免除知情同意签字申请表
- 格式:doc
- 大小:35.00 KB
- 文档页数:2

伦理审查申请指南为指导研究者/申办者提交药物、医疗器械、体外诊断试剂临床试验项目的伦理审查申请/报告,制定本指南。
一、提交伦理审查的研究项目范围根据国家食品药品监督管理总局《药物临床试验质量管理规范》(2003年)、《药物临床试验伦理审查工作指导原则》(2010年)、《体外诊断试剂临床试验技术指导原则》(2014年)、《医疗器械临床试验质量管理规范》(2016年)、《医疗器械临床试验设计指导原则》(2018年),国家卫生和计划生育委员会《涉及人的生物医学研究伦理审查办法》(2016年),下列范围的研究项目应依据本指南提交伦理审查报告:1.药物临床试验2.医疗器械临床试验3.体外诊断试剂二、伦理审查申请/报告的类别1.初始审查:初始审查申请:符合上述范围的临床试验项目,应在研究开始前提交伦理审查申请,经批准后方可实施。
“初始审查申请”是指首次向伦理委员会提交的审查申请。
2.跟踪审查:1)修正案审查:研究过程中若变更主要研究者,对临床研究方案、知情同意书、招募材料等的任何修改,应向伦理委员会提交修正案审查申请,经批准后执行。
为避免研究对受试者的即刻危险,研究者可在伦理委员会批准前修改研究方案,事后应将修改研究方案的情况及原因,以“修正案审查申请”的方式及时提交伦理委员会审查。
2)研究进展报告:按照伦理审查批件,意见规定的年度,定期跟踪审查频率,在截止日期前1个月提交研究进展报告;申办者应当向组长单位伦理委员会提交各中心研究进展的汇总报告;当出现任何可能显著影响研究进行或增加受试者危险的情况时,应以“研究进展报告”的方式,及时报伦理委员会审查。
如果伦理审查批件有效期到期,而项目没有启动,需重新提交伦理申请并获得批准后方可实施。
3)严重不良事件报告:严重不良事件是指临床研究过程中发生需住院治疗、延长住院时间、伤残、影响工作能力、危及生命或死亡、导致先天畸形等事件。
发生严重不良事件,应在获知24小时内向伦理委员会报告。

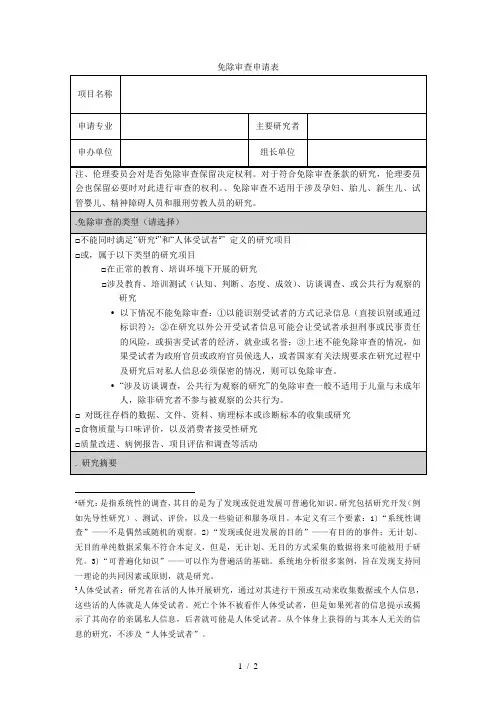
免除审查申请表
1研究:是指系统性的调查,其目的是为了发现或促进发展可普遍化知识。
研究包括研究开发(例如先导性研究)、测试、评价,以及一些验证和服务项目。
本定义有三个要素:1)“系统性调查”——不是偶然或随机的观察。
2)“发现或促进发展的目的”——有目的的事件;无计划、无目的单纯数据采集不符合本定义,但是,无计划、无目的方式采集的数据将来可能被用于研究。
3)“可普遍化知识”——可以作为普遍活的基础。
系统地分析很多案例,旨在发现支持同一理论的共同因素或原则,就是研究。
2人体受试者:研究者在活的人体开展研究,通过对其进行干预或互动来收集数据或个人信息,这些活的人体就是人体受试者。
死亡个体不被看作人体受试者,但是如果死者的信息提示或揭示了其尚存的亲属私人信息,后者就可能是人体受试者。
从个体身上获得的与其本人无关的信息的研究,不涉及“人体受试者”。
申请人签名日期
审查记录
□同意免除审查
请说明免除审查的类别:
□ 研究符合伦理要求,研究风险已经最小化,公平选择受试者,知情同意过程符合要求,受试者隐私与数据保密措施合适。
□不能免除审查
□属于免除审查范畴,但伦理委员会需要保留审查权利
□暂时不能确定是否免除审查(研究内容尚未明确,提请注意:一旦研究确定涉及人体受试者,必须及时咨询伦理委员会。
)
审查委员签名日期
主任委员签名日期。
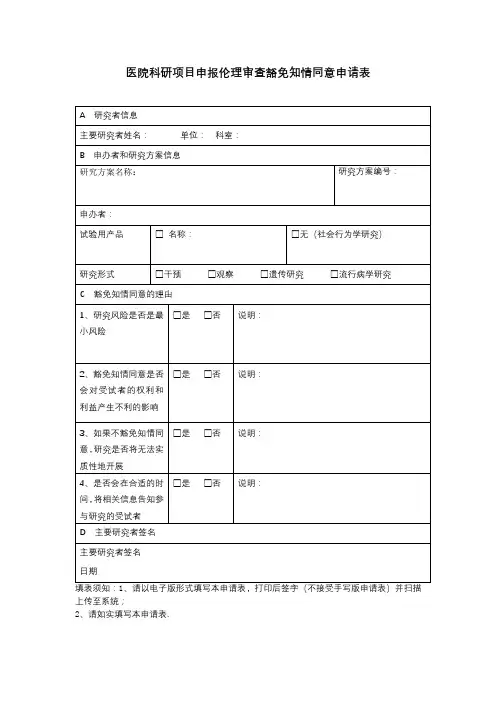

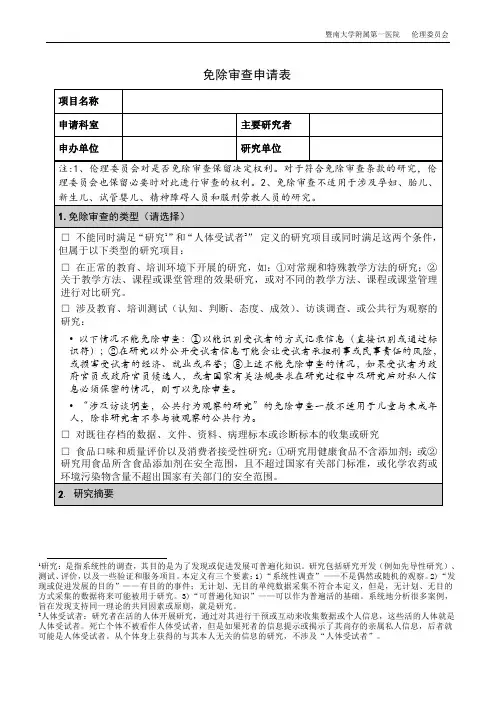
免除审查申请表
1研究:是指系统性的调查,其目的是为了发现或促进发展可普遍化知识。
研究包括研究开发(例如先导性研究)、测试、评价,以及一些验证和服务项目。
本定义有三个要素:1)“系统性调查”——不是偶然或随机的观察。
2)“发现或促进发展的目的”——有目的的事件;无计划、无目的单纯数据采集不符合本定义,但是,无计划、无目的方式采集的数据将来可能被用于研究。
3)“可普遍化知识”——可以作为普遍活的基础。
系统地分析很多案例,旨在发现支持同一理论的共同因素或原则,就是研究。
2人体受试者:研究者在活的人体开展研究,通过对其进行干预或互动来收集数据或个人信息,这些活的人体就是人体受试者。
死亡个体不被看作人体受试者,但是如果死者的信息提示或揭示了其尚存的亲属私人信息,后者就可能是人体受试者。
从个体身上获得的与其本人无关的信息的研究,不涉及“人体受试者”。
主要研究者签名日期
审查记录
□同意免除审查
请说明免除审查的类别:
□研究符合伦理要求,研究风险已经最小化,公平选择受试者,知情同意过程符合要求,受试者隐私与数据保密措施合适。
□不能免除审查
□属于免除审查范畴,但伦理委员会需要保留审查权利
□暂时不能确定是否免除审查(研究内容尚未明确,提请注意:一旦研究确定涉及人体受试者,必须及时咨询伦理委员会。
)
审查委员签名日期。

北京大学人民医院医学伦理委员会初始审查申请研究信息•方案设计类型✧□试验性研究(干预性研究)✧□观察性研究(非干预性研究):□回顾性分析;□前瞻性研究•研究信息✧资金来源:□企业,□政府,□学术团体,□本单位,□自筹✧数据与安全监察委员会:□有,□无✧其他伦理委员会对该项目的否定性或提前中止的决定:□无,□有→请提交相关文件✧研究需要使用人体生物标本(包括但不限于体液、组织等):□否,□是→填写下列选项采集生物标本:□是,□否利用以往保存的生物标本:□是,□否✧研究干预超出产品说明书范围,没有获得行政监管部门的批准:□是,□否(选择“是”,填写下列选项)研究结果是否用于注册或修改说明书:□是,□否研究是否用于产品的广告:□是,□否超出说明书使用该产品,是否显著增加了风险:□是,□否•招募受试者✧谁负责招募:□医生,□研究者,□研究助理,□研究护士,□其他:✧招募方式:□广告,□诊疗过程,□数据库,□中介,□其他:✧招募人群特征:□健康者,□患者,□弱势群体,□孕妇弱势群体的特征(如果选择了弱势群体,填写以下选项):□儿童/未成年人,□因认知障碍或因健康状况而没有能力做出知情同意的成人,□申办者/研究者的雇员或学生,□教育/经济地位低下的人员,□疾病终末期患者,□囚犯或劳教人员,□其他:知情同意能力的评估方式(如果选择了弱势群体,填写该选项):□临床判断,□量表,□仪器涉及孕妇研究的信息(如果选择了孕妇,填写该选项):□没有通过经济利益引诱其中止妊娠,□研究人员不参与中止妊娠的决策,□研究人员不参与新生儿生存能力的判断✧受试者补偿:□有,□无补偿金额:补偿支付方式:□按随访观察时点,分次支付,□按完成的随访观察工作量,一次性支付,□完成全部随访观察后支付•知情同意的过程✧谁获取知情同意:□医生/研究者,□医生,□研究者,□研究护士,□研究助理✧获取知情同意地点:□私密房间/受试者接待室,□诊室,□病房✧知情同意签字:□受试者签字,□法定代理人签字•知情同意的例外:□否,□是→填写下列选项✧□申请开展在紧急情况下无法获得知情同意的研究:研究人群处于危及生命的紧急状况,需要在发病后很快进行干预;在该紧急情况下,大部分病人无法给予知情同意,且没有时间找到法定代理人;缺乏已被证实有效的治疗方法,而试验药物或干预有望挽救生命,恢复健康,或减轻病痛;✧□申请免除知情同意·利用以往临床诊疗中获得的病历/生物标本的研究;✧□申请免除知情同意·研究病历/生物标本的二次利用;✧□申请免除知情同意签字·签了字的知情同意书会对受试者的隐私构成不正当的威胁,联系受试者真实身份和研究的唯一记录是知情同意文件,并且主要风险就来自于受试者身份或个人隐私的泄露;✧□申请免除知情同意签字·研究对受试者的风险不大于最小风险,并且如果脱离“研究”背景,相同情况下的行为或程序不要求签署书面知情同意。

免知情同意申请书尊敬的伦理委员会/相关管理部门:申请人姓名:____________________申请人身份:____________________联系方式:____________________申请日期:____________________一、申请事项本人/我们(以下统称“申请人”)在此申请免除在以下研究项目中的知情同意程序:研究项目名称:____________________研究项目编号:____________________研究项目负责人:____________________研究开始日期:____________________研究结束日期:____________________二、申请理由1. 研究性质:本研究的性质为____________________(描述研究类型,如观察性研究、回顾性研究等),不涉及任何干预措施,对研究对象不会产生直接的生理或心理影响。
2. 风险评估:经过充分的风险评估,本研究对研究对象可能产生的风险极低,且不会对研究对象的生活质量造成影响。
3. 信息获取:本研究所需的数据和信息将通过____________________(描述数据获取方式,如公共数据库、匿名化病历资料等)获得,无需直接与研究对象接触。
4. 隐私保护:本研究将严格遵守相关隐私保护法规,确保研究过程中不会泄露任何可能识别研究对象身份的信息。
5. 法律法规:根据____________________(引用相关法律法规或政策文件)的规定,本研究符合免除知情同意的条件。
三、承诺与保证1. 申请人承诺,本研究将严格遵守国家有关法律法规、伦理准则和科研诚信原则。
2. 申请人保证,研究过程中将采取一切必要措施保护研究对象的隐私和权益。
3. 申请人同意,如有必要,伦理委员会/相关管理部门可以要求提供额外的信息和文件,以支持本申请。
4. 申请人承诺,在研究过程中如遇到任何可能影响研究对象权益的情况,将立即向伦理委员会/相关管理部门报告,并采取相应措施。

知情同意书豁免申请书English Answer:Informed consent is a legal and ethical requirement in research involving human subjects. It ensures thatpotential participants are fully informed about the risks and benefits of the study before they agree to participate. In some cases, however, it may be necessary to waive the requirement for informed consent.A waiver of informed consent may be granted if the following criteria are met:The research poses no more than minimal risk to the subjects.The research could not practicably be carried out without a waiver.The research is intended to benefit the subjects orothers.The decision to waive informed consent must be made by an Institutional Review Board (IRB). The IRB is a committee of experts that reviews research proposals to ensure that they meet ethical and regulatory standards.If you are planning to conduct research that involves human subjects, you should contact your IRB to determine whether a waiver of informed consent is necessary.中文回答:知情同意书是涉及人类受试者的研究中的一项法律和伦理要求。

伦理初审审查材料清单(科研)
1.伦理审查申请表;用本心模板
2.横向课题提项目方案(研究者签字);纵向科研课题,提项目申请
标书(版本号及日期,研究者签字);自行发起项目提供项目方案(研究者签字)。
3.知情同意书(版本号及日期)参考本心模板;申请免知情请填写免
除知情同意书申请表(用本心模板)
4. 招募受试者地材料(如有,版本号及日期);
5. 病例报告表(如有,版本号及日期);
6. 研究者手册(如有,版本号及日期);
7.研究者履历(研究者签字);用本心模板
8. 科研项目批文/任务书(如有);
9. 试验药物/器械/试剂盒上市许可证(如有);
10. 组长单位批件(如有);
11. 保险单(如有,版本号及日期);
12.研究者利益冲突声明;用本心模板
13.文件一致说明;用本心模板
14.设置安慰剂组请填写安慰剂对照说明;(如有)用本心模板
15. 其它文件。
(如有)(类遗传资源有关材料等)。

免除知情同意申请表项目名称申请专业主要研究者申办单位组长单位注: 对于以下两种情况之一,伦理委员会可以批准免除知情同意。
但是,请注意:免除知情同意,伦理委员会也可以要求研究者向受试者提供研究告知信息。
1.利用以往临床诊疗中获得的病历/生物标本的研究,申请免除知情同意✉本研究使用的病历或生物标本是以往临床诊疗中获取的。
请说明:本项研究所使用的肿瘤标本、血液标本及脑脊液标本均为患者标本的二次利用。
分别取自于:送检常规病理检查后剩余的肿瘤组织;送检常规血液检查后剩余的血液;患者因自身病情需要,送检脑脊液检查后剩余的脑脊液。
✉本研究对受试者的风险不大于最小风险1。
请说明:本研究使用的标本均为正常诊疗过程中剩余的组织标本,对患者无附加风险。
✉免除知情同意不会对受试者的权利和健康产生不利的影响。
请说明:患者只需接受疾病的正常诊疗过程,任何医疗待遇与权益不会受到影响✉受试者的隐私和个人身份信息得到保护。
请说明:患者在试验中的个人资料均属保密,组织标本将以编号数字而非姓名加以标示。
可以识别患者身份的信息不会透露给研究小组以外的成员。
所有研究成员和研究申办方都被要求对患者身份保密。
患者档案将仅供研究人员查阅。
为确保研究按照规定进行,必要时,政府管理部门或伦理审查委员会成员按规定可以在研究单位审查患者资料。
这项研究成果发表时,将不会披露患者个人的任何资料✉若规定需获取知情同意,研究将无法进行(病人有权知道其病历/标本可能用于研究,其拒绝或不同意参加研究,不是研究无法实施、免除知情同意的证据)。
请说明:患者有权知道其组织标本可能用于研究,有权拒绝参加研究。
1最小风险(Minimal Risk):指试验中预期风险的可能性和程度不大于日常生活、或进行常规体格检查或心理测试的风险。
本研究不利用病人/受试者以前已明确地拒绝利用的医疗记录和标本。
2. 研究病历/生物标本的二次利用,申请免除知情同意以往研究已获得受试者的书面同意,允许其他的研究项目使用其病历或标本。
免除知情同意签字申请表
项目名称
申请专业主要研究者
申办单位组长单位
注: 对于批准的免除知情同意书面签字,伦理委员会一般要求研究者向受试者提供书面的研究告知信息并获得口头知情同意。
1.研究属于以下类别之一:
□签署的知情同意书会对受试者的隐私构成不正当的威胁;联系受试者真实身份和研究
的唯一记录是知情同意文件,并且主要风险就来自于受试者身份或个人隐私的泄露。
请说明:
1,如果脱离“研究”背景,相同情况下的□本研究对受试者造成的风险不大于最小风险
行为或程序不要求签署书面知情同意(如访谈研究,邮件/电话调查)。
请说明:
2. 如果免除知情同意签字,本研究会做到:
□向受试者或其法定代理人提供书面告知信息。
□获得受试者或其法定代理人的口头知情同意。
□口头知情同意记录在案。
1最小风险(Minimal Risk):指试验中预期风险的可能性和程度不大于日常生活、或进行常规体格检查或心理测试的风险。
申请人签名日期。