
残留溶剂的鉴别、控制、定量
【注意-在下列各方法中指定的无有机物水在色谱上应没有明显的杂质干扰峰】
1类和2类残留溶剂
水溶性样品
步骤A
一类标准贮备溶液:转移1.0mL USP一类残留溶剂混合溶液标准品于100mL容量瓶中,加9mL二甲基亚砜,以水稀释至刻度,混匀。转移1.0 mL此溶液于100mL容量瓶中,以水稀释至刻度,混匀。转移1.0 mL该溶液于10mL容量瓶中,以水稀释至刻度,混匀。
一类标准溶液:转移1.0 mL一类标准贮备溶液于适合的顶空进样样品瓶中,加入5.0 mL 水,压上瓶塞和瓶盖,混匀。
二类标准贮备溶液:转移1.0mL USP二类残留溶剂混合溶液A标准品于100mL容量瓶中,以水稀释至刻度,混匀。此为二类标准贮备溶液A。转移1.0mL USP二类残留溶剂混合溶液B标准品于100mL容量瓶,以水稀释到刻度,混匀。此为二类标准贮备溶液B。
二类混合溶液A标准溶液:转移1.0 mL二类标准贮备溶液A于适当的顶空进样样品瓶中,加入5.0 mL水,压上瓶塞和瓶盖,混匀。
二类混合溶液B标准溶液:转移5.0 mL二类标准贮备溶液B于适当的顶空进样样品瓶中,加入1.0 mL水,压上瓶塞和瓶盖,混匀。
供试贮备溶液:精确称取250mg供试样品,转引至25 mL容量瓶中,溶于水并以水稀释至刻度,混匀。
供试溶液:转移5.0 mL供试贮备溶液于适当的顶空进样样品瓶中,加入1.0 mL水,压上瓶塞和瓶盖,混匀。
一类系统适应性溶液:转移1.0 mL一类标准贮备溶液于适当的顶空进样样品瓶中,加入5.0 mL供试贮备溶液,压上瓶塞和瓶盖,混匀。
色谱系统(见色谱<621>):该气相色谱仪装备一个火焰离子化(FID)检测器,一个0.32mm ×30m 毛细管柱(fused-silica column)内部涂以1.8-μm 厚的phaseG43涂层或者一个0.53mm ×30m 大孔径毛细管柱以3.0-μm 厚的phaseG43为涂层。载气为氮气或氦气,线速度为每秒35cm,分流比为1:5。柱温在40°C保持20分钟,再以每分钟10°C 的速度升至240°C,
在240°C保持20分钟。进样口和检测器温度分别保持在140°C和250°C。按照步骤项下规定,将一类标准溶液、一类系统适应性溶液、二类混合溶液A标准溶液进样,并记录出峰的响应值:在一类标准溶液中1,1,1-三氯甲烷的信噪比不小于5;在一类系统适应性溶液中每个峰的信噪比不小于3;在二类混合溶液A标准溶液中,乙腈和二氯甲烷的分离度分离度(R)不小于1.0。
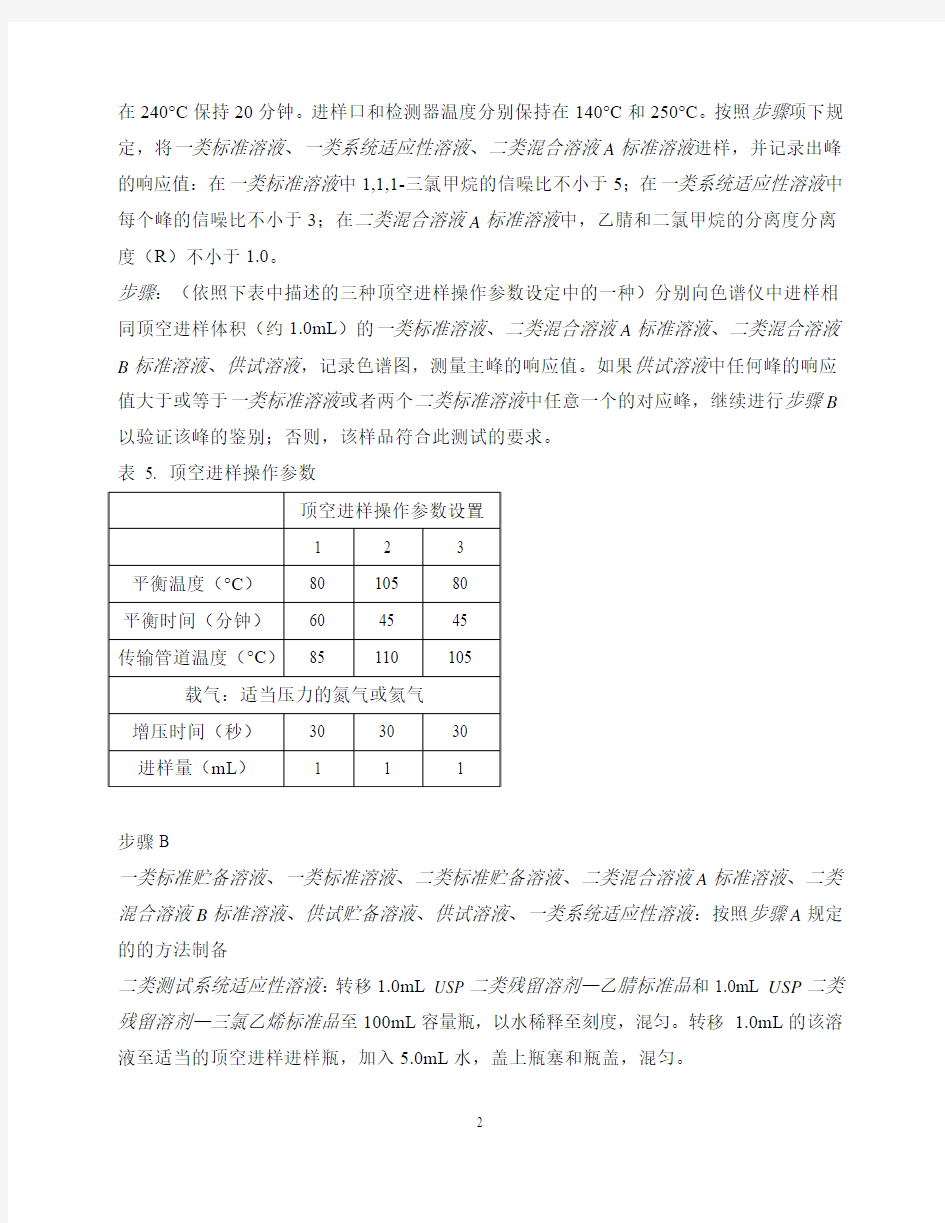
步骤:(依照下表中描述的三种顶空进样操作参数设定中的一种)分别向色谱仪中进样相同顶空进样体积(约1.0mL)的一类标准溶液、二类混合溶液A标准溶液、二类混合溶液B标准溶液、供试溶液,记录色谱图,测量主峰的响应值。如果供试溶液中任何峰的响应值大于或等于一类标准溶液或者两个二类标准溶液中任意一个的对应峰,继续进行步骤B 以验证该峰的鉴别;否则,该样品符合此测试的要求。
表 5. 顶空进样操作参数
步骤B
一类标准贮备溶液、一类标准溶液、二类标准贮备溶液、二类混合溶液A标准溶液、二类混合溶液B标准溶液、供试贮备溶液、供试溶液、一类系统适应性溶液:按照步骤A规定的的方法制备
二类测试系统适应性溶液:转移1.0mL USP二类残留溶剂—乙腈标准品和1.0mL USP二类残留溶剂—三氯乙烯标准品至100mL容量瓶,以水稀释至刻度,混匀。转移1.0mL的该溶液至适当的顶空进样进样瓶,加入5.0mL水,盖上瓶塞和瓶盖,混匀。
色谱系统(见色谱<621>):该气相色谱仪装备一个火焰离子化(FID)检测器,一个0.32mm ×30m 毛细管柱(fused-silica column)涂以0.25-μm 厚的phaseG16涂层或者一个0.53mm ×30m 大孔径毛细管柱涂以0.25-μm 厚的phaseG16涂层。载气为氮气或氦气,线速度35cm/秒,分流比为1:5。柱温在50°C下保持20分钟,然后以每分钟6°C的速率升至165,并在165°C保持20分钟。按照步骤项下规定,将一类标准溶液、一类系统适应性溶液、二类系统适应性溶液进样,并记录出峰的响应值:一类标准溶液中苯的信噪比不小于5;一类系统适应性溶液中每个峰的信噪比不小于3;二类系统适应性溶液中乙腈和三氯乙烯的分离度(R)不小于1.0。
步骤:(依照表5中描述的三种顶空进样操作参数设定中的一种)分别向色谱仪中进样相同顶空进样体积(约1.0mL)的一类标准溶液、二类混合溶液A标准溶液、二类混合溶液B标准溶液、供试溶液,记录色谱图,测量主峰的响应值。如果在步骤A中发现的超标峰的响应值在供试溶液中仍然大于或等于一类标准溶液或者两个二类标准溶液中任意一个的对应峰,继续进行步骤C,对该峰进行定量检测;否则,该样品符合此测试的要求。
步骤C-
一类标准贮备溶液、一类标准溶液、二类标准贮备溶液A、二类混合溶液A标准溶液、供试贮备溶液、供试溶液、一类系统适应性溶液:按照步骤A规定的的方法制备。
标准溶液:【注意:为通过步骤A和步骤B识别和确认的每一个峰单独配制一份标准溶液】根据通过步骤A和步骤B识别和证实的每一个残留溶剂峰,精确量取一定体积的每一种对应USP标准品,转移到适合的容器中,以水定量稀释,如果必要可以逐级稀释,以得到最终浓度为表1或表2(浓度限度项下)规定值1/20的溶液。转移1.0 mL该溶液至适当的顶空进样样品瓶中,加入5.0 mL水,压上瓶塞和瓶盖,混匀。
增敏供试溶液:【注意:为通过步骤A和步骤B识别和证实的每一个峰单独配制一份增敏供试溶液】转移5.0mL供试贮备溶液至适当的顶空进样进样瓶,加入1.0mL标准溶液,压上瓶塞和瓶盖,混匀。
色谱系统(见色谱<621>):【注意:如果发现A步骤中色谱仪的结果比B步骤中发现差,B步骤的色谱系统可以被取代】该气相色谱仪装备一个火焰离子化(FID)检测器,一个
0.32mm×30m 毛细管柱(fused-silica column)涂以1.8-μm 厚的phaseG43涂层或者一个0.53mm×30m 大孔径毛细管柱涂以3.0-μm 厚phaseG43涂层。载气为氮气或氦气,线速度35cm/S,分流比为1:5。柱温在40°C保持20min,然后以每分钟10°C的速度升至240°C,并在240°C保持20分钟。进样口和检测器温度分别保持在140°C和250°C。按照步骤项下规定,将一类标准溶液、一类系统适应性溶液、二类混合溶液A标准溶液进样,并记录出峰的响应值:在一类标准溶液中1,1,1-三氯甲烷的信噪比不小于5;在一类系统适应性溶液中每个峰的信噪比不小于3;在二类混合溶液A标准溶液中,乙腈和二氯甲烷的分离度分离度(R)不小于1.0。
步骤:(依照表5中描述的三种顶空进样操作参数设定中的一种)分别向色谱仪中进样相同顶空进样体积(约1.0mL)的标准溶液、供试溶液、增敏供试溶液,记录色谱图,测量主峰的响应值。按照下列公式,计算供试样品中发现的每一个残留溶剂的量(ppm):
5(C/W)[r U /(r ST–r U)]
其中,C为在标准溶液中的适当的USP标准品的浓度(ppm);W为用于配制供试贮备溶液的样品重量(g);r
r ST分别为供试溶液和增敏供试溶液中得到的残留溶剂色谱峰的响
U和
应值。
疏水样品
步骤A
一类标准贮备溶液、一类标准溶液、一类系统适应性溶液、二类标准贮备溶液A、二类标准品溶液B、二类混合溶液A标准溶液、二类混合溶液B标准溶液、色谱系统:按照水溶性样品项下的步骤A中的规定进行
二类标准贮备溶液C:转移1.0mL USP二类残留溶剂混合溶液C标准品至100mL容量瓶,以1,3-二甲基-2-眯唑啉酮(以下简称DMI)稀释至刻度,混匀。
二类混合溶液C标准溶液:【注意:此溶液是用于鉴别和定量供试品中的二甲基酰胺(以下简称DMF)和/或N,N-二甲基乙酰胺】转移1.0mL二类标准贮备溶液C至适当的顶空进样样品瓶,加入5.0mL的DMI,压上瓶塞和瓶盖,混匀。
供试贮备溶液:精密称取供试样品约250mg,转移至25 mL容量瓶中,以DMF溶解并稀释到刻度,混匀。
供试溶液1:转移5.0 mL供试贮备溶液至适当的顶空进样样品瓶中,加入DMF1.0 mL,压上瓶塞和瓶盖,混匀。
供试溶液2:【注意:此溶液是用于鉴别供试品中的DMF和/或N,N-二甲基乙酰胺】精密称取供试样品约250mg,转移至25 mL容量瓶中,以DMI溶解并稀释,定容制刻度,混匀。转移5.0 mL此溶液至适当的顶空进样样品瓶中,加入1.0 mL的DMI,压上瓶塞和瓶盖,混匀。
步骤:(依照表5中描述的三种顶空进样操作参数设定中的一种)分别向色谱仪中进样相同顶空进样体积(约1.0mL)的一类标准溶液、二类混合溶液A标准溶液、二类混合溶液B标准溶液、二类混合溶液C标准溶液、供试溶液1、供试溶液2(如果需要),记录色谱图,测量主峰的响应值。如果供试溶液1中任何峰的响应值大于或等于一类标准溶液或者三个二类标准溶液中任意一个的对应峰,继续进行步骤B以验证该峰的鉴别;否则,该样品符合此测试的要求。如果在供试溶液2中的DMF或者N,N-二甲基乙酰胺的峰响应值大于或等于二类混合溶液C标准溶液中对应峰,继续进行步骤B,以验证该峰的鉴别;否则,该样品符合此项检测的要求。
步骤B
一类标准贮备溶液、一类标准溶液、一类系统适应性溶液、二类标准贮备溶液A、二类标准贮备溶液B、二类混合溶液A标准溶液、二类混合溶液B标准溶液:按照水溶性样品项下步骤A中的规定制备
二类标准贮备溶液C、二类混合溶液C标准溶液、供试贮备溶液、供试溶液1、供试溶液2:按照步骤A的规定进行
二级系统适应性溶液和色谱系统:按照水溶性样品项下步骤B中规定的进行
步骤:(依照表5中描述的三种顶空进样操作参数设定中的一种)分别向色谱仪中进样相同顶空进样体积(约1.0mL)的一类标准溶液、二类混合溶液A标准溶液、二类混合溶液B标准溶液、二类混合溶液C标准溶液、供试溶液1、和/或者供试溶液2(如果需要),记录色谱图,测量主峰的响应值。如果在步骤A中发现的超标峰的响应值在供试溶液1中仍然大于或等于一类标准溶液或者三个二类标准溶液中任意一个的对应峰,继续进行步骤C以对该峰进行定量检测;否则,该样品符合此测试的要求。如果在供试溶液2中的DMF
或N,N-二甲基乙酰胺的峰响应值大于或等于在二类混合溶液C标准溶液中的对应峰,继续进行步骤C,对该峰进行定量检测;否则,该样品符合此项测试的要求。
步骤C-
一类标准贮备溶液、一类标准溶液、一类系统适应性溶液、二类标准贮备溶液A、二类混合溶液A标准溶液:按照水溶性样品项下步骤A中的规定进行
标准溶液1:【注意:为通过步骤A和步骤B识别和确认的每一个峰单独配制一份标准溶液】根据通过步骤A和步骤B识别和证实的每一个残留溶剂峰,精确量取一定体积的每一种对应USP标准品,转移到适合的容器中,以DMF定量稀释,如果必要可以逐级稀释,以得到最终浓度为表1或表2(浓度限度项下)规定值1/20的溶液。转移1.0 mL该溶液至适当的顶空进样样品瓶中,加入5.0 mL的DMF,压上瓶塞和瓶盖,混匀。
标准溶液2:【注意:此溶液是用于对供试品中的DMF和/或N,N-二甲基乙酰胺进行定量检测】精确量取一定体积的USP二类残留溶剂—N,N-二甲基酰胺标准品和/或者一定体积的USP二类残留溶剂—N,N-二甲基乙酰胺标准品至适当的容器,以DMI进行定量稀释,如有必要可以逐级操作,以得到最终浓度为表2(浓度限度项下)数值的1/20的溶液。转移1.0mL 该溶液至适当的顶空进样进样瓶,加入5.0mL的DMI,压上瓶塞和瓶盖,混匀。
供试贮备溶液、供试溶液1、供试溶液2:按照步骤A中规定的进行
增敏供试溶液1:【注意:为通过步骤A和步骤B识别和确认的每一个峰单独配制一份增敏供试溶液】转移5.0mL供试贮备溶液至适当的顶空进样进样瓶中,加入1.0mL标准溶液1,压上瓶塞和瓶盖,混合。
增敏供试溶液2:【注意:为通过步骤A和步骤B识别和确认的每一个峰单独配制一份增敏供试溶液】转移5.0mL供试溶液2至适当的顶空进样进样瓶中,加入1.0mL标准溶液2,压上瓶塞和瓶盖,混合。
色谱系统:依据水溶性样品项下的步骤C进行
步骤:(依照表5中描述的三种顶空进样操作参数设定中的一种)分别向色谱仪中进样相同顶空进样体积(约1.0mL)的标准溶液、供试溶液1和/或供试溶液2、增敏供试溶液1和/或增敏供试溶液2,记录色谱图,测量主峰的响应值。按照下列公式,计算供试样品中发现的每一个残留溶剂的量(ppm):
5(C/W)[r U /(r ST –r U)]
其中,C为在标准溶液中的适当的USP标准品的浓度(ppm);W为用于配制供试贮备溶液的样品重量(g);r
r ST分别为供试溶液1或供试溶液2和增敏供试溶液1或增敏供试U和
溶液2中得到的残留溶剂色谱峰的响应值。
3类残留溶剂
如果只存在3类残留溶剂,依照干燥失重<731>项的规定测定残留溶剂的水平。如果干燥失重值大于0.5%,应当按照水分测定<921>项的规定进行水分测定。除非在专著中明确指定其他方法,水分测定应当按照方法1a进行。如果专著中3类溶剂的限度大于每天50mg (相当于选项1下的5000ppm或0.5%),那么该残留溶剂必须要鉴别和定量,并且上述步骤,以及对于标准溶液的适当修正,应当被应用于所有可能的地方。否则,须采用经过适当认证的程序。这样的程序将被递交USP进行评估。只要是USP提供的标准品,就应当在这些程序中使用USP标准品。关于残留溶剂限度检测的应用的流程图,请见图1所示。
图1:与残留溶剂鉴别和实施限度检测有关流程图
残留溶剂测定法
残留溶剂测定法 1 简述 药品中的残留溶剂系指在原料药或辅料的生产中,以及在制剂制备过程中使用过,但在工艺过程中未能完全去除的有机溶剂。药物中常见的残留溶剂及限度参照《中国药典》2015年版四部通则0861附表1的规定,除另有规定外,第一、第二、第三类溶剂的残留量应符合其规定;对其他溶剂,应根据生产工艺的特点,制订相应的限度,使其符合产品质量标准的要求。本法照气相色谱法(《中国药典》2015年版四部通则0521测定。 本测定方法适用于对各论项下未收载残留溶剂检测方法的品种中残留溶剂的检验,也可用于指导建立各论项下具体品种的残留溶剂检查方法。 2 仪器和用具 2.1 气相色谱仪,带FID检测器,顶空进样器。 2.2 计算机,安装工作站软件。 2.3 色谱柱 2.3.1 毛细管柱除另有规定外,极性相近的同类色谱柱之间可以互代使用。2.3.1.1 非极性色谱柱固定液为100%的二甲基聚硅氧烷的毛细管柱。 2.3.1.2 极性色谱柱固定液为聚乙二醇(PEG-20M)的毛细管柱。 2.3.1.3 中极性色谱柱固定液为(35%)二苯基-(65%)二甲基聚硅氧烷,(50%)二苯基-(50%)二甲基聚硅氧烷,(35%)二苯基-(65%)二甲基亚芳基聚硅氧烷,(14%)氰丙基苯基-(86%)二甲基聚硅氧烷,(6%)氰丙基苯基-(94%)二甲基聚硅氧烷的毛细管柱。 2.3.1.4 弱极性色谱柱固定液为(5%)苯基-(95%)甲基聚硅氧烷,(5%)二苯基-(95%)二甲基亚芳基硅氧烷共聚物的毛细管柱。 2.3.2 填充柱以直径为0.18~0.25mm的二乙烯苯-乙基乙烯苯型高分子多孔小球或其他适宜的填料作为固定相。 3 供试品溶液和对照品溶液的制备 3.1 供试品溶液的制备 3.1.1 顶空进样除另有规定外,精密称取供试品0.1~1g;通常以水为溶剂;对于非水溶性药物,可采用N,N-二甲基甲酰胺、二甲基亚砜或其他适宜溶剂;
方案批准 注:在方案批准部分签字表明签字者同意方案中规定的检测项目检测方法和记录要求。在执行本方案的过程中可能会出现影响严格执行本方案的偏差,对较小的偏差将通过偏差报告的形式来解决,对于关键性偏差,如对方法的调整、对参数或接受标准的调整必须制定出增补方案并按照原方案批准程序得到批准才能进行。所有的偏差报告和增补方案必须在提交验证报告供批准时一同提交。
目录 1.概述 (3) 2.参考资料 (4) 3. 职责 (4) 4. 色谱系统及色谱条件 (4) 5. 器材与试剂 (5) 6. 验证试验 (5) 6.1系统适应性 (5) 6.2专属性 (6) 6.3耐用性 (7) 6.4定量限 (8) 6.5检测限 (8) 6.6线性与围 (8) 6.7准确度 (9) 6.8精密度 (11) 7.再验证周期 (12) 8.偏差及纠正措施 (13) 9.最终审核和批准 (13) 药品残留溶剂顶空分析方法草案 (14)
1.概述 1.1根据ICH对药品中残留溶剂含量的要求及盐酸噻氯匹定生产工艺,必须控制盐酸噻氯匹定生产工艺中使用到的溶剂乙醇、丁酮、甲苯、N,N-二甲基甲酰胺(DMF)的残留量。限度分别为:乙醇≤5000ppm、丁酮≤5000ppm、甲苯≤890ppm、DMF≤880ppm。 1.2分析方法草案见附件。 1.3本分析方法属于杂质定量分析,因此需要验证的项目有:系统适应性、专属性、线性、 准确度、检测限、定量限、精密度、耐用性,具体参数及接受标准要求见下表:
2.参考资料 ICH Q3C (R3), November 2005. ICH Q2 (R1), November 2005. <467> Residual Solvents, United States Pharmacopoeia 31, November 2007. <20424> Residual Solvents, European Pharmacopoeia 6.0, June 2007. 3. 职责 4.1色谱系统
1. 目的:建立残留溶剂测定法(二部)检验标准操作规程,并按规程进行检验,保证检验操作规范化。 2. 依据: 2.1. 《中华人民共和国药典》2010年版二部。 3.范围:适用于所有用残留溶剂测定法(二部)测定的供试品。 4. 责任:检验员、质量控制科主任、质量管理部经理对本规程负责。 5.正文: 5.1. 药品中的残留溶剂系指在原料药或辅料的生产中,以及在制剂制备过程中使用的,但在工艺过程中未能完全去除的有机溶剂。药品中常见的残留溶剂及限度见附表1,除另有规定外,第一、第二、第三类溶剂的残留限度应符合表1中的规定;对其他溶剂,应根据生产工艺的特点,制定相应的限度,使其符合产品规范、药品生产质量管理规范(GMP)或其他基本的质量要求。 5.2. 本法照气相色谱法(附录ⅤE)测定。 5.3. 色谱柱 5.3.1. 毛细管柱:除另有规定外,极性相近的同类色谱柱之间可以互换使用。 5.3.1.1. 非极性色谱柱:固定液为100%的二甲基聚硅氧烷的毛细管柱。 5.3.1.2. 极性色谱柱:固定液为聚乙二醇(PEG-20M)的毛细管柱。
5.3.1.3. 中性色谱柱:固定液为(35%)二苯基-(65%)甲基聚硅氧烷、(50%)二苯基-(50%)二甲基聚硅氧烷、(35%)二苯基-(65%)二甲基聚硅氧烷、(14%)氰丙基苯基-(86%)二甲基聚硅氧烷、(6%)氰丙基苯基-(94%)二甲基聚硅氧烷的毛细管柱等。 5.3.1.4. 弱极性色谱:柱固定液为(5%)苯基-(95%)甲基聚硅氧烷、(5%)二苯基-(95%)二甲基硅氧烷的毛细管柱等。 5.3.2. 填充柱:以直径为0.18~0.25mm 的二乙烯苯-乙基乙烯苯型高分子多孔小球或其他适宜的填料作为固定相。 5.4. 系统适用性试验。 5.4.1. 用待测物的色谱峰计算,毛细管色谱柱的理论板数一般不低于5000;填充柱的理论板数一般不低于1000。 5.4.2. 色谱图中,待测物色谱峰与其相邻色谱峰的分离度应大于1.5。 5.4.3. 以内标法测定时,对照品溶液连续进样5次,所得待测物与内标物峰面积之比的相对标准偏差(RSD)不大于5%;若以外标法测定,所得待测物峰面积的RSD 应不大于10%。 5.5. 供试品溶液的制备。 5.5.1. 顶空进样:除另有规定外,精密称取供试品0.1~1g,通常以水为溶剂;对于非水溶性药物,可采用N,N-二甲基甲酰胺、二甲基亚砜或其他适宜溶剂;根据供试品
原料药或制剂中有机溶剂的残留量一般要求控制在几个至几千个ppm之间,属于微量或痕量测定,与常量测定有着不同的特点。残留溶剂检查方法的选择对测定结果有着重要的影响,有时采用不同的方法测定同一个样品会得到截然不同的结果。 通过对最近一段申报资料的审评,经常发现在残留溶剂的检查方法尚不合理的情况下,若样品的色谱图中未出现溶剂峰,也未经其它系统验证,研究者就简单地作出样品无该溶剂残留的结论,进而不将其残留定入质量标准,药检所也不再进行复核。针对这种情况,从审评的角度出发,就如何评价残留溶剂检查方法的合理性谈自己的一些认识,与各位业内同仁交流。 有机残留溶剂检查一般采用气相色谱法,评价色谱系统的适用性和方法学验证资料遵循与液相色谱方法评价相同的原则,不再赘述。与液相方法不同的是,气相色谱有多种进样方式,残留溶剂检查常用直接进样法或顶空进样法。针对这两种进样方法不同的特点,评价方法合理性的要点应有所不同。对于直接进样法,应着重评价方法的灵敏度和重复性。目前已普遍用毛细管柱取代填充柱,因为毛细管柱的柱效高,其灵敏度也较填充柱大为提高。但由于毛细管柱直接进样的体积小,一般仅几微升,即使提高供试溶液的浓度,对于测定限量极低的溶剂(如:苯、四氯化碳、1,2-二氯乙烷等)及对FID检测器响应低的溶剂(如:含氯的溶剂),其检测限一般接近或高于限量,灵敏度难以满足测定的需要。测定此类溶剂最好采用顶空进样法,对含卤素的溶剂可改用电子捕获检测器(ECD)。进样量小也易造成进样重复性差,采用内标法较外标法的结果更为准确。 顶空进样法是将大量样品中的残留溶剂富集在顶空瓶上层的气体中,对绝大多数有机溶剂而言,灵敏度较直接进样法大为提高,但顶空进样系统中存在气液两相的平衡问题,对结果准确性的影响因素增多。评价方法是否合理,应着重关注以下三个方面:1)顶空条件:顶空瓶的平衡温度和时间是最重要的参数,根据溶解样品的溶剂和待测溶剂的不同性质,达到气液平衡所需的温度和时间可能不同,应有试验数据作为选择的依据,但在申报资料中一般都未提及。判断顶空条件是否适用,一般的规律是:顶空瓶的平衡温度应低于溶解样品所用溶剂的沸点10℃以下,能满足检测灵敏度即可;对于沸点过高的溶剂,如DMF、DMSO、聚乙二醇等,用顶空进样测定的灵敏度不如直接进样,不适宜采用顶空法;顶空瓶的平衡时间一般应为30至60分钟,才能保证气液两相达到稳态平衡。 2)供试品和对照品是否平行:由于供试品和对照品的液体部分状态不完全一致而造成的基质效应会直接影响到结果的准确性。采用标准加入法可以消除基质效应,但目前在国内的申报资料中较少见到,其原因可能是方法较为繁琐,且需要消耗大量的样品,对新药研发初期样品量较少的情况或一些贵重的药品不太适用。如果申报资料中提供了回收率数据,就容易判断基质效应的大小,但由于目前对此没有强制要求,大多数资料都未对回收率进行研究。因此在评价方法时,至少应要求对照品和供试品采用相同的溶剂溶解,且液体部分的体积应完全一致。 3)重复性:由于顶空进样法存在气液平衡和气体进样的问题,粗放度较大,中国药典2005年版的要求是:内标法连续五次进样的相对标准偏差小于5%,外标法的相对标准偏差小于10%;欧洲药典则要求相对标准偏差小于15%,因此重复性应密切关注。 此外,无论是直接进样或顶空进样,都应尽量使供试液中的样品完全溶解,否则当残留溶剂被包裹在样品晶格中时就不能被检测出来,可能造成结果与实际情况完全不符。对溶解性差的样品,可采用不挥发性酸或碱的溶液、高沸点的有机溶剂、混合溶剂等来溶解样品,即使样品在加热的条件下可能被破坏,只要待测的残留溶剂不被破坏(如:测定酯类溶剂不
***产品残留溶剂分析方法验证方案 20**年**月
验证方案的起草与审批 方案实施日期:
目录 1.验证目的 (4) 2.方法简介与确认范围 (4) 3.标准品、供试品 (4) 4.风险评估 (4) 5.验证的可接受标准 (5) 6.验证步骤 (6) 6.1系统适应性 (6) 6.2专属性 (6) 6.3定量限与检测限 (7) 6.4线性 (7) 6.5准确度 (8) 6.6精密度 (9) 6.7范围 (9) 6.8耐用性 (9) 6.9样品测定 (10) 7.偏差 (10) 8.风险的接收与评审 (10) 9.再验证 (10) 10.确认结果评审和结论 (10) 11.更改历史 (10) 12. 附录 (10)
1.验证目的 根据法规的要求,分析方法应进行验证,证明采用的方法适合于相应的检测要求。 这个验证方案的目的是为验证提供具体方法参数、可接受标准和研究步骤。 2.方法简介与确认范围 ***产品生产过程中用到有机溶剂乙醇、丙酮、二氯甲烷、乙酸乙酯、四氢呋喃,为了准确测定溶剂在成品中的残留量,现对该测定方法进行验证,验证包括方法的专属性、检测限与定量限、线性、范围、准确度、精密度及耐用性。 3.标准品、供试品 3.1标准品 3.2供试品 4.风险评估 按照《质量风险管理规程》,质量控制部和质量管理部共同对分析方法进行了风险评
风险评估人: 评估日期: 5.验证的可接受标准
6.验证步骤 6.1系统适应性 精密称取乙醇200mg、丙酮200mg、二氯甲烷24mg、乙酸乙酯200mg、四氢呋喃28.8mg,置于已加入10ml二甲基亚砜的50ml量瓶中,用二甲基亚砜溶解并稀释至刻度,作为对照溶液储备液;精密移取对照液储备液5ml,置于一100ml量瓶中,用二甲基亚砜稀释至刻度;精密移取5ml,置于20ml顶空瓶中,密封。同法配制6份,连续顶空进样,记录色谱图,相邻组份之间的分离度R均应不小于1.5,各组份峰面积的相对标准偏差(RSD A)均应不大于10%。 6.2专属性 a) 空白:精密移取二甲基亚砜5ml,置于20ml顶空瓶中,密封。顶空进样,记录色谱图,确定二甲基亚砜出峰位置,且溶剂对测定应无干扰。 b) 乙醇定位溶液:取乙醇约10mg,置于50ml量瓶中,用二甲基亚砜溶解并稀释至刻度(含乙醇约200μg/ml);精密移取5ml,置于20ml顶空瓶中,密封。顶空进样,记录色谱图,确定乙醇出峰位置,并不得有干扰峰,且与二甲基亚砜之间的分离度应不小于1.5。 c) 丙酮定位溶液:取丙酮约10mg,置于50ml量瓶中,用二甲基亚砜溶解并稀释至刻度(含丙酮约200μg/ml);精密移取5ml,置于20ml顶空瓶中,密封。顶空进样,记录色谱图,确定丙酮出峰位置,并不得有干扰峰,且与二甲基亚砜之间的分离度应不小于1.5。 d) 二氯甲烷定位溶液:取二氯甲烷约12mg,置于50ml量瓶中,用二甲基亚砜溶解并稀释至刻度,移取1.0ml,置于10ml量瓶中,用二甲基亚砜稀释至刻度(含二氯甲烷约24μg /ml);精密移取5ml,置于20ml顶空瓶中,密封。顶空进样,记录色谱图,确定二氯甲烷出峰位置,并不得有干扰峰,且与二甲基亚砜之间的分离度应不小于1.5。 e) 乙酸乙酯定位溶液:取乙酸乙酯约10mg,置于50ml量瓶中,加二甲基亚砜溶解并稀释至刻度(含乙酸乙酯约200μg/ml);精密移取5ml,置于20ml顶空瓶中,密封。顶空进样,记录色谱图,确定乙酸乙酯出峰位置,并不得有干扰峰,且与二甲基亚砜之间的分离度应不小于1.5。 f) 四氢呋喃定位溶液:取四氢呋喃约14.4mg,置于50ml量瓶中,用二甲基亚砜溶解并稀释至刻度,移取1.0ml,置于10ml量瓶中,用二甲基亚砜稀释至刻度(含四氢呋喃约28.8μg/ml);精密移取5ml,置于20ml顶空瓶中,密封。顶空进样,记录色谱图,确定四氢呋喃出峰位置,并不得有干扰峰,且与二甲基亚砜之间的分离度应不小于1.5。 i) 对照液:精密称取乙醇200mg、丙酮200mg、二氯甲烷24mg、四氢呋喃28.8mg、
USP[467]有机挥发性杂质 残留溶剂限度 Ⅰ级需避免的溶剂 已知人体实验致癌物质; 强烈疑似人体实验致癌物质;环境危害物质 Ⅱ级需被限制的溶剂 动物实验非生殖毒性(遗传);动物实验致癌物质或其它可能的非可逆致病因子;产生毒性如神经毒性或致畸性; 其它疑似重大但可逆毒性 Ⅲ级具有低潜在毒性溶剂 对人体存在低毒性溶剂;无人体摄入量规定(注:Ⅲ级残留溶剂PDEs上限为≥50mg/天) 表1 Ⅰ级残留溶剂 溶剂浓度限度(ppm)不良反应 苯 2 致癌物四氯化碳 4 中毒和外周脑组织损害1,2-二氯乙烷 5 中毒 1,1-二氯乙烷8 中毒 1,1,1-三氯乙烷1500 环境危害 Ⅱ级残留溶剂 表2 Ⅱ级残留溶剂 溶剂PDE(mg/天)浓度限度(ppm) 乙腈Acetonitrile 4.1 410 氯苯Chlorobenzene 3.6 360 氯仿Chloroform0.6 60 环己烷Cyclohexane38.8 3880 1,2-二氯乙烯 1,2-Dichloroethene 18.7 1870 1,2-二甲氧乙烷 1.0 100 N,N-二甲基乙酰胺 N,N-Dimethylacetamide 10.9 1090 N,N-二甲基甲酰胺 N,N-Dimethylformamide 8.8 880 1,4-二氧六环 1,4-Dioxane 3.8 380 2-乙氧基乙醇 1.6 160 乙二醇Ethylene glycol 6.2 620 甲酰胺Formamide 2.2 220
己烷Hexane 2.9 290 甲醇Methanol30.0 3000 2-甲氧基乙醇0.5 50 甲基丁基(甲)酮 0.5 50 2-己酮 甲基环己烷11.8 1180 二氯甲烷 6.0 600 N-甲基吡咯烷酮 5.3 530 硝酸甲烷0.5 50 吡啶 2.0 200 环丁砜 1.6 160 四氢呋喃7.2 720 1,2,3,4-四氢化萘 1.0 100 甲苯8.9 890 三氯乙烯0.8 80 二甲苯* 21.7 2170 *通常含有60%间-二甲苯,14%对-二甲苯,9%邻-二甲苯和17%乙苯 Ⅲ级残留溶剂 表3 Ⅲ级残留溶剂 (GMP或其它原料药、赋形剂和药物制剂质量标准规定) 醋酸Acetic acid 庚烷Heptane 丙酮Acetone 乙酸异丁酯 苯甲醚乙酸异丙酯 正丁醇乙酸甲酯 2-丁醇3-甲基-1-丁醇 乙酸丁酯甲基乙基酮 叔丁基甲基醚甲基异丁基酮 异丙基苯2-甲基-1-丙醇 二甲基亚砜戊烷 乙醇1-戊醇 乙酸乙酯1-丙醇 乙醚2-丙醇 甲酸乙酯乙酸丙酯 甲酸 表4 其它残留溶剂 (缺乏足够的毒物学方面的资料) 1,1-二乙氧基丙烷甲基异丙基酮 1,1-二甲氧基甲烷甲基四氢呋喃 2,2-二甲氧基丙烷溶剂己烷 异辛烷三氯醋酸 异丙醚三氟醋酸除另有规定外,样品中残留的有机挥发性杂质不得超过下表中规定限度:
残留溶剂的指导原则 1.介绍 本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理学上的可接受水平。 药物中的残留溶剂在此定义为在原料药或赋形剂的生产中,以及在制剂制备过程中产生或使用的有机挥发性化合物,它们在工艺中不能完全除尽。在合成原料药中选择适当的溶剂可提高产量或决定药物的性质,如结晶型。纯度和溶解度。因此.有时溶剂是合成中非常关键的因素。本指导原则所指的溶剂不是谨慎地用作赋形剂的溶剂,也不是溶剂化物,然而在这些制剂中的溶剂含量也应进行测定,并作出合理的判断。 出于残留溶剂没有疗效,故所有残留溶剂均应尽可能.去,以符合产品规范、GMP或其他基本的质量要求。制剂所含残留溶剂的水平不能高于安全值,已知一些溶剂可导致不接受的毒性(第一类,表1),除非被证明特别合理,在原药、赋形剂及制剂生产中应避免使用。一些溶剂毒性不太大(第二类,表2)应限制使用,以防止病人潜在的不良反应。使用低毒溶剂(第三类,表3)较为理想。附录1中列出了指导原则中的全部溶剂。 表中所列溶剂并非详尽无遗,其他可能使用的溶剂有待日后补充列人。第一、二类溶剂的建议限度或溶剂的分类会随着。新的安全性资料的获得而调整。含有新溶剂的新药制剂、其上市申请的安全性资料应符合本指导原则或原料药指导原则(Q3A新原料药中的杂质)或新药制剂(Q3B新药制剂中的杂质)中所述的杂质控制原则,或者符合上述三者。 2. 指导原则的范围 指导原则范围包括原料药、赋形剂或制剂中所含残留溶剂.因此,当生产或纯化过程中会出现这些溶剂时。应进行残留溶剂的检验。也只有在上述情况下,才有必要作溶剂的检查。虽然生产商可以选择性地测定制剂,但也可以从制剂中各成分的残留溶液水平来累积计算制剂中的残留溶剂。如果计算结果等于或低于本原则的建议水平,该制剂可考虑不检查残留溶剂,但如果计算结果高于建议水平则应进行检测,以确定制剂制备过程中是否降低了有关溶剂的量以达到可接受水平。果制剂生产中用到某种溶剂,也应进行测定。 本指导原则不适用于临床研究阶段的准新原料药、准赋形剂和准制剂。也不适用于已上市的药品。 本指导原则适用于所有剂型和给药途径。短期(如30天或更短)使用或局部使用时,允许存在的残留溶剂水平可以较高。应根据不同的情况评判这些溶剂水平。 有关残留溶剂的背景附加说明见附录2。 3.通则 3.1 根据危害程度对残留溶剂分类 “可耐受的日摄人量”(TDI)是国际化学品安全纲要(IPCS)用于描述毒性化合物接触限度的术语。“可接受的日摄人量”(ADI)是WHO及一些国家和国际卫生组织所用的术语。新术语“允许的日接触量”(PDE)是本指导原则中用于定义药物中可接受的有机溶剂摄人量,以避免与同一物质的ADI混淆。 本原则中残留溶剂的评价以通用名和结构列于附录1,根据它们对人体可能造成的危害分为以下三类; (1)第一类溶剂:应避兔的溶剂 为人体致癌物、疑为人体致癌物或环境危害物。 (2)第二类溶剂。应限制的溶剂 非遗传毒性动物致癌或可能导致其他不可逆毒性测神经毒性或致畸性)的试剂。 可能具其他严重的但可逆毒性的溶剂。
残留溶剂测定法 药品中的残留溶剂系指在原料药或辅料的生产中,以及在制剂制备过程中使用的,但在工艺过程中未能完全去除的有机溶剂。药品中常见的残留溶剂及限度见附表1,除另有规定外,第一,第二,第三类溶剂的残留限度应符合附表1中的规定;对其他溶剂,应根据生产工艺的特点,制定相应的限度,使其符合产品规范,药品生产质量管理规范(GMP)或其他基本的质量要求。 本法照气相色谱法(通则0521)测定。 色谱柱 1,毛细管柱 除另有规定外,极性相近的同类色谱柱之间可以互换使用。 (1)非极性色谱柱固定液为100%的二甲基聚硅氧烷的毛细管柱。 (2)极性色谱柱固定液为聚乙二醇(PEG-20M)的毛细管柱。 (3)中极性色谱柱固定液为(35%)二苯基-(65%)甲基聚硅氧烷、(50%)二苯基-(50%)二甲基聚硅氧烷、(35%)二苯基-(65%)二甲基聚硅氧烷、(14%)氰丙基苯基-(86%)二甲基聚硅氧烷、(6%)氰丙基苯基-(94%)二甲基聚硅氧烷的毛细管柱等。 (4)弱极性色谱柱固定液为(5%)苯基-(95%)甲基聚硅氧烷、(5%)二苯基-(95%)二甲基聚硅氧烷共聚物的毛细管柱等。 2,填充柱 以直径为0.18~0.25mm的二乙烯苯-乙基乙烯苯型高分子多孔小球或其他适宜的填料作为固定相。 系统适用性试验 (1)用待测物的色谱峰计算,毛细管色谱柱的理论板数一般不低于5000,;填充柱的理论板数一般不低于1000。 (2)色谱图中,待测物色谱峰与其相邻色谱峰的分离度应大于1.5。 (3)以内标法测定时,对作品溶液连续进样5次,所得待测物与内标物峰面积之比的相对标准偏差(RSD)应不大于5%;若以外标法测定,所得待测物峰面积的RSD应不大于10%。 供试品溶液的制备 1,顶空进样 除另有规定外,精密称取供试品0.1~1g;通常以水为溶剂;对于非水溶性药物,可采用N,N-二甲基甲酰胺,二甲基亚砜或其他适宜溶剂;根据供试品和待测溶剂的溶解度,选择适宜的溶剂且应不干扰待测溶剂的测定。根据各品种项下残留溶剂的限度规定配制供试品溶液,其浓度应满足系统定量测定的需要。 2,溶液直接进样 精密称取供试品适量,用水或合适的有机溶剂使溶解;根据各品种项下残留溶剂的限度规定配制供试品溶液,其浓度应满足系统定量测定的需要。 对照品溶液的制备 精密称取各品种项下规定检查的有机溶剂适量,采用与制备供试品溶液相同的方法和溶剂制备对照品溶液;如用水作溶剂,应先将待测有机溶剂溶解在50%二甲基亚砜或N,N-二甲基甲酰胺溶液中,再用水逐步稀释。若为限度检查,根据残留溶剂的限度规定确定对照品溶液的浓度;若为定量测定,为保证定量结果的准确性,应根据供试品中残留溶剂的实际残留量确定对照品溶液的浓度;通常对
四、附录 16
1、残留溶剂表示方法 1.1允许日接触量 允许日接触量(permitted daily exposure, PDE)是指某一有机溶剂被允许摄入而不产生毒性的日平均最大剂量,单位为mg/天。某一具体有机溶剂的PDE值是由不产生反应量、体重调整系数、种属之间差异的系数、个体差异、短期接触急性毒性研究的可变系数等推算出的。部分有机溶剂的PDE 值见附录。由于国内目前尚未对此有系统的研究,附录中所列出的数据均是参考ICH残留溶剂研究指导原则中的数据。 1.2浓度限度 在PDE 表示方法的基础上,为了更加便于计算,引入了浓度限度(%)表示方法,其计算公式为浓度限度(%)=PDE(mg/天)9 /(1000×剂量(g/天))×100%,其中剂量初步定为10g/天。部分有机溶剂的浓度限度见附录。 1.3两种表示方法的比较 以上两种表示方法在残留溶剂研究中均可行,但需要指出的是,PDE值是绝对值,也就是说无论原料药、辅料和制剂,只要能明确各成分的溶剂残留量,以PDE值来计算是很精确的;而对于某一具体制剂来说,由于很难确定处方中各活性成分和各辅料的残留溶剂水平,因此以浓度限度来计算更为简便,只要日摄入总量不超过10g,就无需进一步计算。综合以上情况并考虑目前国内的实际情况,由于大多数药物的日摄入量不会超过10g(包括活性成分和辅料),浓度
限度表示方式是目前更为简便可行的。当然,在某些原料、辅料或制剂的残留溶剂不符合浓度限度时,可根据实际测定的各种残留溶剂量及用法用量计算实际日接触量,并与PDE值比较,如符合限量要求则也属可行。 此外,虽然本指导原则采用浓度限度的表示方式,但由于PDE 值的精确性,药物研发者可采用适当的PDE 值的方式进行残留溶剂研究。
医药中常用有机溶剂分类及残留限度 医药中常用有机溶剂分类及残留限度 药品的残留溶剂无治疗作用并可能对人体的健康和环境造成危害,本文对国际协调大会(ICH)制订的指导原则及各国执行情况作了较为详尽的介绍。 药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。药品还可被来自包装、运输、仓储中的有机溶剂污染。药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。 各国药监部门曾使用不同的药品残留溶剂指导原则,为此国际组织展开了协调工作。经相关程序讨论和审查后,国际协调大会的指导原则于1997年7月17日获得通过,被推荐至国际协调大会(ICH)的指导委员会采用。该指导原则要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进行检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。如果累加量低于或等于指导原则中的推荐量,则该药品无需进行残留溶剂检测;如果累加量高于推荐量,则必须对该药品进行残留溶剂检测。该指导原则适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。在某些情况如短期(小于30天)或局部应用下,视具体情况,溶剂的高残留量也可接受。 按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估): 第一类溶剂 是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。在可能的情况下,应避免使用这类溶剂。如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如: 苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。
残留溶剂方法学验证方案
阿莫西林残留溶剂分析方法验证方案文件编号: VP-01-06-00-041 方案起草 方案审核 方案批准
生效日期:年月日 1、概述 在阿莫西林制备工艺中,使用了甲醇与丙酮两种对人体具有危害的二类溶剂,我公司为了确保阿莫西林原料中甲醇与丙酮的残留在国家要求范围内,开发了阿莫西林胶囊中甲醇与丙酮残留的检测方法,按照《中国药典》2015年版的要求对此检测方法进行方法学验证。 2、验证目的 证明本方法能满足阿莫西林原料中甲醇与丙酮的残留溶剂测定,确保阿莫西林原料中甲醇与丙酮的残留溶剂检测方法准确、重现并耐用,检测结果数据真实可靠。 3、验证范围 本验证方案适用于阿莫西林中甲醇与丙酮的残留溶剂检验方法验证。 4、确认小组成员及职责
5、验证前的风险评估 5.1 验证小组人员按照《质量风险管理规程》,对分析方法进行了风险评估,确定了需进行方法确认的项目。 5.1.1严重性(S ): 危害可能产生后果的程度。严重程度分为五个等级。 5.1.2可能性(P ) 影响检测结果的事件发生的可能性频率或概率,建立以下五个等级:
5.1.3可检测性(D)检测到异常情况存在的能力的程度,定义如下: 5.2风险优先数量等级判定(RPN) 5.2.1风险等级判定标准的确定 RPN是事件发生的严重程度、可能性和可探测性三者乘积,用来衡量可能的仪器缺陷,以便采取可能的预防措施。 RPN = Severity(严重程度)×Possibility(发生的可能性)×Detection(可探测性) 5.2.2风险评价和处理
注:当RPN≤8,但严重性S为5时,仍需按中等以上风险进行后续控制。
本方法用石英毛细色谱柱GC检测,氮气为载气,FID检测器,依据GB/T 5009.37—1996方法并作改动、调整,使准确度提高,检出值可到1ppm,且重复性好,能为食用油产品准确定级提供有效、准确的结果。 关键词:GC 食用油溶剂残留检测 为了提高产品的检测、控制能力,配合国家对五种食用油新标准的制定和落实,更准确的得出食用油脂产品中溶剂残留结果,正确分级。 实验 试剂:N-N-二甲基乙酰胺(DMA)上海精化科技研究所 六号溶剂油宜春石油公司 设备:瓦里安GC 3900 检测器:FID 色谱柱:VB-5(30m×0.32×0.5um) 色谱条件: 进样器温度:250℃ 流速:2.5ml/min 检测器温度:280℃ 柱温箱温度:85℃(1min)20℃/min 130℃(2min) 2、试剂: N-N-二甲基乙酰胺(DMA):吸取1毫升放入100毫升洗好干燥的带胶塞的玻璃瓶中,在50摄氏度放置30分钟,取液上气1ul注入气相色谱仪在10分钟内无干扰即可使用。如有干扰用超声波处理30分钟。 3、六号溶剂标准溶液:称取洗净干燥的10毫升气化瓶的质量为A,瓶中放入比气化瓶体积少0.5毫升的DMA密塞后称量为B(M5),用50ul的注射器取约0.2毫升六号溶剂标准通过塞注入瓶中,混匀,准确称量为C。用下式计算六号溶剂的浓度:X7=(M5-M6)/(M6-M7)/0.935×1000 4、标准曲线的绘制 取预先在气相色谱仪测试无溶剂的成品油(新机榨毛油),分别称取25克放入以试过漏的6只气化瓶中,密塞。通过塞子注入六号溶剂标准液0、20ul 、40ul 、60ul、80ul、100ul。放入50摄氏度烘箱中,平衡30分钟,分别取液上气体注入色谱,各响应扣除空白后,绘制标准曲线。 5、测定 称取25克食用油样,密塞后于50摄氏度恒温烘箱加热30分钟,取出后立即用微量注射器吸取15ul液上气体注入色谱,记录组分测量峰高,与标准曲线比较,求出液上六号溶剂的含量。 6、计算 六号溶剂含量=测定气化瓶六号溶剂的质量/样品质量
Annexes to: CPMP/ICH/283/95 Impurities: Guideline for residual solvents & CVMP/VICH/502/99 Guideline on impurities: residual Solvents EMA关于残留溶剂指南CPMP/ICH/283/95 杂质-残留溶剂指南和CVMP/VICH/502/99 杂质指南-残留溶剂的附录 Annex I: specifications for class 1 and class 2 residual solvents in active Substances 附录I:API中I类和II类残留溶剂质量标准 Annex II: residues of solvents used in the manufacture of finished products 附录II:制剂生产中使用溶剂的残留 Discussion at Quality Working Party 质量工作组讨论会议January 2003 to June 2004 2003.01~2004.06 Adoption by CVMP CVMP 采纳July 2004 2004.07 Adoption by CHMP CHMP 采纳July 2004 2004.07 Date for coming into operation 生效时间January 2005 2005.01 Rev. 01 Adoption by Quality Working Party 质量工作组采纳的01版本22 November 2012 2012.11.22 Rev. 01 Adoption by CVMP CVMP采纳的01版本7 February 2013 2013.02.07 Rev. 01 Adoption by CHMP CHMP采纳的01版本11 February 2013 2013.02.11 Rev. 01 Date for coming into operation 01版本生效1 March 2013 2013.03.01
残留溶剂分析方法验证方案报告**制药股份有限公司 版本号: 第1版验证名称 XX产品残留溶剂分析方法验证方案验证编号 YZ-FX-13-001(P) 页码:Page 1 of 12 ***产品残留溶剂分析方法验证方案 20**年**月 **制药股份有限公司 版本号: 第1版验证名称 XX产品残留溶剂分析方法验证方案验证编号 YZ-FX-13-001(P) 页码:Page 2 of 12 验证方案的起草与审批 验证小组成员 部门人员职责 验证小组组长 职责部门人员 负责组织起草验证方案并按批准方案组织实 施;督促验证人员做好记录;负责各阶段验证 结果汇总及评价、组织起草验证报告;组织相 关培训 方案审核 审核签名及日期 验证委员会 方案批准 批准人批准日期
方案实施日期: **制药股份有限公司 版本号: 第1版验证名称 XX产品残留溶剂分析方法验证方案 验证编号 YZ-FX-13-001(P) 页码:Page 3 of 12 目录 1. 验证目 的 ..................................................................... ............................................................. 4 2. 方法简介与确认范围...................................................................... ........................................ 4 3. 标准品、供试 品 ..................................................................... ................................................. 4 4. 风险评 估 ..................................................................... ............................................................. 4 5. 验证的可接受标准...................................................................... ............................................ 5 6. 验证步 骤 ..................................................................... ............................................................. 6 6.1系统适应 性 ..................................................................... .......................................................... 6 6.2专属 性 .....................................................................
[467]有机挥发性杂质 残留溶剂限度 根据药典要求,药品中残留溶剂定义:在药物或赋形剂制造或使用过程中,或药物制剂生产过程中残存的有机挥发性物质。目前制药技术不能完全去除残留溶剂。药物或赋形剂合成中选择适当的溶剂可提高得率,或获得某些特性如晶型、纯度和溶解性。因此,合成中所用溶剂有时可能是危险因素。本章节不包括涉及用于组分的溶剂或溶剂化物。尽管如此,以上产品仍应标明所含溶剂含量并证明对人体安全。 由于残留溶剂不用于治疗用途,因此应尽可能除去,以符合药物、赋形剂和产品规格要求、GMP或其他质量标准。药物制剂中所含溶剂不得高于安全性评价规定限度。众所周知,药物、赋形剂、药物制剂不应含能引起不可逆毒副作用(Ⅰ级,见表1)的残留溶剂,除非证明有很好的风险-效益比。应对引起次级严重毒副作用的残留溶剂(Ⅱ级,见表2)规定限度,以防止病人发生潜在的不良反应。理想状态下,应尽可能使用低毒性溶剂(Ⅲ级,见表3)。本章节所有溶剂列表见附录1。下述表格及限度并不代表全部。当医药工业发展需使用其他溶剂情况下,应将新溶剂添加到列表中。 当药物、赋形剂、药物制剂生产和纯化过程中残留已知有机溶剂时,应依法检查溶剂限度。本限度检查仅检查用于制造和纯化加工所用的溶剂。虽然制造商可能选择测试药物,我们可采用累积法从制造工艺水平计算产品中残留溶剂。如残留溶剂计算结果等于或低于本章节规定限度,可不进行残留溶剂检查。如残留溶剂计算结果高于本章节规定限度,仍需检查残留溶剂限度以确定是否精加工降低了有关溶剂水平至可接受量。如残留溶剂为制造所用溶剂,必须检查药物制剂残留溶剂限度。 附录2为有关残留溶剂的相关背景资料。 根据安全评估残留溶剂分类 国际化学安全机构用术语“可忍受日摄食量”(TDI)描述有毒化学物质残留限度。世界卫生组织和其他国家和国际卫生机构用术语“可接受日摄食量”(ADI)来描述残留限度。术语“允许日接触量”(PDE)定义为根据药效学残留溶剂可接受摄入量,避免与ADIs混淆。 本章节所列残留溶剂按通用名和结构列于附录1。它们用于评价对人类卫生可能存在的危险等级,共分成3个等级,见下表: Ⅰ级残留溶剂:不得残留 已知人体实验致癌物质;强烈疑似人体实验致癌物质;环境危害物质 Ⅱ级残留溶剂:应在限度内 动物实验非生殖毒性(遗传);动物实验致癌物质或其它可能的非可逆致病因子;产生毒性如神经毒性或致畸性;其它疑似重大但可逆毒性 Ⅲ级残留溶剂:低潜在毒性溶剂 对人体存在低毒性溶剂;无人体摄入量规定(注:Ⅲ级残留溶剂PDEs上限为≥50mg/天) *有关PDEs上限≥50mg/天的Ⅲ级残留溶剂,见残留溶剂限度下Ⅲ级讨论章节
溶剂残留量检测方法 气相色谱仪检测器(氢火焰离子检测器) 色谱柱:25%PEG-1500,301有机担体,柱长2m,内径2mm(也可以采用专业的毛细管柱) 条件:柱室温度90℃检测器温度:150℃气化室温度:150℃ 1.包装材料溶剂残留量的检测 采用气相色谱仪或等同原理的仪器,按生产实际使用溶剂的种类配制标准溶剂样品,用微升注射器取0.5μl、1μl、2μl、 3μl和4μl样品,换算成质量。将样品分别注入用硅橡胶密封好的清洁干燥的500ml三角瓶中,送入80±2℃恒温烘箱中放置30 分钟后,用5ml注射器从瓶中取1ml气体,迅速注入色谱仪中测定。以其出峰总面积值分别与对应的样品质量做出标准曲线。 裁取0.2m2样品,将样品迅速裁成10mm×30mm碎片,放入清洁的、在80℃条件下预热的500ml 三角瓶中,用硅胶塞密封,送入80±2℃恒温烘箱中加热30分钟后,用5ml注射器取1ml瓶中气体注入色谱仪中测定。以出峰总面积值在标准曲线上查出对应的溶剂残留量,试验结果以mg/m2表示。 2.油墨溶剂残留量的检测 采用气相色谱仪或等同原理的仪器,按产品标准要求的溶剂种类配制标准溶剂,将每种溶剂用10μl进样器通过密封胶塞向300ml输液瓶中注入1μl标准溶剂,放入80±1℃恒温烘箱中20分钟后取出,隔日再放入50±1℃恒温烘箱中20小时以上,取出后用1ml注射器分别从瓶中抽取0.2、0.6、0.8、1.0ml的气体进行测试,做出标准曲线。 将油墨在双向拉伸聚丙烯薄膜上制成印样,悬空放置2小时,将试样裁切成4条,规格为5cm ×10cm,总面积为200cm2,立即置于300ml输液瓶中塞紧瓶口,置于80±1℃恒温烘箱中30分钟,取出后用1ml注射器抽取气体,注入色谱仪测定,以出峰总面积值在标准曲线上查出对应的溶剂残留量,试验结果以mg/m2表示。 中心以化工行业技术需求和科技进步为导向,以资源整合、技术共享为基础,分析测试、技术咨询为载体,致力于搭建产研结合的桥梁。以“专心、专业、专注“为宗旨,致力于实现研究和应用的对接,从而推动化工行业的发展。 科标化工分析检测中心致力于推动化工产业发展,欢迎各行同仁前来洽谈、合作。
杂质:残留溶剂的指导原则 1.介绍 本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理学上的可接受水平。 药物中的残留溶剂在此定义为在原料药或赋形剂的生产中,以及在制剂制备过程中产生或使用的有机挥发性化合物,它们在工艺中不能完全除尽。在合成原料药中选择适当的溶剂可提高产量或决定药物的性质,如结晶型。纯度和溶解度。因此.有时溶剂是合成中非常关键的因素。本指导原则所指的溶剂不是谨慎地用作赋形剂的溶剂,也不是溶剂化物,然而在这些制剂中的溶剂含量也应进行测定,并作出合理的判断。 出于残留溶剂没有疗效,故所有残留溶剂均应尽可能.去,以符合产品规范、GMP或其他基本的质量要求。制剂所含残留溶剂的水平不能高于安全值,已知一些溶剂可导致不接受的毒性(第一类,表1),除非被证明特别合理,在原药、赋形剂及制剂生产中应避免使用。一些溶剂毒性不太大(第二类,表2)应限制使用,以防止病人潜在的不良反应。使用低毒溶剂(第三类,表3)较为理想。附录1中列出了指导原则中的全部溶剂。
表中所列溶剂并非详尽无遗,其他可能使用的溶剂有待日后补充列人。第一、二类溶剂的建议限度或溶剂的分类会随着。新的安全性资料的获得而调整。含有新溶剂的新药制剂、其上市申请的安全性资料应符合本指导原则或原料药指导原则(Q3A新原料药中的杂质)或新药制剂(Q3B新药制剂中的杂质)中所述的杂质控制原则,或者符合上述三者。 2. 指导原则的范围 指导原则范围包括原料药、赋形剂或制剂中所含残留溶剂.因此,当生产或纯化过程中会出现这些溶剂时。应进行残留溶剂的检验。也只有在上述情况下,才有必要作溶剂的检查。虽然生产商可以选择性地测定制剂,但也可以从制剂中各成分的残留溶液水平来累积计算制剂中的残留溶剂。如果计算结果等于或低于本原则的建议水平,该制剂可考虑不检查残留溶剂,但如果计算结果高于建议水平则应进行检测,以确定制剂制备过程中是否降低了有关溶剂的量以达到可接受水平。果制剂生产中用到某种溶剂,也应进行测定。 本指导原则不适用于临床研究阶段的准新原料药、准赋形剂和准制剂。也不适用于已上市的药品。 本指导原则适用于所有剂型和给药途径。短期(如30天或更短)使用或局部使用时,允许存在的残留溶剂水平可以较高。应根据不同的情况评判这些溶剂水平。 有关残留溶剂的背景附加说明见附录2。