间质性肺病进展
- 格式:doc
- 大小:1.24 MB
- 文档页数:10

间质性肺疾病的治疗研究进展摘要:间质性肺疾病属于一组弥漫性疾病,它主要影响肺泡腔和肺间质,出现慢性炎症和肺间质纤维化。
近年来,间质性肺病的发病率有所增加,新的治疗方法的范围也有所扩大。
间质性肺病包括范围广泛的疾病,病因复杂,治疗方案有限。
间质性肺病包括范围广泛的疾病,病因复杂,治疗方案有限。
本研究回顾了治疗间质性肺病的临床进展,目的是有效和安全地治疗间质性肺病。
关键词:间质性肺疾病;治疗进展;肺康复;综述Abstract: Interstitial lung diseases are a group of diffuse diseases that primarily involve the alveolar cavity and interstitial spaces of the lungs and are characterised by chronic inflammation as well as interstitial fibrosis. In recent years, due to the increasing incidence of interstitial lung disease, the scope for research intonew treatments for interstitial lung disease has expanded.Interstitial lung disease involves a large number of diseases with complex etiologies and limited therapeutic options. This articlereviews advances in the clinical management of interstitial lung disease with the aim of treating it effectively and safely.Keywords:interstitial lung disease; treatment advances; pulmonary rehabilitation; review间质性肺疾病(Interstitial lung disease,ILD )属于一组异质性和弥漫性的疾病,主要侵犯肺泡单位和肺泡周围组织,主要影响肺间质。


间质性肺病治疗的研究进展何渝娇1,李风森2*(1.新疆医科大学第四临床医学院,新疆乌鲁木齐830000;2.新疆医科大学附属中医医院,新疆乌鲁木齐830000)摘要:间质性肺病的病因繁多病机复杂,患病率逐年升高,可因弥漫性肺部病变导致肺功能下降,呼吸衰竭,而导致死亡。
目前中医、西医治疗取得了一定疗效。
本文总结归纳了该病中西医治疗的研究现状,以期为中西医治疗本病提供临床思路。
关键词:间质性肺病;中医治疗;西医治疗;研究进展间质性肺疾病(Interstitial lung disease,ILD)是一组肺异质性疾病,包括200多个以肺实质广泛纤维化和/或炎性异常为特征的肺实质病变[1]。
发病原因是肺泡上皮细胞的异常修复造成细胞外基质的大量分泌,进而造成肺组织结构和功能的不可逆损伤,形成肺纤维化。
肺纤维化主要累及肺泡和肺间质的弥散性、致死性的肺部疾病,是所有ILD的终末期。
呼吸衰竭是ILDs晚期或急性恶化后的常见并发症,其预后是呼吸系统疾病中相对较差的。
目前对该病病因、诊断及治疗的研究也在增加,引起越来越多医者对该病的重视,但仍未给出明确指南。
近年来,由于环境恶化,ILD的发病率、患病率正在逐年增高[2]。
ILD其病因繁多病机复杂,分类及亚型多,其中特发性肺纤维化(IPF)是预后最差的,而结缔组织疾病相关的ILD(CTD-ILD)预后相对更好[3]。
尽管所有ILD患者急性呼吸衰竭的死亡率都很高,但与其他纤维化ILD患者相比,IPF因急性呼吸衰竭住院后的一年死亡率更高(分别为87%和71%)[4],其预计生存期为2~3年[5],特发性肺纤维化急性发作(AE-IPF)的中位生存期在22天至4.2个月之间[6]。
本文将对间质性肺病在国内外流行病学的发病特点、诊断的更新、中医及西医治疗的研究进展等做一综述。
1间质性肺病在国内外的流行病学特点随着呼吸系统疾病谱的变化以及对弥漫性间质性肺疾病(DILD)诊治水平的提高,肺间质纤维化的研究日益深入。

间质性肺疾病的诊治进展【中图分类号】r563 【文献标识码】a 【文章编号】1672-3783(2011)09-0425-01【摘要】间质性肺疾病的发病率近几年有所升高,患者可出现肺容量减少、限制性通气功能障碍;最终出现低氧血症和呼吸衰竭。
目前对其诊断较困难,现有的药物在改进病人的肺功能和存活率方面也有限。
故综述近几年的诊断以及治疗进展。
【关键词】间质性肺疾病,特发性间质性肺炎,糖皮质激素间质性肺疾病(interstitial lung disease,ild)是下呼吸道以肺泡壁破坏为主要病变的弥漫性疾病的总称。
主要病理表现为肺泡壁广泛的破坏、肺泡毛细血管的功能丧失、胶原疤痕组织堆积。
故在临床上可出现肺的顺应性降低、肺容量减少和限制性通气功能障碍;并可因细支气管炎变以及肺小血管的闭塞而引起通气血流比例失调和弥散功能的降低,最终发生低氧血症和呼吸衰竭。
目前ild 大约有200多种,特发性间质性肺炎( idiopathic interstitialpneumonias, iips)是间质性肺疾病中最常见和最重要的疾病。
1 ild的诊断进展ild的诊断主要依赖于临床症状、体征、影像学(常规x线检查、hrct)检查、肺功能检查、肺活检组织病理学检查和肺泡灌洗液检查等。
1.1 ild患者大多有进行性呼吸困难、咳嗽、肺部爆裂音(velcro 啰音) 、肺部弥漫性阴影和限制性通气功能障碍,对诊断有帮助,但缺乏特异性。
胸部hrct能够帮助临床医师最大程度地从临床接近病理诊断。
经支气管镜肺活检和经胸壁肺穿刺活检对于肿瘤、结节病和肺泡蛋白沉积症等有较高的诊断价值,但在区分iips的病理类型方面由于标本太小而难于判断,其作用有限。
肺活检是确定iips 病理类型的最佳选择,诊断率可达92% ,但患者往往难以接受[1]。
1.2 ild的诊断步骤如下:首先,确定是否为iips,如有相关的病因可寻,则可能不是iips;若无原因可查, 则可能是iips, 需要行hrct。

基金项目:基金名称:吴阶平医学基金会 课题名称:吉非替尼联合恩度一线治疗EGFR 突变晚期肺腺癌的疗效及机制研究 课题编号:320.6750.17570。
·综述·具有自身免疫特征的间质性肺炎(IPAF)的研究进展刘磊1,高俊珍2(通信作者*)(1.内蒙古医科大学,内蒙古 呼和浩特 010030;2.内蒙古医科大学附属医院,内蒙古 呼和浩特 010030)摘要:间质性肺疾病(ILD )是一组以肺泡单位的炎症和间质纤维化为基本病变的异质性非肿瘤和非感染性肺部疾病的总称,又称为弥漫性实质性肺疾病,在临床中,有很大一部分患者表现出自身免疫特征,但不符合明确的结缔组织病(CTD )的诊断标准。
2015年,美国胸科协会/欧洲呼吸学会(ATS/ERS )首次提出了具有自身免疫特征的间质性肺炎(IPAF )一词[1],IPAF 可以认为是特发性间质性肺炎(IIP ),尤其是特发性肺间质纤维化(IPF )和CTD-ILD 的重叠,IPF 在临床中预后最差,所以将二者区别开来显得格外重要,目前针对IPAF 的临床研究较少,不同地域的临床医生对其认知参差不齐,故本文将近年来针对IPAF 展开的研究做一综述,以期指导临床工作。
关键词:自身免疫特征的间质性肺炎;间质性肺疾病;结缔组织相关的间质性肺疾病中图分类号:R563.1 文献标识码:A DOI :10.3969/j.issn.1671-3141.2021.23.026本文引用格式:刘磊,高俊珍.具有自身免疫特征的间质性肺炎(IPAF)的研究进展[J].世界最新医学信息文摘,2021,21(23):80,86.1 流行病学早在上世纪90年代,就有报道提出多达25%的自身免疫性疾病患者不符合美国风湿病学会(ACR)的CTD 分类标准[2]。
近些年,根据IPAF 的标准来报道的间质性肺疾病的报道较少,有N.O 等人报道IPAF 在间质性肺炎中的发生频率从7.3%-34%不等[3]。

临床肺科杂志2007年1月第12卷第l期・专家论坛・间质性肺疾病的诊断和治疗进展朱元珏间质性肺疾病也被称为ILD,即interstitiallungdisease,由于这一大组疾病所侵犯的并不仅限于肺的间质,近来也有学者把他们称为弥漫性肺实质性疾病(D讧-fuseparenchymallungdjsease,DPLD),可能更贴近这组疾病,但是,间质性肺病ILD已应用多年,为大多数学者所熟悉,仍常与DPLD通用。
DPIJD涵盖许多疾病,各有其临床特点,诊断有其难处,治疗和预后又因诊断而异。
DPLD的诊断目前,一般呼吸界均采用2002年由ArI'S和ERS推荐的DPLD和ⅡP的分类…,即是(1)已知原因的DPLD:如药物,和结缔组织病相关和环境相关的间质性肺病等;(2)肉芽肿性DPLD,如结节病,外源过敏性肺泡炎即HP等;(3)罕见的但具有临床病理特征的DPLD,如淋巴管平滑肌瘤病即LAM,朗格罕细胞肉芽肿病即LcG,肺泡蛋白沉着症即PAP等,(4)特发性间质性肺炎即IIPs四大类,IIP再进一步分为两组(7种),寻常性/特发性即uIP/IPF和非uIP两组,在后一组中又分为DIP,RBILD,AIP,c0P,uP和NsIP六种间质性肺病,(参见图1)。
DPLD的上述1—3三组就涵盖了上百种疾病,其中很多又较少见,不为临床医师所熟悉,诊断本身就较为困难。
但是,若要诊断特发性间质性肺炎,又必须在排除上述三组疾病基础上才能初步判定不是继发性间质性肺炎而是IIP。
为达到此目的除了详尽的临床检查以外还常常需要侵入性检查,如经支气管肺活检,经皮或开胸肺活检,取得肺活检组织,最后通过组织病理学检查,才能判定。
当考虑为IIP时,就需要区分是哪一种IIP。
特发性问质性肺炎和特发性肺纤维化的分类一、IPF的传统认识长期以来,IPF指的是一组原因不明的肺间质病,以进展性的肺纤维化为特征。
但是概念含混,诊断标准不一。
在欧洲,IPF常被另一个名称所替代,即隐原性致纤维化性肺泡炎(cryptogenic舶msingalveolitis,CFA)。




间质性肺炎病的治疗进展从间质性肺炎的症状入手新式中医疗法。
间质性肺炎的出现会打破人们的正常生活,会给患者的健康带来极大的威胁。
间质性肺炎前期还都比较容易控制,但是一旦发展到肺纤维后就已经十分的严重了,给患者带来的不只是身体的痛苦,还让患者心里蒙受了极大的阴影。
因此我们要学会通过一种疾病的症状,知道间质性肺炎病的治疗进展,这样才能有效的控制疾病。
间质性肺炎的概述:间质性肺炎大多由于病毒所致,主要为腺病毒、呼吸道合胞病毒、流感病毒、副流感病毒、麻疹病毒等,其中以腺病毒和流感病毒引起的间质性肺炎较多见,也较严重,常形成坏死性支气管炎及支气管肺炎,病程迁延易演变为慢性肺炎。
或是导致出现间质性肺纤维化,因此不得不让人多加以注意。
而肺间质纤维化则是间质性肺炎的一个阶段。
治疗肺间质纤维化也和治疗其它难度大的疾病一样是一个工程。
治疗目的:争取可逆部份和时间,控制病情发展,改善症状,提高生存质量。
临床最常见的是与自身免疫性疾病相关的肺泡炎和肺间质纤维化。
可以先于自免疫病出现,也可以在自身免疫病发病数年之后出现。
早期常常被作为肺部感染治疗。
间质性肺炎病的治疗进展?间质性肺炎的前期症状轻微甚至不明显,随病程逐渐加重:1、时有呼吸困难,干咳等是间质性肺炎的主要症状。
2、常因感冒、急性呼吸道感染而诱发和加重,且呈进行性加重。
3、逐渐出现呼吸增快但无喘鸣,刺激性咳嗽或有咳痰,少数有发烧、咯血或胸痛。
4、严重后动则出现气喘,心慌出虚汗,全身乏力,体重减轻,唇甲紫绀及杵状指(趾),这些是间质性肺炎症状较严重的时候出现的。
5、作体检时在下肺野可听到湿罗音。
6、在并发肺原性心脏病时有肺动脉第二音亢进,颈静脉怒张,肝肿大和下肢浮肿。
间质性肺炎病的治疗进展就是使用现代中医的新疗法:磁药叠加调节免疫疗法是以中医腧穴理论为基础,通过拔罐、药物熏蒸、针灸、外贴等多种手法对疾病进行综合的治疗和调理。
通过这种特殊的治疗方法,以绿色(不产生副作用,不伤及其他器官功能)、自然(按照事物的变化规律)为治疗原则,让患者在治疗疾病的同时又达到了“治未病”的目的。
2023间质性肺疾病年度进展摘要本文系统复习了2022年11月至2023年10月国内外权威杂志中关于间质性^疾病诊疗方面的文献,着重关注有关特发性肺纤维化、结缔组织疾病相关性间质性^疾病、结节病、进展性肺纤维化以及罕见间质性肺疾病等的发病机制、诊断和治疗进展以及其诊疗共识、指南。
同时也关注了有关特发性肺纤维化和结节病的流行病学以及卫生经济学方面的文献。
特发性肺纤维化(idiopathic pulmonary fibrosis , IPF )、结缔组织病相关性间质性肺疾病(connective tissue disease associated interstitial lung disease , CTD-ILD )、结节病、进展性肺纤维化(progressive pulmonary fibrosis , PPF )等的发病机制、诊断和治疗进展仍是2023 年国内外间质性肺疾病(interstitial lung disease z ILD )专家重点关注的话题、相关研究成果斐然。
在IPF方面,遗传(基因)缺陷与IPF发病、预后相关,很遗憾在IPF 的新药探索中出现挫折、部分有潜力的新药研究宣告失败[L 2, 3 ];抗纤维化药物花费以及IPF共病的诊疗费用是IPF患者的主要花费所在,且肺功能越差的IPF患者医疗花费越多[4, 5 ]o CTD-ILD领域,推出美国风湿协会系统性硬化症相关间质性肺疾病(systemic sclerosis-associated interstitial lung disease , SSc-ILD ) 的美国专家共识,评价了生物和靶向改善病情的抗风湿药(biologic and targeted synthetic disease-modifying antirheumaticdrug , b/tsDMARD )后类风湿关节炎患者ILD的发生率,以及比较了利妥昔单抗与环磷酰胺治疗CTD-ILD的疗效[6, 7, 8 ]o结节病诊疗上,报道了美国退伍军人中结节病的流行病学数据,发现新药——Efzofitimod (ATYR1923 )能有效辅助肺结节病患者的激素减量[9, 10 ]o此外,本文还关注了淋巴管肌瘤病(IymPhangiC)Ieie)myomatosis , LAM \肺泡蛋白沉积症(pulmonary alveolar proteinosis ,PAP 蹲罕见ILD[ 11,12 ]0 非常遗憾,受限于篇幅,本文仅针对上述几个方面进行汇总,未能纳入2023年度间质性肺疾病领域的所有卓越文献。
间质性肺疾病分类进展中文摘要:间质性肺疾病(interstitial lung disease, ILD)是一组主要累及肺间质和肺泡腔,导致肺泡-毛细血管功能单位丧失的弥漫性肺疾病。
临床主要表现为进行性加重的呼吸困难、限制性通气功能障碍伴弥散功能降低、低氧血症及影像学上的双肺弥漫性病变。
ILD可最终发展为弥漫性肺结构的损害,导致呼吸衰竭而死亡。
ILD的不同病因及分类,其治疗和预后可能不同,故而对ILD进行准确的分类尤为重要。
英文摘要:Interstitial lung disease (ILD) is a group diffuse lung disease of mainly involving the pulmonary interstitial and alveolar cavity, causing the alveolar-capillary function units loss. The main clinical manifestations are the progression of the difficulty in breathing, restrictive ventilation with dispersion function reducing, hypoxemia, and imaging of pulmonary diffuse lesions. ILD can eventually develop into diffuse lung structure damage, leading to respiratory failure and even death. Different etiology and classification of ILD, its treatment and prognosis may be different, so the ILD accurate classification is particularly important..关键词:间质性肺疾病(ILD),特发性间质性肺炎(IIP),特发性肺纤维化(IPF),呼吸性细支气管炎伴间质性肺病(RB-ILD),脱屑性间质性肺炎(DIP),隐源性机化性肺炎(COP),急性间质性肺炎(AIP),淋巴细胞性间质性肺炎(LIP),特发性胸膜间质弹力纤维化(PPFE),急性纤维素性机化性肺炎(AFOP)第一节、间质性肺疾病的概念和分类一、间质性肺疾病的概念间质性肺疾病(interstitial lung disease, ILD)是一组以肺泡单位的炎症和间质纤维化为基本病变的异质性非肿瘤和非感染性肺部疾病的总称。
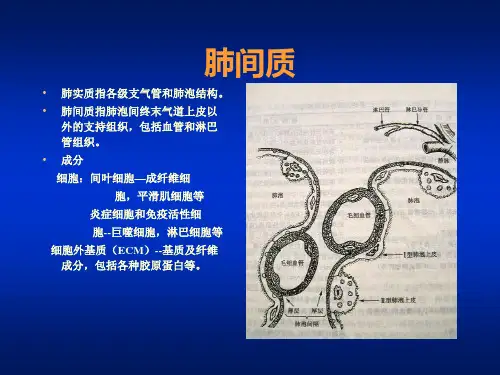
肺间质,从解剖学上看,是指肺泡上皮细胞和毛细血管内皮细胞基底膜之间的间隙,含肺泡间隔内的血管和淋巴周围组织,包括细支气管和支气管周围组织。
ILD病变目前认为起始部位与肺泡的上皮细胞和肺泡炎有关,但是病变可能并不仅仅累及肺泡与毛细血管基膜之间的间质,同时也可累及细支气管、肺泡实质、相关的淋巴管和血管以及胸膜等。
因此,有学者提出将ILD称为弥漫性实质性肺疾病(diffuse parenchymal lung disease, DPLD),2002年美国胸科学会/欧洲呼吸学会选用DPLD,视其为ILD的同义词。
DPLD也逐渐成为这类疾病使用更多的术语,但我国学者多使用ILD作为术语。
我国学者认为,间质性肺疾病是由各种病因所导致的弥漫性肺“间质-实质”病变的总称。
间质性肺疾病的概念也应该是包含两个方面:(1)不包括肿瘤性和感染性病因所致的“类”间质性肺疾病的临床及放射的表现,在临床中需要予以排除;(2)病变累及肺组织的全部:间质和实质。
也因此,将间质性肺疾病简单的认为是弥漫性实质性肺疾病,确有不足之处而造成对疾病认识的不全面。
间质性肺疾病更准确的范畴和概念,相信在续后的研究及临床工作中,会日臻完善。
二、间质性肺疾病的分类1935年Hamman和Rich首次描述弥漫性肺间质纤维化以来,已有200多种以上的相关疾病被囊括于ILD以下。
如何对ILD进行合理的归纳分类,一直也是临床医生和研究学者多年面临的挑战和努力。
ILD的分类涉及广泛,且分类的不同会有着不同的病因、病理、生理、临床、治疗及疾病的预后。
本章节引用2013年ATS/ERS及2014年德国发布的ILD的最新分类方法(见图1):分为四类,分别是已知病因的ILD(如,药物、胶原血管性疾病CVD等)、特发性间质性肺炎(IIP)、肉芽肿性ILD(如,结节病)和其他ILD(如,肺朗格汉斯组织细胞增生症);其中IIP再分为3类,分别为主要IIP(包括特发性肺纤维化IPF (图2)、非特异性间质性肺炎NSIP(图3)、呼吸性细支气管炎伴间质性肺病RB-ILD(图4)、脱屑性间质性肺炎DIP、隐源性机化性肺炎COP(图5)和急性间质性肺炎),少见IIP (包括淋巴细胞性间质性肺炎LIP、特发性胸膜间质弹力纤维化PPFE和急性纤维素性机化性肺炎AFOP)和未分类IIP。
图1.间质性肺疾病的分类图2. 典型IPF的HRCT表现:双下肺胸膜下可见蜂窝肺影(箭头处)(图片出处:Kekevian A, Gershwin ME, Chang C. Diagnosis and classification of idiopathic pulmonary fibrosis. Autoimmunity Reviews, 2014 (13): 508-512.)图3.NSIPA/B.肺病CT显示双下肺磨玻璃影、明显的支气管扩张和双下肺容积的减少;C/D.病理显示弥漫性肺泡间隔的增厚,肺泡结构仍保存、未见蜂窝改变或成纤维细胞的聚集。
(图片来源:Travis WD, Costabel U, Hansell DM, et al. Am J Respir Crit Care Med. 2013 Sep 15; 188(6):733-48.)间质性肺疾病如何合理的分类,仍然存在诸多的争论。
迄今为止,ILD的分类仍在不断的变化和修订着,也反映了对ILD的认识处于不断的发展及完善中。
其中以IIP在ILD中是呼吸内科常常所遇到的类型。
三、间质性肺疾病中IIP(特发性间质性肺炎)的最新进展(一)IIP分类进展的主要纲要:间质性肺疾病中,以IIP为其重要的一部分。
自2002年A TS/ERS的指南后,直至2014年,只有IIP有相关的进展的再次更新。
指南中的纲要如下:1. NSIP目前被认为是一种特殊的临床病理类型。
在临床中常常表现为明显的异质性,部分研究表明,一部分患者可进展为终末期肺纤维化。
故而对于NSIP标准的定义有助于我们对疾病的诊断。
2. 增加了对SR-ILD的认识,包括肺气肿合并间质纤维化的患者。
临床中,越来越多的RB-ILD患者不再行外科活检,而仅从患者的吸烟史、CT的影像特征(磨玻璃样影和小叶中心性结节)以及肺泡灌洗液(含棕褐色颗粒的巨噬细胞和缺乏淋巴细胞的增多)的特征就可进行诊断。
3. IPF是公认的异质性疾病,部分患者可以保持长时间的稳定,但一部分患者可以在快速的进展,甚至死亡。
4. 对于“急性加重期”有了更好的定义,且常出现在慢性纤维性IIP患者(IPF和NSIP)。
5. 部分IIP患者可表现为复合型肺部损伤而很难进行具体分类。
6. 对IIP进行一个合理的分类管理,有助于对ILD一个更好的认识,尤其对于一些无法取得活检标本以及HRCT不能进行诊断的患者。
7. PPFE是一类少见IIP类型,通常为特发性,缺乏典型的组织类型。
8.分子标志物,为IIP的诊断及对药物治疗的反应提供了有用的依据和帮助。
相关基因的研究可能对IIP的诊断及分类带来新的革新。
(二)自2002年A TS/ERS后IIP的基本进展1.多学科联合诊断方法:(1)IIP多学科的合作:IIP常呈现为动态发展,诊断IIP往往需要多学科(临床医师、放射科医师及病理科医师)的合作。
临床的病史(症状、特殊暴露、吸烟史、基础疾病、肺功能及实验室检查)和影像学的改变,对于IIP的诊断非常重要。
但是多学科联合的方法并不会削弱肺活检在诊断IIP中的地位,一旦病理学医师认定了某种病理模式(如,NSIP或COP),临床医生就必须重新考虑潜在的病因(如,外源性过敏性肺泡炎、胶原血管性疾病和药物暴露。
)(2)IIP的诊断是在排除其他已知原因的间质性肺疾病(如吸入剂的暴露和胶原血管性疾病)的基础上。
(3)IIP诊断者的一致性:临床医师、放射科医师及病理科医师依据临床经验并整合所有病情资料,对疾病进行一致性的诊断、验证、修改和证实。
2.遗传性间质性肺炎(familial interstitial pneumonias,FIPs):近年的研究发现,部分IIP病例与家族有关(约2-20%),但目前这部分病案仍被归类为IIP中。
约20%的FIPs病例是以TERC、SFTPA2、TERT和TERC杂合子突变为主。
散发的家族性IPF,主要是与端粒酶的缩短相关。
80%的FIP患者都是垂直性遗传,提示可能是常染色体的发病机制。
全基因组检测发现MUCB基因可能与遗传性及散发性IPF均相关。
所以,对于疑似FIP的患者,均应该进行家族性疾病的调查及基因的检测。
(三)自2002年A TS/ERS后IIP的最新进展1.慢性纤维化IIP:(1)IPF(特发性肺纤维化)IPF是一种少见的,慢性、进行性且不可逆转的肺纤维化疾病,其诊断困惑了临床医生很多年。
IPF是公认的异质性疾病,部分患者可以保持长时间的稳定,但一部分患者可以在快速的进展,甚至死亡。
总体而言,IPF的预后不佳,甚至比部分肺癌患者都差(中位生存期2-5年);IPF的诊断和治疗在很长的时间都没有明显的进展。
IPF好发于60-70岁老年吸烟/曾吸烟的男性,欧洲的发病率是1-23/10万。
2011年最新指南提出IPF的诊断标准:①除外其他可能原因的间质性肺疾病,②典型的UIP表现及典型的HRCT表现(见图2.和表1.)③HRCT上表现为“可能UIP”和肺活检为UIP表现。
表1. HRCT表现:可能UIP胸膜下,基底部多见;网格状改变;缺乏任何一个与UIP表现不相符合的征象aUIP表现胸膜下,基底部多见;网格状改变;缺乏任何一个与UIP表现不相符合的征象a蜂窝肺a与UIP表现不相符合的征象:中上肺病变为主,支气管血管周围为主,广泛的磨玻璃影,弥漫性微结节,多发囊状改变(远离蜂窝区),气体陷闭,支气管肺段实变目前IPF的治疗策略有了一定的变化,具体指南如表2:表2. IPF治疗指南药物推荐推荐级别级别证据单独糖皮质激素秋水仙碱环孢菌素A糖皮质激素+免疫抑制剂糖皮质激素+硫唑嘌呤+乙酰半胱氨酸乙酰半胱氨酸γ-干扰素波生坦依那西普抗凝剂吡非尼酮不推荐不推荐不推荐不推荐多数IPF患者不使用多数IPF患者不使用不推荐不推荐不推荐多数IPF患者不使用多数IPF患者不使用强强强强弱弱强强强弱弱低低低低低低高中等中等非常低低-中等(2)非特发性间质性肺炎ATS研讨会中总结了NSIP的诊断标准。