
有机反应机制的研究方法
有机化学中用来解释反应机理的传统方法主要集中在Kinetics和Dynamics两方面,即理解势能面、深入研究分子运动和碰撞、测定活化参数、测定速率常数、确定某个反应机理中一系列化学步骤的顺序、确定反应限速步骤和决速步骤。
研究机理的关键目的是反应机理知识可以对如何在原子或分子水平上操纵物质给出最快速的洞察,而不是依靠运气来获得偶然性的变化从而获得想要的结果。由于动力学在辨别机理方面起着关键作用,所以动力学是整个有机反应机理研究领域中最重要的分支之一。
传统的反应机理研究方法除了动力学分析之外,还有同位素效应、结构-功能分析等。这些都是研究有机反应机理的标准实验工具,然后实验化学家可以根据其想象力和化学创造性,设计出一些完全不同于之前出现过的研究方法。因此,本文总结了一些最为常见的方法。首先分析最简单的实验,例如产物和中间体的鉴定。但也会分析一些更为微妙、精细的实验,如交叉和同位素置乱(cross-over and isotope scrambling)实验。
1.改变反应物结构以转变或捕获预想的中间体
有时可以通过合成一种类似于所研究的反应物的新反应物来破译中间体的性质,但是这需要所预测的中间体能以一种可预想的方式进行反应。没有标准的方式来处理这一类实验,所以实验者必须根据具体实验情况来设计实验。下面以酶反应作为此方法的应用实例。
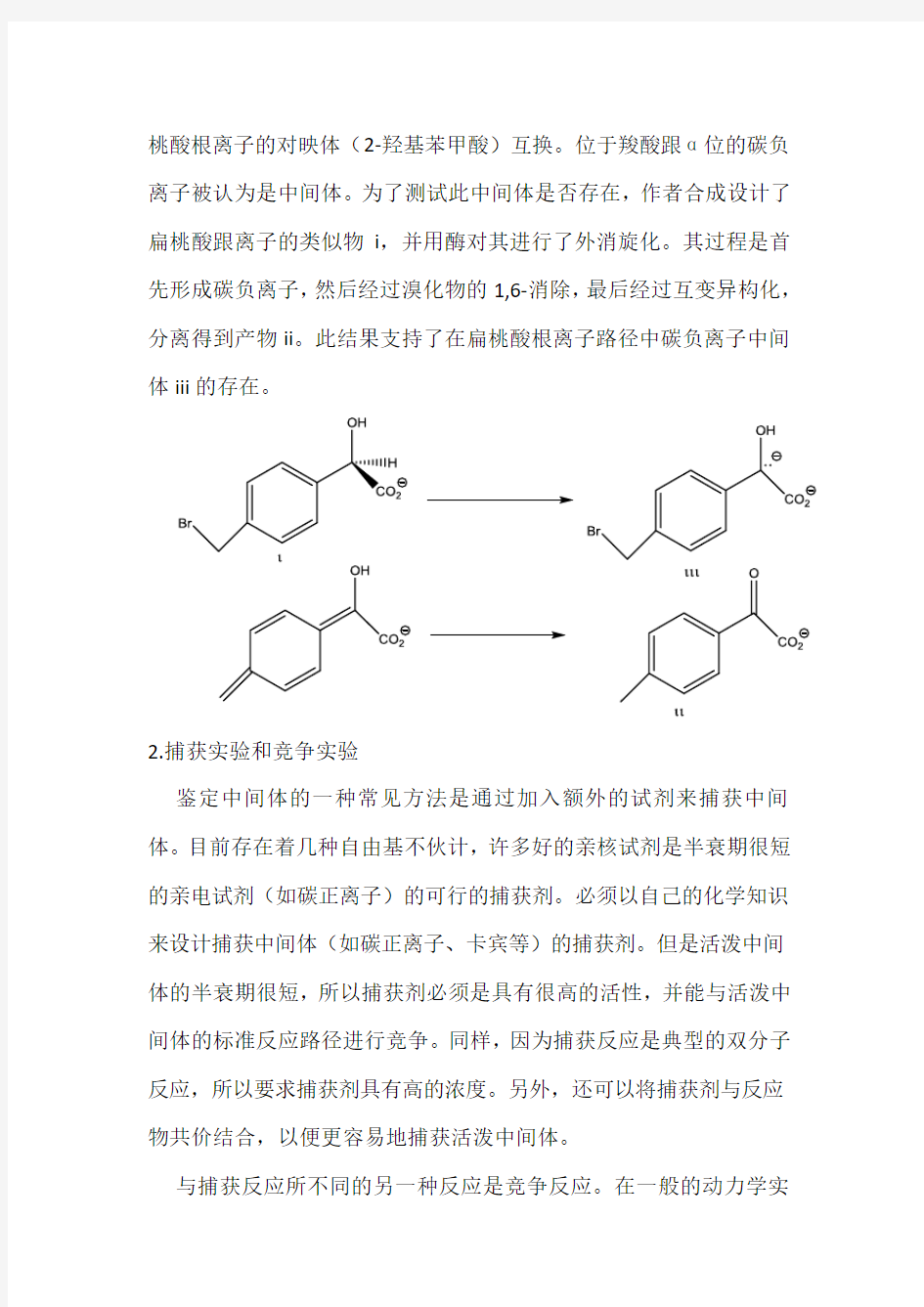
Lin[1]等人设计了一种转变中间体的方法。扁桃酸消旋化酶可使扁
桃酸根离子的对映体(2-羟基苯甲酸)互换。位于羧酸跟α位的碳负离子被认为是中间体。为了测试此中间体是否存在,作者合成设计了扁桃酸跟离子的类似物i,并用酶对其进行了外消旋化。其过程是首先形成碳负离子,然后经过溴化物的1,6-消除,最后经过互变异构化,分离得到产物ii。此结果支持了在扁桃酸根离子路径中碳负离子中间体iii的存在。
2.捕获实验和竞争实验
鉴定中间体的一种常见方法是通过加入额外的试剂来捕获中间体。目前存在着几种自由基不伙计,许多好的亲核试剂是半衰期很短的亲电试剂(如碳正离子)的可行的捕获剂。必须以自己的化学知识来设计捕获中间体(如碳正离子、卡宾等)的捕获剂。但是活泼中间体的半衰期很短,所以捕获剂必须是具有很高的活性,并能与活泼中间体的标准反应路径进行竞争。同样,因为捕获反应是典型的双分子反应,所以要求捕获剂具有高的浓度。另外,还可以将捕获剂与反应物共价结合,以便更容易地捕获活泼中间体。
与捕获反应所不同的另一种反应是竞争反应。在一般的动力学实
验的分析中,除了决速步意外的其他步骤对动力学没有显著影响,因此关于这些其他步骤的数据都不能得到。这种缺乏动力学依赖性常常使得在动力学分析中,很大一部分的机理没能弄清楚。一种解决这个问题的有效办法是运用竞争性实验,竞争性实验包含了两种或者多种试剂来竞争与一个或多个中间体反应。这是捕获反应的一种派生办法,它所用的捕获剂不止一种。不同的捕获剂所得到的产物的比率代表了不同的捕获剂与中间体反应速率常数的比率。根据这一比率,可以得到有关中间体的性质。只有在捕获反应遵循动力学控制时,实验才是可行的。
Sarma[2]等人报道的实验中捕获了正膦以证明它的存在。五配位物种(正膦)被认为是RNA和DNA水解过程的中间体。这一物种在被广泛接受前,化学家研究了磷酸酯作为模型体系的化学性质,磷酸能够环化生成正膦,但是室温下根本观察不到中间体的存在。但是往反应溶液中加入乙酰氯后,就可能分别将两种中间体捕获。
3.核对共同中间体
通常,相类似的
[1].Lin,D.T.,J.Am.Chem.Soc.,110,323(1988).
[2].Sarma,R.J.Am.Chem.Soc.,100,5391(1978).
反应历程(机理):Reaction Mechanism
有机反应按化学反应所经历的全部过程:即为反应机理。反应机理是由反应物转变为产物的途径,若为基元反应,则为一步反应得到产物;若不为基元反应,则可以分解为多步基元反应过程。
反应机理(或反应历程)就是对反应物到产物所经历过程的详细描述和理论解释,特别是对中间体杂化状态、能量变化等的描述。
目前关于反应机理的描述主要是根据一些实验结果及观察到的实验现象和模型而做出的合理的理论假设和判断。限于检测手段及理论研究的局限,迄今为止,尚未有一个反应机理被真正证明过。
依据的实验事实越多,则由此作出的理论解释越可靠。一个反应的历程应经得起实验事实的考验,并应有一定的预见性。
b)反应类型:按反应机理分为自由基反应(自由基加成、自由基
取代)、离子型反应(亲电加成、亲电取代、消除反应、亲核加成、亲核取代)、协同反应;按反应底物和产物的因果关系
分类:取代反应(亲核取代、亲电取代、自由基取代)、加成反应(亲核加成、亲电加成、自由基加成)、消除反应、氧化还原反应
北京大学的徐光宪院士总结了20世纪化学的三大理论成就:
1.化学热力学。
它可以判断化学反应的方向,提出化学平衡和相平衡理论,可以预见化学反应的可能性,为合成化学指明方向。
2.量子化学和化学键理论,以及结构和性能关系的初步规律。这
对设计合成具有优良性能的化合物是至关重要的。
3.化学动力学和分子反应动态学的研究,特别是催化理论的发展
和计算机设计合成方法的推广,大大推动了合成化学。
热力学:解决反应进行的方向和程度,即反应可进行到哪里的问题。
动力学:解决反应进行快慢的问题,对反应速率的处理。研究主要有碰撞理论和过渡态理论两种学说。
3.3 有机反应的热力学控制和动力学控制
在一定反应条件下,一个从同一反应物出发的反应可能由于竞争反应而产生不同产物,此时应分析出生成目的产物途径的诸因素,以便控制反应条件,使反应趋向目的产物而获得最佳收率。
如果两个反应都不可逆,则速率快的反应生成的产物多,即C的产量较多。这种反应产物的比率决定于反应速率的过程称之为动力学控制。
反应可逆时,当反应平衡完全建立后产物的比例取决于它们的相对热力学稳定性的大小,称为热力学控制或平衡控制的反应。
1903年,Lapworth在研究丙酮与氰化氢的反应时,发现反应速率因加碱而加快,因加酸而变慢,提出了第一个有机反应机理。
反应机理的给出需从实验结果出发,通过合理的电子转移图示将底物转化为产物,提出机理时需考虑以下几个方面:
?产物中的每个原子在反应物中都要有来源,即产物和包括底物、试剂甚至溶剂在内的反应物上的原子有对应关系。
?找出导致产物形成的反应途径,确定哪些基团或键发生了变化。
?底物的化学结构有无原子重排。
α-羟基氰
C
O
H3C CH3HCN
OH-
HO CN
+
? 运用众所周知的基元反应机理,避免不合理的电子转移或出现高能量的过渡态和中间体。
例:鲁宾逊(Robinson )增环反应
含活泼亚甲基的环酮与α,β-不饱和羰基化合物在碱存在下反应,形成一个二并六员环的环系。
? 观察产物与反应物的原子数,确定反应是否增加碳原子。 ? 将产物中相关的碳原子进行编号并与反应物相对应,以便跟踪反应物中碳原子的去向和产物中碳原子的来源。
? 观察旧键的断裂和新键的形成,推测可能的反应机理。
其反应机理为:本反应分为两步,第一步是迈克尔(Micheal )加成反应,第二步是羟醛缩合反应。
a) Micheal 加成反应
b) 羟醛缩合反应
O
O O O O
+EtOH EtO -1674
32
8512345678
反应机理合理性原则
●提出的机理应明确解释所有已知的事实,同时又尽可能简单,
易于重复和证明。
●基元反应应是单分子或双分子的,通常情况不必考虑三个分
子以上的其他反应。
●机理中每一反应步骤在能量上是允许的,化学上则是合理的,
合乎通用性的一般规则,如正性部分总是和负性部分结合。
●机理要有一定的预见性,当反应条件或反应物结构变化时,
应能对新反应的速率和产物变化作出正确的预测。
反应机理的研究步骤
①提出一个与已有实验事实及理论知识相符的假设。
②设计并进行实验以检验所提出的历程假设。
③根据实验结果对假设进行修正或推断。
通常上述步骤要重复多次。对同一实验的解释可能有多种选择,与试验事实相符的反应机理也可能不止一条,为此要认
真细心、去伪存真、反复推敲,以得到较为满意又令人信服的机理历程。
例如: -羟基丙腈
推测机理1
羰基碳易受亲核试剂的进攻,半缩醛上的碳不宜接受亲核试剂的进攻,另外,EtO-亲核性极强,不易离去。推测机理1不合理。 推测机理2:
OC 2H
OH
CN +C 2H 5O -
OC 2H 5OH
CN
HCN C 2H 5OH +
+
醛羰基易于接受亲核试剂的进攻
上述四步反应过程化学上合理,能量上有利,因此认为其机理推测合理。
还有同位素、动力学等研究机理的方法。
(《第三章_有机反应机理和测定方法-1》)
3.3确定有机反应机理的方法
1、产物的鉴定
OC 2H 5OH
OH - or CN -OC 2H 5O -H 2O or HCN
+OC 2H 5O -H O
+C 2H 5O -H O CN -CN
O -CN O -CN OH
HCN OH -
++CN -
CH4 + Cl2CH
3
Cl CH3. +CH3.CH3CH3 2、中间体的确定---
?中间体的分离
N-溴代酰胺
异氰酸酯
R-N=C=O
?中间体的检测:IR、NMR、ESR(EPR)
?中间体的捕获:
3、立体化学方法
4、同位素标记
用标记的水(H2O18)水解丁二酸甲酯,得到普通的甲醇和含有O18的丁二酸,令人信服的证明水解反应是通过酰氧键断裂进行的。
5、同位素效应(Isotope Effect)
?同位素效应:当反应物分子中的氢被重氢置换后,再发生取代、消除等C—H键断裂反应时,所表现出的H与D的反应速度不
同的现象。常以KH/KD之比来表示。
?C—H键断裂所需活化能比C-D断裂活化能小,当键断裂发生在决速步时就会出现同位数效应,KH/KD=1-9
?同位素效应有第一同位素效应和第二同位素效应两种。所谓第二同位素效应是指尽管连接同位素的键不参与反应,但却表现
有同位素效应现象。一般说,第二同位素效应比第一同位素效
应值小,最高只有1.5 。
?14C,18O也存在同位素效应,但数值很小。
?通过同位素效应,可以确定反应机理中的决速步骤。
K H/K D≌12是第一同位素效应,K H/K D’≌1.11是第二同位素效应。?在大多数芳香族亲电取代反应中,不存在同位素效应,K H/K D=1.0,这就提供了一个明确的指示,在定速步骤中不涉及C—H键的断裂,氢没有失去。因此,反应至少包括两个步骤和一个中间体:
6、化学热力学方法
?热力学方法通过研究一个反应的热效应是放热还是吸热,焓、熵以及自由能的变化来求得相关机理方面的许多信息。
H 键能(生成)键能(断裂) CH 3CH=CH -CH 3
CH 3CH 2CH 2CH 3生成键能 断裂键能
2C —H 47.0 KJ.mol -1 H 2 24.7 KJ.mol -1
C —C 19.3 KJ.mol -1 C=C 34.7 KJ.mol -166.3 KJ.mol
-159.4 KJ.mol -1H 59.4 - 66.3 = -6.9KJ.mol -1
7、动力学方法
? 通过动力学的研究可以获得有哪些分子和有多少个分子参与了决定速率步骤的信息。
? 自催化反应 H 3C C C H 3
O
3C C C H O H
+H 3C C C H 2O H r 2H 3C C C H 2B r O H 3C C C H 2O
H 3C C C H 2O -
(《第三章_有机化学反应机理的研究》)
反应机理包括反应中:
①试剂的进攻
②化学键的断裂与形成、断键顺序、
③活性中间体的生成、
④反应的相对速率
分子振动和碰撞的速率10-12~10-14,观测分子和原子的手段尚不完善,主要根据反应中观测的现象来推断反应历程。
飞秒化学:快速闪光照相机:fs(10-15s),Zewail 1999Nobelist 慢动作观察化学键的断裂与形成。
一. 有机合成反应的基本类型
绝大多数有机化合物是以共价键结合的。
有机化合物反应的本质是:
即为旧的共价键的断裂和新的共价键生成的过程。因此化学反应可以按新键形成分类:
(一)按新键形成分类可分为:
C-H键形成反应C-O键形成反应
C-C键形成反应C-N键形成反应
C-X键形成反应C-Si键形成反应
此分类方法对于剖析有机化合物结构,研究新键的形
成,创造性提出新的合成路线,具有一定的指导意
义。但此种分类法还仅限于宏观过程的理解与归纳,
缺乏深入的微观分析与理解。
(二)按引入原子和基团或采用的试剂分类
?经化学反应后,
?产物的化学结构中仅引入了卤素原子——称卤化反应
?如引入NO2——硝化反应;
?采用氧化剂——氧化反应
还原剂——还原反应
?采用水、醇、胺做溶剂进行的分解反应——分别称
为水解反应、醇解反应、胺解反应
?采用催化剂Catalyst存在下,氢化还原NO2、N2+基、
不饱和键、羰基等官能团的反应——催化氢化反应
(三)按合成反应的机理分类
按机理分类:
亲电取代反应、亲核取代反应、
亲电加成反应、亲核加成反应、
电性消除反应、自由基反应、
重排反应、周环反应。
按机理分类法,可将众多化学反应,从其本质上归纳起来。对于现实中某一反应,则能从理论上给予判断和预计,对于控制化学反应,研究新的合成方法起到指导作用。
二、有机合成反应机理与分类
一)有机反应机理--根据键断裂方式分类
(有三种基本类型)
1. 离子反应历程
化学键断裂后,形成正离子或负离子中间体,经过正负离子中间体所进行的反应。此种反应过程称为离子反应历程(Ionic Mechanism)
or 异裂历程(Heterolytic mechanism)
or 极性反应历程(Polar mechanism)
?X:Y→X:- + Y+
?2. 自由基反应历程
化学键断裂后,两个断片各带一个电子形成自由基,通过自由基中间体而进行的反应历程——称自由基历程(Free radical mechanism)
or 均裂历程(Homolytic mechanism)
X:Y →X· + Y·
3. 协同反应历程(一步反应历程)
断键与成键同时进行,有离子或自由基中间体生成(离子或自由基)。即电子在封闭的环中运动,通常为6个电子,有时为4个电子,通过环状过渡态进行的反应历程——协同反应历程(Concerted mechanism)
(二)有机反应机理——反应结果分类
有机反应数目和范围广,按反应的结果可分为5
类,每一类都可在不同条件下,按上述历程进行反应。
1. 取代反应
(1) 亲电取代反应(Electrophilic substitution reaction )
? 通式: A X
+Y
A-Y
+X ? 如芳烃的硝化、磺化、卤代、Friedel-Crafts 反应(烃化、酰化)
H
NO 2NO 2H
Br X R ++RCO H +
? (2)亲核取代反应(Nucleolhilic substitution reaction ) ? 如卤烃的水解,醇的卤代
? 通式: A X
+Y
A-Y +X R X
+OH ROH +X
? (3)自由基取代反应(Free radical substitution reaction )
? 如烷烃的卤代,NBS 的溴化丙烯型和溴化苄型化合物的制备 ? 通式
:A X +Y
A-Y +X
? 2. 加成反应(addition reaction )
(1) 亲电加成反应(electrophilic addition reaction )基本类型
Y +W
A B Y W A Y W +A=B
? 如C=C 双键的加成 CH 2CH 2+HBr CH 2-CH 3Br CH 2Br-CH 3+
(2) 亲核加成(nucleophilic addition reaction )如羰基加成、Michael 加成:
(3)自由基加成反应
(free radical addition reaction)
如过氧化物存在下烯烃与溴化氢的加成
(4) 一步加成反应(环化加成反应)
Cycloaddition reaction
? 主要指2分子烯烃加成形成稳定的环状化合物的反应,如Diels-alder 反应
? 有时也将电环化反应——共轭多烯转变成环烯烃反应视为分子内部环加成
3. β-消除反应(β-elimination Reaction )
(1)异裂消除反应(Heterolgtic elimination Reaction )
如醇脱水 卤烃脱卤化氢 Hofmann 消除反应等
(2)一步消除反应
(concerted elimination reaction 协同消除反应)如脂的热裂:
4. 重排反应(Rearrangement reaction )
?重排反应通常是指一个原子或基团从同一分子中的一个原子转
移到另一个原子的反应。
(分子内迁移反应)
?根据迁移原子或基团在迁移时所带电子的多少分为4种类:
(1) 带一对电子的转移:
(缺电子重排或亲核重排反应)
Nucleophilic rearrangement reaction
(3)不带电子转移(亲电重排)
(4)一步重排反应(σ重排)
Claisen重排
?烯丙基苯基醚类化合物的热重排反应
5. 氧化还原反应
许多氧化还原反应按上述反应历程进行,
有些则不是
(三)按参加反应的分子或质子数分类
?1.单分子反应历程
Unimolecular reaction mechanism
?化学反应常常不是一步反应,而是分既不完成。每一步反应速
率不同,其中最慢一步决定反应速率,称之为决速步骤。
?当此步速率只与一种分子或质子的浓度有关时,(形成活化中间
体时只涉及一个分子)即为单分子反应历程(Unimolecular reaction mechanism)
有机化学试卷 班级姓名分数 一、机理题 ( 共44题 288分 ) 1. 8 分 (2701) 2701 为下述实验事实提出合理的、分步的反应机理(用弯箭头表示电子对的转移,用鱼钩箭头表示单电子的转移,并写出各步可能的中间体)。 邻苯二甲酰亚胺用Br2-NaOH处理获得邻氨基苯甲酸。 2. 8 分 (2702) 2702 为下述实验事实提出合理的、分步的反应机理(用弯箭头表示电子对的转移,用鱼钩箭头表示单电子的转移,并写出各步可能的中间体。 2,4-二硝基氟苯(A)及2,4-二硝基溴苯(B)分别用C2H5NH2处理,都获得N-乙基-2,4-二硝基苯胺,但A比B速率快。 3. 8 分 (2703) 2703 为下述实验事实提出合理的、分步的反应机理(用弯箭头表示电子对的转移,用鱼钩箭头表示单电子的转移,并写出各步可能的中间体)。 异丙苯过氧化氢用酸处理,获得苯酚和丙酮(石油化工生产)。 4. 8 分 (2704) 2704 为下述实验事实提出合理的、分步的反应机理(用弯箭头表示电子对的转移,用鱼钩箭头表示单电子的转移,并写出各步可能的中间体)。 用14C标记的2-甲基-6-烯丙基苯酚的烯丙醚(A)加热发生Claisen重排反应,生成的2-甲基-4,6-二烯丙基苯酚中有一半以上含14C的烯丙基在对位。
5. 6 分 (2705) 2705 为下述实验事实提出合理的、分步的反应机理(用弯箭头表示电子对的转移,用鱼钩箭头表示单电子的转移,并写出各步可能的中间体)。 邻位和对位的羟基苯甲酸容易失羧,而间位异构体无此特征。 6. 6 分 (2706) 2706 为下述实验事实提出合理的、分步的反应机理(用弯箭头表示电子对的转移,用鱼钩箭头表示单电子的转移,并写出各步可能的中间体)。 旋光的苯基二级丁基酮在碱性溶液中发生外消旋化;这个酮失去旋光性的速率正好和它在碱性条件下溴化的速率相等。 7. 6 分 (2707) 2707 为下述实验事实提出合理的、分步的反应机理。 旋光的扁桃酸乙酯[C6H5CH(OH)CO2C2H5]在碱性条件下易外消旋化。 8. 6 分 (2708) 2708 为下述实验事实提出合理的、分步的反应机理。 旋光的扁桃酸[C6H5CH(OH)CO2H]在碱中的外消旋化比其酯慢得多。 9. 6 分 (2709) 2709 为下述实验事实提出合理的、分步的反应机理(用弯箭头表示电子对的转移,用鱼钩箭头表示单电子的转移,并写出各步可能的中间体)。 苯基二级丁基酮进行酸催化的外消旋化与其碘代反应的速率常数相等。 *. 6 分 (2710) 2710 为下述实验事实提出合理的、分步的反应机理(用弯箭头表示电子对的转移,用鱼钩箭头表示单电子的转移,并写出各步可能的中间体)。 醇醛缩合(aldol)反应亦可酸催化,如乙醛在酸催化下可生成-羟基丁醛。
1.Arbuzov 反应 卤代烷反应时,其活性次序为:R'I >R'Br >R'Cl。除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a- 或 b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。 本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得: 一般认为是按 S N2 进行的分子内重排反应: 2.Arndt-Eister 反应 酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。 重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。 3.Baeyer----Villiger 反应
过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。因此,这是一个重排反应 具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排: 不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为: 4.Beckmann 重排 肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺: 在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
1.A rndt-Eister 反应 酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。 反应机理 重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。 反应实例
2.Baeyer----Villiger 反应 反应机理 过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。因此,这是一个重排反应 具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排: 不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为: 醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。 反应实例
酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。这类氧化剂的特点是反应速率快,反应温度一般在10~40℃之间,产率高。 3.Beckmann 重排 肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:
反应机理 在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。 迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如: 反应实例
基础有机化学反应总结 一、烯烃 1、卤化氢加成 (1) CH CH 2 R HX CH CH 3R X 【马氏规则】在不对称烯烃加成中,氢总是加在含碳较多的碳上。 【机理】 CH 2 C H 3+ CH 3 C H 3X + CH 3 C H 3 +H + CH 2 +C 3X + C H 3X 主 次 【本质】不对称烯烃的亲电加成总是生成较稳定的碳正离子中间体。 【注】碳正离子的重排 (2) CH CH 2 R CH 2CH 2 R Br HBr ROOR 【特点】反马氏规则 【机理】 自由基机理(略) 【注】过氧化物效应仅限于HBr 、对HCl 、HI 无效。 【本质】不对称烯烃加成时生成稳定的自由基中间体。 【例】 CH 2 C H 3Br CH CH 2Br C H 3CH + CH 3 C H 3HBr Br CH 3CH 2CH 2Br CH CH 3 C H 3 2、硼氢化—氧化 CH CH 2 R CH 2CH 2R OH 1)B 2H 62)H 2O 2/OH -
【特点】不对称烯烃经硼氢化—氧化得一反马氏加成的醇,加成是顺式的,并且不重排。 【机理】 2 C H 33H 32 3H 32 CH CH 2C H 3 2 CH CH=CH (CH 3CH 2CH 2)3 - H 3CH 2CH 2C 22CH 3 CH 2O CH 2CH 2CH 3 3CH 2CH 2C 2CH 2CH 3 + O H - O H B - OCH 2CH 2CH 3CH 2CH 2CH 3 H 3CH 2CH 2B OCH 2CH 2CH 3 CH 2CH 2CH 32CH 2CH 3 HOO -B(OCH 2CH 2CH 3)3 B(OCH 2CH 2CH 3)3 + 3NaOH3NaOH 3HOCH 2CH 2CH 33 + Na 3BO 3 2 【例】 CH 3 1)BH 32)H 2O 2/OH -CH 3 H H OH 3、X 2加成 C C Br /CCl C C Br Br 【机理】
1.Arndt-Eister 反应 酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。 重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。 2.Baeyer----Villiger 反应 过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。因此,这是一个重排反应
具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排: 不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为: 醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。
酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。这类氧化剂的特点是反应速率快,反应温度一般在10~40℃之间,产率高。 重排 肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺: 在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如: 例 还原 芳香化合物用碱金属(钠、钾或锂)在液氨与醇(乙醇、异丙醇或仲丁醇)的混合液中还原,苯环可被还原成非共轭的1,4-环己二烯化合物。
有机化学复习总结 一.有机化合物的命名 1. 能够用系统命名法命名各种类型化合物: 包括烷烃,烯烃,炔烃,烯炔,脂环烃(单环脂环烃和多环置换脂环烃中的螺环烃和桥环烃),芳烃,醇,酚,醚,醛,酮,羧酸,羧酸衍生物(酰卤,酸酐,酯,酰胺),多官能团化合物(官能团优先顺序:-COOH >-SO3H >-COOR >-COX >-CN >-CHO >>C =O >-OH(醇)>-OH(酚)>-SH >-NH2>-OR >C =C >-C ≡C ->(-R >-X >-NO2),并能够判断出Z/E 构型和R/S 构型。 2. 根据化合物的系统命名,写出相应的结构式或立体结构式(伞形式,锯架式,纽曼投影式,Fischer 投影式)。 立体结构的表示方法: 1 )伞形式: COOH OH H 3 2)锯架式:CH 3 OH H H OH C 2H 5 3) 纽曼投影式: H H 4)菲舍尔投影式:COOH CH 3 OH H 5)构象(conformation) (1) 乙烷构象:最稳定构象是交叉式,最不稳定构象是重叠式。 (2) 正丁烷构象:最稳定构象是对位交叉式,最不稳定构象是全重叠式。 (3) 环己烷构象:最稳定构象是椅式构象。一取代环己烷最稳定构象是e 取代的椅 式构象。多取代环己烷最稳定构象是e 取代最多或大基团处于e 键上的椅式构象。 立体结构的标记方法 1. Z/E 标记法:在表示烯烃的构型时,如果在次序规则中两个优先的基团在同一侧,为Z 构型, 在相反侧,为E 构型。 CH 3 C H C 2H 5CH 3C C H 2H 5Cl (Z)-3-氯-2-戊烯 (E)-3-氯-2-戊烯 2、 顺/反标记法:在标记烯烃和脂环烃的构型时,如果两个相同的基团在同一侧,则为顺式; 在相反侧,则为反式。
3.Baeyer----Villiger 反应拜耳-维立格氧化重排反应 过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。因此,这是一个重排反应 具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排: 不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为: 4.Beckmann 重排 肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺: 在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变。 7.Cannizzaro 反应 凡α位碳原子上无活泼氢的醛类和浓NaOH或KOH水或醇溶液作用时,不发生醇醛缩合或树脂化作用而起歧化反应生成与醛相当的酸(成盐)及醇的混合物。此反应的特征是醛自身同时发生氧化及还原作用,一分子被氧化成酸的盐,另一分子被还原成醇: 脂肪醛中,只有甲醛和与羰基相连的是一个叔碳原子的醛类,才会发生此反应,其他醛类与强碱液,作用发生醇醛缩合或进一步变成树脂状物质。 醛首先和氢氧根负离子进行亲核加成得到负离子,然后碳上的氢带着一对电子以氢负离子的形式转移到另一分子的羰基不能碳原子上。 9.Claisen 酯缩合反应
含有α-氢的酯在醇钠等碱性缩合剂作用下发生缩合作用,失去一分子醇得到β-酮酸酯。如2分子乙酸乙酯在金属钠和少量乙醇作用下发生缩合得到乙酰乙酸乙酯。 乙酸乙酯的α-氢酸性很弱(pK a-24.5),而乙醇钠又是一个相对较弱的碱(乙醇的 pK a~15.9),因此,乙酸乙酯与乙醇钠作用所形成的负离子在平衡体系是很少的。但由于最后产物乙酰乙酸乙酯是一个比较强的酸,能与乙醇钠作用形成稳定的负离子,从而使平衡朝产物方向移动。所以,尽管反应体系中的乙酸乙酯负离子浓度很低,但一形成后,就不断地反应,结果反应还是可以顺利完成。 10.Claisen 重排 烯丙基芳基醚在高温(200°C)下可以重排,生成烯丙基酚。 当烯丙基芳基醚的两个邻位未被取代基占满时,重排主要得到邻位产物,两个邻位均被取代基占据时,重排得到对位产物。对位、邻位均被占满时不发生此类重排反应。
有机化学 一、烯烃 1、卤化氢加成 (1) CH CH 2 R HX CH CH 3R X 【马氏规则】在不对称烯烃加成中,氢总是加在含碳较多的碳上。 【机理】 CH 2 C H 3+ CH 3 C H 3X + CH 3 C H 3 +H + CH 2 +C 3X + C H 3X 主 次 【本质】不对称烯烃的亲电加成总是生成较稳定的碳正离子中间体。 【注】碳正离子的重排 (2) CH CH 2 R CH 2CH 2 R Br HBr ROOR 【特点】反马氏规则 【机理】 自由基机理(略) 【注】过氧化物效应仅限于HBr 、对HCl 、HI 无效。 【本质】不对称烯烃加成时生成稳定的自由基中间体。 【例】 CH 2 C H 3Br CH CH 2Br C H 3CH + CH 3 C H 3HBr Br CH 3CH 2CH 2Br CH CH 3 C H 3 2、硼氢化—氧化 CH CH 2 R CH 2CH 2R OH 1)B 2H 62)H 2O 2/OH - 【特点】不对称烯烃经硼氢化—氧化得一反马氏加成的醇,加成是顺式的,并且不重排。 【机理】
2 C H3 3 H3 2 3 H3 2 CH CH2 C H3 2 CH CH=CH (CH3CH2CH2)3 - H3CH2CH2C 22 CH3 CH2 B O CH2CH2CH3 3 CH2CH2C 2 CH2CH3 +O H- O H B-OCH2CH2CH3 CH2CH2CH3 H3CH2CH2 B OCH2CH2CH3 CH2CH2CH3 2 CH2CH3 HOO- B(OCH2CH2CH3)3 B(OCH2CH2CH3)3+3NaOH3NaOH3HOCH2CH2CH33+Na3BO3 2 【例】 CH3 1)BH 3 2)H 2 O 2 /OH- CH3 H H OH 3、X2加成 C C Br 2 /CCl 4 C C Br Br 【机理】 C C C C Br Br C Br +C C Br O H2+ -H+ C C Br O H
大学有机化学反应方程 式总结较全 Document serial number【NL89WT-NY98YT-NC8CB-NNUUT-NUT108】
有 机化学 一、烯烃 1、卤化氢加成 (1) CH CH 2 R HX CH CH 3R X 【马氏规则】在不对称烯烃加成中,氢总是加在含碳较多的碳上。 【机理】 CH 2 C H 3CH + CH 3 C H 3X + CH 3 C H 3 +H + CH 2 +C 3X + C H 3X 主 次 【本质】不对称烯烃的亲电加成总是生成较稳定的碳正离子中间体。 【注】碳正离子的重排 (2) CH CH 2 R CH 2CH 2 R Br HBr ROOR 【特点】反马氏规则 【机理】 自由基机理(略) 【注】过氧化物效应仅限于HBr 、对HCl 、HI 无效。 【本质】不对称烯烃加成时生成稳定的自由基中间体。 【例】 CH 2 C H 3Br CH CH 2Br C H 3CH + CH 3 C H 3HBr Br CH 3CH 2CH 2Br CH CH 3 C H 3 2、硼氢化—氧化 CH CH 2 R CH 2CH 2R OH 1)B 2H 62)H 2O 2/OH - 【特点】不对称烯烃经硼氢化—氧化得一反马氏加成的醇,加成是顺式的,并且不重排。
【机理】 2 C H3 3 H3 2 3 H3 2 CH CH2 C H3 2 CH CH=CH (CH3CH2CH2)3 - H3CH2CH2C 22 CH3 CH2 O CH 2 CH2CH3 H3CH2CH2C 2 CH2CH3 +O H- O H B-OCH2CH2CH3 CH2CH2CH3 H3CH2CH2C B OCH2CH2CH3 CH2CH2CH3 2 CH2CH3 HOO- B(OCH2CH2CH3)3 B(OCH2CH2CH3)3+3NaOH3NaOH3HOCH2CH2CH33+Na3BO3 2 【例】 CH3 1)BH 3 2)H 2 O 2 /OH- CH3 H H OH 3、X 2 加成 C C Br/CCl C C Br Br 【机理】 C C C C Br Br C Br +C C Br O H2+ -H+ C C Br O H
欢迎阅读 有机化学复习总结 一.有机化合物的命名 1.能够用系统命名法命名各种类型化合物: 包括烷烃,烯烃,炔烃,烯炔,脂环烃(单环脂环烃和多环置换脂环烃中的螺环烃和桥环烃),芳烃,醇,酚,醚,醛,酮,羧酸,羧酸衍生物(酰卤,酸酐,酯,酰胺),多官能团化合物(官能团优先顺序:-COOH >-SO3H >-COOR >-COX >-CN >-CHO >>C =O >-OH(醇)>-OH(酚)>-SH >-NH2>-OR >C =C >-C ≡C ->(-R >-X >-NO2),并能够判断出Z/E 构型和R/S 构型。 2.根据化合物的系统命名,写出相应的结构式或立体结构式(伞形式,锯架式,纽曼投影式,Fischer 投影式)。 立体结构的表示方法: 1)伞形式:C COOH OH H 3C H 2)锯架式:CH 3 OH H H OH C 2H 5 3)纽曼投影式: H H H H H H H H H H H H 4)菲舍尔投影式:COOH CH 3 OH H 5)构象(conformation) (1) 乙烷构象:最稳定构象是交叉式,最不稳定构象是重叠式。 (2) 正丁烷构象:最稳定构象是对位交叉式,最不稳定构象是全重叠式。 (3) 环己烷构象:最稳定构象是椅式构象。一取代环己烷最稳定构象是e 取代的椅式构象。多取代环己烷最稳定构象是e 取代最多或大基团处于e 键上的椅式构象。 立体结构的标记方法 1. Z/E 标记法:在表示烯烃的构型时,如果在次序规则中两个优先的基团在同一侧,为Z 构型,在相反侧,为E 构型。 2、 顺/反标记法:在标记烯烃和脂环烃的构型时,如果两个相同的基团在同一侧,则为顺式;在相反侧,则为反式。 3、 R/S 标记法:在标记手性分子时,先把与手性碳相连的四个基团按次序规则排序。然后将最不优先的基团放在远离观察者,再以次观察其它三个基团,如果优先顺序是顺时针,则为R 构型,如果是逆时针,则为S 构型。 注:将伞状透视式与菲舍尔投影式互换的方法是:先按要求书写其透视式或投影式,然后分别标出其R/S 构型,如果两者构型相同,则为同一化合物,否则为其对映体。 二.有机化学反应及特点 1.反应类型 还原反应(包括催化加氢):烯烃、炔烃、环烷烃、芳烃、卤代烃
本文整理出常见的有机人名反应80多个,共计约100页,大部分内容在竞赛考察范围之内。全国初赛有机难度虽然有所降低,但有能力冲刺决赛的选手对于有机反应必须熟练掌握,熟记反应实例与机理。熟记有机人名反应不仅是化学竞赛的要求,也是考研的重要内容,更是对化学先驱们的尊重与缅怀。 索引: Arbuzov反应 Arndt-Eister反应 Baeyer-Villiger 氧化 Beckmann 重排 Birch 还原 Bischler-Napieralski 合成法 Bouveault-Blanc还原 Bucherer 反应 Cannizzaro 反应 Chichibabin 反应 Claisen 酯缩合反应 Claisen-Schmidt 反应 Clemmensen 还原 Combes 合成法 Cope 重排 Cope 消除反应 Curtius 反应 Dakin 反应 Darzens 反应 Demjanov 重排 Dieckmann 缩合反应 Elbs 反应 Eschweiler-Clarke 反应 Favorskii 反应 Favorskii 重排 Friedel-Crafts烷基化反应 Friedel-Crafts酰基化反应 Fries 重排 Gabriel 合成法 Gattermann 反应 Gattermann-Koch 反应 Gomberg-Bachmann 反应 Hantzsch 合成法 Haworth 反应 Hell-V olhard-Zelinski 反应 Hinsberg 反应 Hofmann 烷基化 Hofmann 消除反应 Hofmann 重排(降解)
Houben-Hoesch 反应Hunsdiecker 反应 Kiliani 氰化增碳法Knoevenagel 反应 Knorr 反应 Koble 反应 Koble-Schmitt 反应Leuckart 反应 Lossen反应 Mannich 反应 Meerwein-Ponndorf 反应Meerwein-Ponndorf 反应Michael 加成反应Norrish I和II 型裂解反应Oppenauer 氧化 Paal-Knorr 反应 Pictet-Spengler 合成法Pschorr 反应Reformatsky 反应 Reimer-Tiemann 反应Reppe 合成法 Robinson 缩环反应Rosenmund 还原 Ruff 递降反应Sandmeyer 反应Schiemann 反应 Schmidt反应 Skraup 合成法Sommelet-Hauser 反应Stephen 还原 Stevens 重排 Strecker 氨基酸合成法Tiffeneau-Demjanov 重排Ullmann反应 Vilsmeier 反应 Wagner-Meerwein 重排Wacker 反应 Williamson 合成法 Wittig 反应 Wittig-Horner 反应 Wohl 递降反应 Wolff-Kishner-黄鸣龙反应Yurév 反应 Zeisel 甲氧基测定法
有机化学反应机理 弯箭头代表一对电子的转移,弯钩意味着一个电子的转移,后者适用于自由基反应
1 有机反应机理入门 1.1 画路易斯结构式 先画出分子的骨架,环和pi键应准确无误,然后用氢原子完成其余的化学键。对于有机分子,骨架有时以简化形式给出。 画出孤对电子,使每个原子核外满足充满电子的结构:氢2个;硼、铝和镓6个;其它原子8个。最后结构式中的每个原子总的成键电子数可以通过数其核外的成键电子获得(包括共享电子)。 提示:画路易斯结构式可参考以下结构特征: (1) 氢原子永远在构的外围,因为它只能成一个共价键; (2) 碳、氮和氧有特定的键合模式。 在以下的示例中R代表氢、烷基、芳基或它们的组合,这种变化并不影响成键模 式。 ①中性的碳原子为4键。这4个键可以都是sigma键,也可以是sigma键与pi
键的组合(如双键和三键)。 ②带有单个正电荷或负电荷的碳有3个键。 ③中性的氮原子(氮烯除外)有3个键和一对未成对电子。 键,带有一个正电荷。4正电荷的氮成④. ⑤负电荷的氮成2键,带有一个负电荷和2对未成键电子。 ⑥中性的氧原子成2键,带有2对孤对电子。 ⑦带正电荷的氧成3键,带有1对孤对电子。 (3) 有时磷原子和硫原子可有10个成键电子,这是因为磷和硫具有d轨道,可以扩展而容纳10个电子。 Lewis结构式是价键理论的重要内容,也是学习反应机理的基础。 1.2 电负性 多数有机反应依赖于带有正电荷(或部分正电荷)的分子与带有负电荷(或部分负电荷)的分子的相互作用而发生。在中性有机分子中,部分电荷的产生依赖于电负性的差异。 电负性的数值最初由Linus Pauling在1960年确定。其数值越大,表明其吸电子能力越强,所以氟是吸电子能力最强的元素,见表: 成键后,电负性大的元素的原子拥有部分负电荷,而且,双键结构的部分电荷比单键结构的部分电荷密度更大,这是因为双键上的pi电子受原子核的束缚小,更易于流动。
基础有机化学人名反应 第四章 狄尔斯–阿尔德反应(Diels–Alder reaction)(140) 1921年,狄尔斯和其研究生巴克(Back)研究偶氮二羧酸二乙酯(半个世纪后因光延反应而在有机合成中大放光芒的试剂)与胺发生的酯变胺的反应,当他们用2-萘胺做反应的时候,根据元素分析,得到的产物是一个加成物而不是期待的取代物。狄尔斯敏锐地意识到这个反应与十几年前阿尔布莱希特做过的古怪反应的共同之处。这使他开始以为产物是类似阿尔布莱希特提出的双键加成产物。狄尔斯很自然地仿造阿尔布莱希特用环戊二烯替代萘胺与偶氮二羧酸乙酯作用,结果又得到第三种加成物。通过计量加氢实验,狄尔斯发现加成物中只含有一个双键。如果产物的结构是如阿尔布莱希特提出的,那么势必要有两个双键才对。这个现象深深地吸引了狄尔斯,他与另一个研究生阿尔德一起提出了正确的双烯加成物的结构。1928年他们将结果发表。这标志着狄尔斯-阿德尔反应的正式发现。他们也因此获得1950年的诺贝尔化学奖。 含有一个活泼的双键或叁键的化合物(亲双烯体)与共轭二烯类化合物(双烯体)发生1,4-加成,生成六员环状化合物: 这个反应极易进行并且反应速度快,应用范围极广泛,是合成环状化合物的一个非常重要的方法。
带有吸电子取代基的亲双烯体和带有给电子取代基的双烯体对反应有利。常用的亲双烯体有: 下列基团也能作为亲双烯体发生反应: 常用的双烯体有: a.反应机理 这是一个协同反应,反应时,双烯体和亲双烯体彼此靠近,互相作用,形成一个环状过渡态,然后逐渐转化为产物分子:
反应是按顺式加成方式进行的,反应物原来的构型关系仍保留在环加成产物中。例如: 正常的Diels-Alder反应主要是由双烯体的HOMO(最高已占轨道)与亲双烯体的LUMO(最低未占轨道)发生作用。反应过程中,电子从双烯体的HOMO“流入”亲双烯体的LUMO。也有由双烯体的LUMO与亲双烯体的HOMO作用发生反应的。 b.反应实例
十、反应和反应机理 有机反应:在一定的条件下,有机化合物分子中的成键电子发生重新分布,原有的键断裂,新的键形成,从而使原分子中原子间的组合发生了变化,新的分子产生。这种变化过程称为有机反应(organic reaction)。 一级反应:在动力学上,将反应速率只取决于一种化合物浓度的反应称为一级反应。 二级反应:在动力学上,将反应速率取决于两种化合物浓度的反应称为二级反应。 按化学键的断裂和生成分类 协同反应:在反应过程中,旧键的断裂和新键的形成都相互协调地在同一步骤中完成的反应称为协同反应。协同反应往往有一个环状过渡态。它是一种基元反应。 自由基型反应:由于分子经过均裂产生自由基而引发的反应称为自由基型反应。自由基型反应分链引发、链转移和链终止三个阶段:链引发阶段是产生自由基的阶段。由于键的均裂需要能量,所以链引发阶段需要加热或光照。链转移阶段是由一个自由基转变成另一个自由基的阶段,犹如接力赛一样,自由基不断地传递下去,像一环接一环的链,所以称之为链反应。链终止阶段是消失自由基的阶段,自由基两两结合成键,所有的自由基都消失了,自由基反应也就终止了。 离子型反应:由分子经过异裂生成离子而引发的反应称为离子型反应。离子型反应有亲核反应和亲电反应,由亲核试剂进攻而发生的反应称为亲核反应,亲核试剂是对原子核有显著亲和力而起反应的试剂。由亲电试剂进攻而发生的反应称为亲电反应。亲电试剂是对电子有显著亲合力而起反应的试剂。 按反应物和产物的结构关系分类 加成反应:两个或多个分子相互作用,生成一个加成产物的反应称为加成反应。 取代反应:有机化合物分子中的某个原子或基团被其它原子或基团所置换的反应称为取代反应。 重排反应:当化学键的断裂和形成发生在同一分子中时,会引起组成分子的原子的配置方式发生改变,从而形成组成相同,结构不同的新分子,这种反应称为重排反应。 消除反应:在一个有机分子中消去两个原子或基团的反应称为消除反应。可以根据两个消去基团的相对位置将其分类。若两个消去基团连在同一个碳原子上,称为1,1-消除或α-消除;两个消去基团连在两个相邻的碳原子上,则称为1,2-消除或β-消除;两个消去基团连在1,3位碳原子上,则称为1,3-消除或γ-消除。其余类推。 氧化还原反应:有机化学中的氧化和还原是指有机化合物分子中碳原子和其它原子的氧化和还原,可根据氧化数的变化来确定。氧化数升高为氧化,氧化数降低为还原。氧化和还原总是同时发生的,由于有机反应的属性是根据底物的变化来确定的,因此常常将有机分子中碳原子氧化数升高的反应为氧化反应,碳原子氧化数降低的反应为还原反应。有机反应中,多数氧化反应表现为分子中氧的增加或氢的减少,多数还原反应表现为分子中氧的减少或氢的增加。
有机反应机制的研究方法 有机化学中用来解释反应机理的传统方法主要集中在Kinetics 和Dynamics两方面,即理解势能面、深入研究分子运动和碰撞、测定活化参数、测定速率常数、确定某个反应机理中一系列化学步骤的顺序、确定反应限速步骤和决速步骤。 研究机理的关键目的是反应机理知识可以对如何在原子或分子水平上操纵物质给出最快速的洞察,而不是依靠运气来获得偶然性的变化从而获得想要的结果。由于动力学在辨别机理方面起着关键作用,所以动力学是整个有机反应机理研究领域中最重要的分支之一。 传统的反应机理研究方法除了动力学分析之外,还有同位素效应、结构-功能分析等。这些都是研究有机反应机理的标准实验工具,然后实验化学家可以根据其想象力和化学创造性,设计出一些完全不同于之前出现过的研究方法。因此,本文总结了一些最为常见的方法。首先分析最简单的实验,例如产物和中间体的鉴定。但也会分析一些更为微妙、精细的实验,如交叉和同位素置乱(cross-over and isotope scrambling)实验。 1.改变反应物结构以转变或捕获预想的中间体 有时可以通过合成一种类似于所研究的反应物的新反应物来破译中间体的性质,但是这需要所预测的中间体能以一种可预想的方式进行反应。没有标准的方式来处理这一类实验,所以实验者必须根据具体实验情况来设计实验。下面以酶反应作为此方法的应用实例。 Lin[1]等人设计了一种转变中间体的方法。扁桃酸消旋化酶可使扁
桃酸根离子的对映体(2-羟基苯甲酸)互换。位于羧酸跟α位的碳负离子被认为是中间体。为了测试此中间体是否存在,作者合成设计了扁桃酸跟离子的类似物i,并用酶对其进行了外消旋化。其过程是首先形成碳负离子,然后经过溴化物的1,6-消除,最后经过互变异构化,分离得到产物ii。此结果支持了在扁桃酸根离子路径中碳负离子中间体iii的存在。 2.捕获实验和竞争实验 鉴定中间体的一种常见方法是通过加入额外的试剂来捕获中间体。目前存在着几种自由基不伙计,许多好的亲核试剂是半衰期很短的亲电试剂(如碳正离子)的可行的捕获剂。必须以自己的化学知识来设计捕获中间体(如碳正离子、卡宾等)的捕获剂。但是活泼中间体的半衰期很短,所以捕获剂必须是具有很高的活性,并能与活泼中间体的标准反应路径进行竞争。同样,因为捕获反应是典型的双分子反应,所以要求捕获剂具有高的浓度。另外,还可以将捕获剂与反应物共价结合,以便更容易地捕获活泼中间体。 与捕获反应所不同的另一种反应是竞争反应。在一般的动力学实
1.A rndt-Eister反应 令狐采学 酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。 反应机理 重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。 反应实例 2.Baeyer----Villiger反应 反应机理 过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。因此,这是一个重排反应 具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排: 不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为:
醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。 反应实例 酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂。这类氧化剂的特点是反应速率快,反应温度一般在10~40℃之间,产率高。 3.Beckmann 重排 肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺: 反应机理 在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。 迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如: 反应实例 4.Birch还原
有机化学复习总结 一.有机化合物的命名 1. 能够用系统命名法命名各种类型化合物: 包括烷烃,烯烃,炔烃,烯炔,脂环烃(单环脂环烃和多环置换脂环烃中的螺环烃和桥环烃),芳烃,醇,酚,醚,醛,酮,羧酸,羧酸衍生物(酰卤,酸酐,酯,酰胺),多官能团化合物(官能团优先顺序:-COOH >-SO3H >-COOR >-COX >-CN >-CHO >>C =O >-OH(醇)>-OH(酚)>-SH >-NH2>-OR >C =C >-C ≡C ->(-R >-X >-NO2),并能够判断出Z/E 构型和R/S 构型。 2. 根据化合物的系统命名,写出相应的结构式或立体结构式(伞形式,锯架式,纽曼投影式,Fischer 投影式)。 立体结构的表示方法: 1)伞形式:C COOH OH H 3C H 2)锯架式:CH 3 OH H H OH C 2H 5 3) 纽曼投影式: H H H H H H H H H H H H 4)菲舍尔投影式:COOH CH 3 OH H 5)构象(conformation) (1) 乙烷构象:最稳定构象是交叉式,最不稳定构象是重叠式。 (2) 正丁烷构象:最稳定构象是对位交叉式,最不稳定构象是全重叠式。 (3) 环己烷构象:最稳定构象是椅式构象。一取代环己烷最稳定构象是e 取代的椅 式构象。多取代环己烷最稳定构象是e 取代最多或大基团处于e 键上的椅式构象。 立体结构的标记方法 1. Z/E 标记法:在表示烯烃的构型时,如果在次序规则中两个优先的基团在同一 侧,为Z 构型,在相反侧,为E 构型。 CH 3 C C H Cl C 2H 5CH 3C C H C 2H 5Cl (Z)-3-氯-2-戊烯 (E)-3-氯-2-戊烯 2、 顺/反标记法:在标记烯烃和脂环烃的构型时,如果两个相同的基团在同一侧, 则为顺式;在相反侧,则为反式。 CH 3C C H CH 3H CH 3C C H H CH 3顺-2-丁烯 反-2-丁烯CH 3 H CH 3 H CH 3 H H CH 3顺-1,4-二甲基环己烷反-1,4-二甲基环己烷 3、 R/S 标记法:在标记手性分子时,先把与手性碳相连的四个基团按次序规则排序。然后将最不优先的基团放在远离观察者,再以次观察其它三个基团,如果优先顺序
Chap 1绪论 一、构造、构型、构象 二、共价键 轨道杂化:C:sp、sp2、sp3杂化方式、空间构型(键角)、未参与杂化p轨道与杂化轨道位置、电负性比较 基本属性:键长:越短键越牢固键能:越大键越牢固σ键能大于п键能 键角:取代基越大键角越大极性和极化性:偶极矩(会判断偶极矩大小:矢 量和) 键断裂方式和反应类型:自由基反应、离子型(亲电、亲核)、周环反应 Lewis酸、碱 氢键、电负性 三、官能团、优先次序(ppt) Chap 2饱和烃——烷烃 一、烃分类 烃:开链烃和环状烃 开链烃:饱和烃和不饱和烃环状烃:脂环烃和芳香烃 二、烷烃通式和构造异构、构象异构(乙烷和丁烷构象) 烷烃通式:C n H2n+2 构造异构体:分子内原子链接顺序不同 σ键形成及特性:电子云重叠程度大,键能大,不易断;可绕轴自由旋转;两核间不能有两个或以上σ键。 乙烷构象:Newman投影式、重叠式(不稳定,因为非键张力大)、交叉式(稳定,各个氢距离远,非键张力小) 丁烷构象:Newman投影式;稳定性(大到小):对位交叉式、邻位交叉式、部分重叠式、全部重叠式 甲烷结构和sp3杂化构型:正四面体型 三、命名 普通命名法(简单化合物):正、异、新 衍生物命名法:以甲烷为母体,选取取代基最多的C为母体C。 系统命名法:①选取最长碳链为主链,主链C标号从距离取代基最近的一端开始标。 ②多取代基时,合并相同取代基,尽量使取代基位次和最小。书写时按照 官能团大小(小在前)命名 ③含多个相同长度碳链时,选取取代基最多的为主链 四、物理性质 沸点(b.p.):直链烷烃随分子量增大而增大(分子间色散力与分子中原子大小和数目成正比,分子量增大,色散力增大,沸点增大) 支链越多,沸点越低(支链多,烷烃体积松散,分子间距离大,色散力小)熔点(m.p.):总趋势:分子量增大,m.p.增大 m.p.曲线(书P48) 相对密度:分子量增大,相对密度增大,接近于0.8 溶解度:不溶于水,易溶于有机溶剂(相似相溶,烷烃极性小)
一、Arbuzow反应(重排) 亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷: 卤代烷反应时,其活性次序为:R'I >R'Br >R'Cl。除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a- 或 b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团。 本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得: 如果反应所用的卤代烷 R'X 的烷基和亚磷酸三烷基酯 (RO)3P 的烷基相同(即 R' = R),则Arbuzow反应如下: 这是制备烷基膦酸酯的常用方法。 除了亚磷酸三烷基酯外,亚膦酸酯 RP(OR')2和次亚膦酸酯 R2POR' 也能发生该类反应,例如:
反应机理 一般认为是按 S N2 进行的分子内重排反应: 反应实例 二、Arndt-Eister 反应 酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。 反应机理
重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。 反应实例 三、Baeyer----Villiger 反应 反应机理 过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂。因此,这是一个重排反应
具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型保持不变,说明反应属于分子内重排: 不对称的酮氧化时,在重排步骤中,两个基团均可迁移,但是还是有一定的选择性,按迁移能力其顺序为: 醛氧化的机理与此相似,但迁移的是氢负离子,得到羧酸。 反应实例
有机化学反应机理
有机化学反应机理 对于均裂反应来说:反应物既提供电子又接受电子注意:提供和接受的电子均为单电子 对于异裂反应来说: 提供和接受的电子为电子对 反应物的分类:亲核试剂:电子云密度高的中性分子或带负电荷的原子、原子团或 分子(又为Lewis 碱)。 亲电试剂:电子云密度低的中性原子、原子团或分子或带正电荷的 任何分子、原子、原子团(Lewis酸)。 ?取代反应:SN1 和SN2 ?伯卤代烃= SN2 ?仲卤代烃= SN1 和SN2 ! ?叔卤代烃= SN1 ?离去基团:大多数是卤素 ?亲核试剂:许多亲核试剂!! 邻基取代:在离去基团的邻位上能够进行邻基参与的基团 酯基、羧基、羟基、苯基、稀基、卤素。 .波谱特征 红外光谱 红外特征吸收峰是C-X键的振动吸收,都在指纹区,其中C-F 键的吸收频率在1400~1000 cm-1,C-Cl键为800~600 cm-1,C-Br 键为600~500 cm-1,而C-I 键的吸收频率在500 cm-1附近。 核磁共振谱 1H-NMR谱中,卤素电负性较大,因此与卤素直接相连的碳上的氢的化学位移移向低场 卤代烃及亲核取代反应 反应活性次序: 叔卤烷>仲卤烷>伯卤。用于卤烷的定性分析.
卤素相同、烃基结构不同的卤代烷,其活性顺序为:1°>2°>3°。 此反应也可用于鉴别卤代烃,反应最快的是伯卤代烷,其次是仲卤代烷,反应最慢的是叔卤代烷。 Saytzeff 规则 如果分子内含有几种β-H 时,主要消除含氢较少的碳上的氢,生成双键碳上连有较多取代基的烯烃,这一经验规则称Saytzeff 规则。 RX AgNO 3 C 2H 5OH RONO 2AgX ++RBr + NaI RI + NaBr RCl + NaI RI + NaCl 丙酮丙酮R-X ROH ROR'R-CN R-NH 2O H 2NaOH ,,NH R-R R'COOR R'C CR CNa R-O-NO 2AgX AgNO 3+ △