正因子的情况下,可用不加校正因子的主成分自身对照法计算杂质含量。
应明确表明单个杂质和杂质总量相当于主成分的重量比(%) 或面积比(% )。
3.中药化学成分测定方法的准确度可用对照品进行加样回收率测定,即向已知被测成分含量的供试品中再精密加人一定量的被测成分对照品,依法测定。
用实测值与供试品中含有量之差,除以加入对照品量计算回收率。
在加样回收试验中须注意对照品的加人量与供试品中被测成分含有量之和必须在标准曲线线性范围之内;加入对照品的量要适当,过小则引起较大的相对误差,过大则干扰成分相对减少,真实性差。
回收率:%= (C - A ) /S X 100%式中:A为供试品所含被测成分量;B 为加入对照品量;C 为实测值。
4.校正因子的准确度对色谱方法而言,绝对(或定量)校正因子是指单位面积的色谱峰代表的待测物质的量。
待测定物质与所选定的参照物质的绝对校正因子之比,即为相对校正因子。
相对校正因子计算法常应用于化学药有关物质的测定、中药材及其复方制剂中多指标成分的测定。
欧洲药典方法验证指导原则简介欧洲药典方法验证指导原则是欧洲药典委员会制定的一套用于验证药品质量的指导原则。
这些原则旨在确保药品在生产和使用过程中的质量和安全性。
本文将详细介绍欧洲药典方法验证指导原则的背景、目的、主要内容以及应用等方面,以便读者对其有更全面和深入的了解。
背景随着现代医学和制药技术的不断发展,对于药品质量和安全性的要求越来越高。
为了确保药品有效性和稳定性,各国纷纷建立了一套严格的质量控制体系。
欧洲药典方法验证指导原则就是其中之一。
目的欧洲药典方法验证指导原则旨在提供一套统一而标准化的方法来验证各种类型的药物。
这些验证方法可以确保生产商能够按照规定进行质量控制,并使监管机构能够评估产品是否符合规定标准。
主要内容1. 方法选择和开发在欧洲药典方法验证指导原则中,首要任务是选择和开发适合的分析方法。
这些方法应该能够准确、可靠地评估药品的质量特性。
2. 方法验证一旦选择和开发了合适的分析方法,就需要对其进行验证。
方法验证是通过实验来证明所选方法可以在一定程度上提供准确和可靠的结果。
3. 方法转移当一个药品生产企业决定采用某种特定的分析方法时,他们需要将该方法从一个实验室转移到另一个实验室。
欧洲药典方法验证指导原则提供了相应的指导原则。
4. 方法更新随着科学技术的不断进步,可能会出现新的分析方法或对现有分析方法进行改进。
因此,欧洲药典委员会会定期审查和更新已有的验证指导原则。
应用欧洲药典方法验证指导原则广泛应用于制药行业和相关领域。
这些指导原则被用于评估各种类型的药物,包括化学药品、生物制剂、植物提取物等。
遵循欧洲药典方法验证指导原则可以帮助制药企业确保其产品的质量和安全性,并与监管机构保持合规。
这些指导原则也有助于促进国际间的药品贸易和技术交流。
结论欧洲药典方法验证指导原则是欧洲药典委员会为了确保药品质量和安全性而制定的一套标准化方法。
这些原则对于制药企业和监管机构来说都是非常重要的。
通过选择、验证和转移合适的分析方法,制药企业可以确保其产品符合规定标准。
注释:红色带中间横线的文字表示在2020版药典删除了的2015版内容;蓝色字表示2020版药典新增加的。
9101 药品质量标准分析方法验证指导原则药品质量标准分析方法验证(analytical method validation)的目的是证明采用建立的方法适合于相应检测要求。
在建立药品质量标准时,分析方法需经验证;在药品生产工艺变更、制剂的组分变更、原分析方法进行修订时,则质量标准分析方法也需进行验证。
在建立药品质量标准、变更药品生产工艺或制剂组分、修订原分析方法时,需对分析方法进行验证。
方法验证的理由、过程和结果均应记载在药品质量标准起草说明或修订说明中。
生物制品质量控制中采用的方法包括理化分析方法和生物学测定方法,其中理化分析方法的验证原则与化学药品基本相同,所以可参照本指导原则进行,但在进行具体验证时还需要结合生物制品的特点考虑;相对于理化分析方法而言,生物学测定方法存在更多的影响因素,因此本指导原则不涉及生物学测定方法验证的内容。
验证的分析项目有:鉴别试验、限量或定量检查、原料药或制剂中有效成分含量测定,以及制剂中其他成分(如防腐剂等,中药中其他残留物、添加剂等)的测定。
药品溶出度、释放度等检查中,其溶出量等的测定方法也应进行必要验证。
鉴别试验、杂质测定(限度或定量分析)、含量测定(包括特性参数和含量/效价测定,其中特性参数如:药物溶出度、释放度等)。
验证的指标有:专属性、准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐用性。
在分析方法验证中,须采用标准物质进行试验。
由于分析方法具有各自的特点,并随分析对象而变化,因此需要视具体方法情况拟订验证的指标。
表1中列出的分析项目和相应的验证指标可供参考。
②如一种方法不够专属,可用其他分析方法予以补充。
③视具体情况予以验证。
方法验证内容如下。
三一、专属性专属性系指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的分析方法能正确测定出被测物的能力。
药品及生物制品的分析方法和方法验证指导原则目录1.介绍...................... (1)2.背景..................... .. (2)3.分析方法开发. ..................... . (3)4.分析程序内容.............................................. ......... ..................................... .. 3A.原则/范围 (4)B.仪器/设备............................................. . (4)C.操作参数.............................................. .. (4)D.试剂/标准............................................. . (4)E.样品制备.............................................. .. (4)F.标准对照品溶液的制备............................................ .. (5)G.步骤......... ....................................... (5)H.系统适应性..... (5)I.计算 (5)J.数据报告 (5)5.参考标准和教材............................................ (6)6分析方法验证用于新药,仿制药,生物制品和DMF (6)A.非药典分析方法............................................. (6)B.验证特征 (7)C.药典分析方法............................................. .. (8)7.统计分析和模型 (8)A.统计 (8)B.模型 (8)8.生命周期管理分析程序 (9)A.重新验证 (9)B.分析方法的可比性研究............................................ . (10)1.另一种分析方法............................................... .. (10)2.分析方法转移的研究 (11)C.报告上市后变更已批准的新药,仿制药,或生物制品 (11)9.美国FDA方法验证............................................... . (12)10.参考文献前言本指导原则草案,定稿后,将代表美国食品和药物管理局(FDA)目前关于这个话题目前的想法。