一、PDB数据库中查找蛋白质结构数据
1.https://www.doczj.com/doc/de10911074.html,/pdb/home/home.do
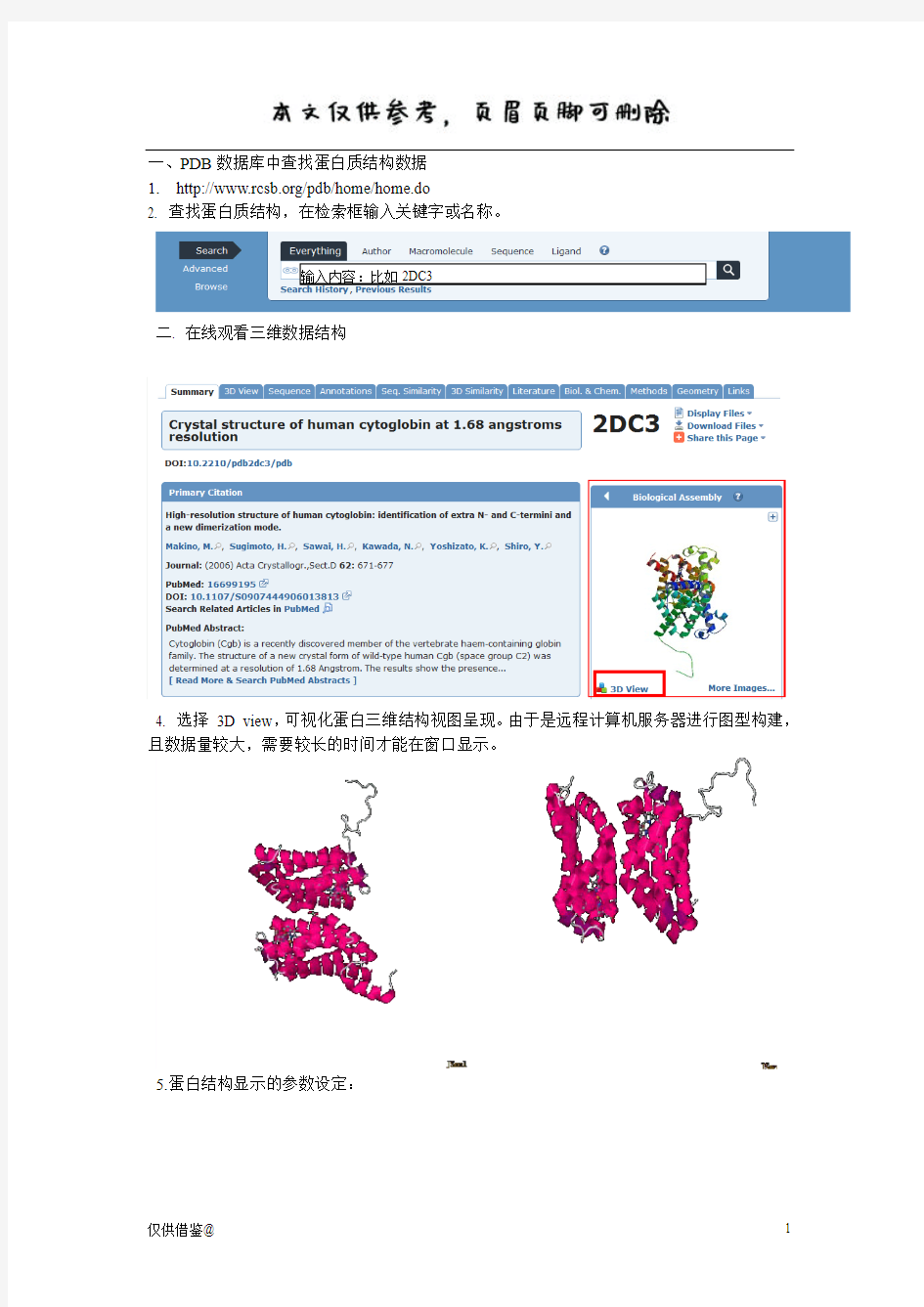
2. 查找蛋白质结构,在检索框输入关键字或名称。
输入内容:比如2DC3
二. 在线观看三维数据结构
且数据量较大,需要较长的时间才能在窗口显示。
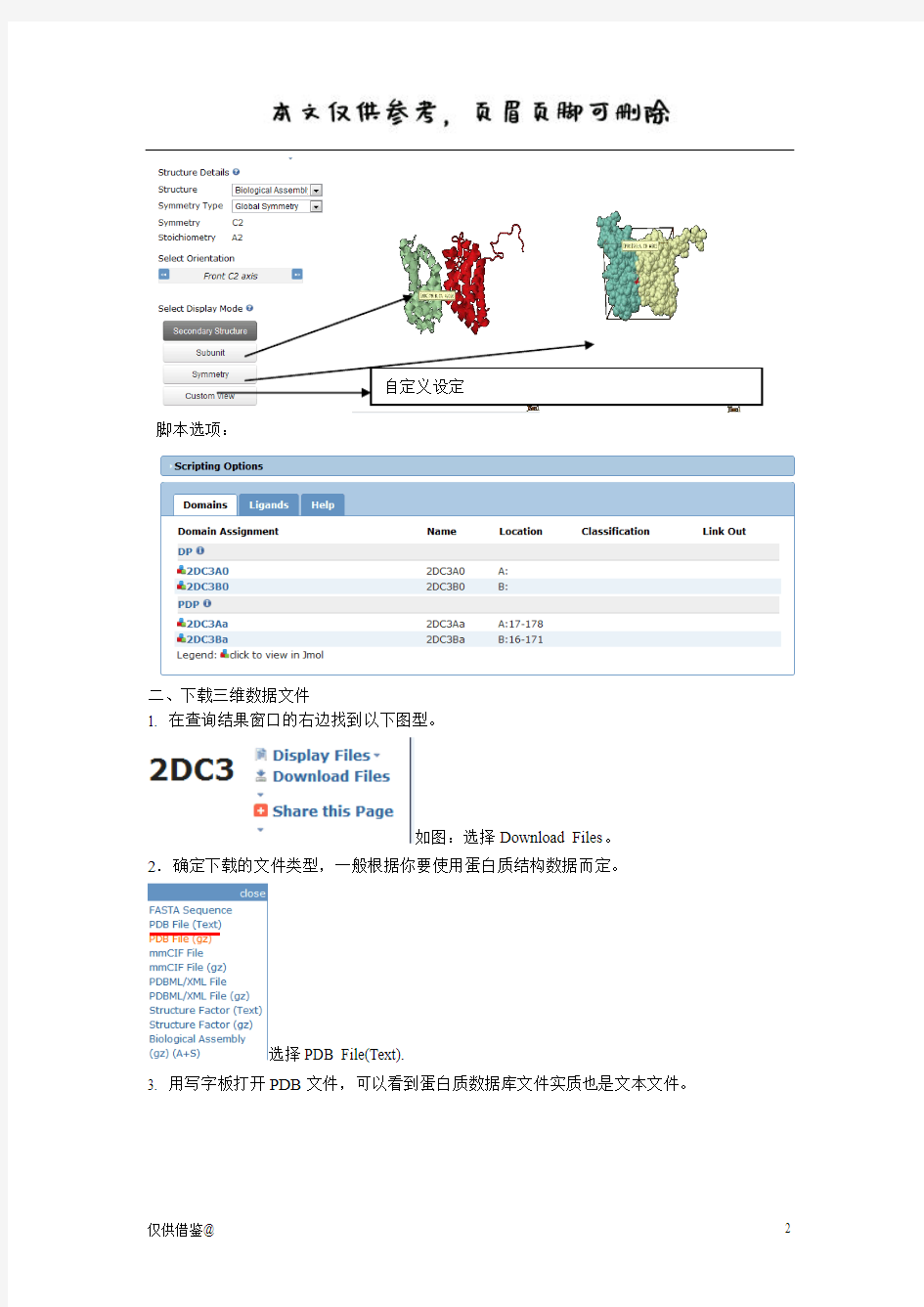
脚本选项:
二、下载三维数据文件
1. 在查询结果窗口的右边找到以下图型。
如图:选择Download Files 。
2.确定下载的文件类型,一般根据你要使用蛋白质结构数据而定。
选择PDB File(Text).
3. 用写字板打开PDB 文件,可以看到蛋白质数据库文件实质也是文本文件。
4. 下载Pymol,并安装。点DOWNLOAD进入下载,选择windows版本,并安装(需要pay money)。
或到此网站下载:https://www.doczj.com/doc/de10911074.html,/~gohlke/pythonlibs/#pymol
选择win32,1.7.1.1版本。并安装,需要python编程软件的支持,可从360软件管件直接安装。安装如图:
使用详情参见教程,注意打开程序需要找到python的安装目录:C:\Python27\PyMOL,找到PyMOL.exe文件,双击启动程序。
打开2DC3.pdb
或使用RasMol软件进行三维视图分析,步骤同上。
蛋白质结构分析原理及工具 (南京农业大学生命科学学院生命基地111班) 摘要:本文主要从相似性检测、一级结构、二级结构、三维结构、跨膜域等方面从原理到方法再到工具,系统地介绍了蛋白质结构分析的常用方法。文章侧重于工具的列举,并没有对原理和方法做详细的介绍。文章还列举了蛋白质分析中常用的数据库。 关键词:蛋白质;结构预测;跨膜域;保守结构域 1 蛋白质相似性检测 蛋白质数据库。由一个物种分化而来的不同序列倾向于有相似的结构和功能。物种分化后形成的同源序列称直系同源,它们通常具有相似的功能;由基因复制而来的序列称为旁系同源,它们通常有不同的功能[1]。因此,推测全新蛋白质功能的第一步是将它的序列与进化上相关的已知结构和功能的蛋白质序列比较。表一列出了常用的蛋白质序列数据库和它们的特点。 表一常用蛋白质数据库 网址可能有更新 氨基酸替代模型。进化过程中,一种氨基酸残基会有向另一种氨基酸残基变化的倾向。氨基酸替代模型可用来估计氨基酸替换的速率。目前常用的替代模型有Point Accepted Mutation (PAM)矩阵、BLOck SUbstitution Matrix (BLOSUM)矩阵[2]、JTT模型[3]。 序列相似性搜索工具。序列相似性搜索又分为成对序列相似性搜索和多序列相似性搜索。成对序列相似性搜索通过搜索序列数据库从而找到与查询序列相似的序列。分为局部联配和全局联配。常用的局部联配工具有BLAST和SSEARCH,它们使用了Smith-Waterman 算法。全局联配工具有FASTA和GGSEARCH,基于Needleman-Wunsch算法。多序列相似性搜索常用于构建系统发育树,这里不阐述。表二列举了常用的成对序列相似性比对搜索工具
考点一组成蛋白质的氨基酸及其种类(5年6考) 组成蛋白质的氨基酸的结构及种类 观察下列几种氨基酸的结构 (1)写出图中结构的名称 a.氨基; b.羧基。 (2)通过比较图中三种氨基酸,写出氨基酸的结构通式 (3)氨基酸的不同取决于R基的不同,图中三种氨基酸的R基依次为 (4)氨基酸的种类:约20种 ■助学巧记 巧记“8种必需氨基酸” 甲(甲硫氨酸)来(赖氨酸)写(缬氨酸)一(异亮氨酸)本(苯丙氨酸)亮(亮氨酸)色(色氨酸)书(苏氨酸) 注:评价蛋白质食品营养价值主要依据其必需氨基酸的种类和含量。
组成蛋白质的氨基酸的种类与结构 1.(海南卷)关于生物体内组成蛋白质的氨基酸的叙述,错误的是() A.分子量最大的氨基酸是甘氨酸 B.有些氨基酸不能在人体细胞中合成 C.氨基酸分子之间通过脱水缩合形成肽键 D.不同氨基酸之间的差异是由R基引起的 解析甘氨酸应是分子量最小的氨基酸,它的R基是最简单的氢。 答案 A 2.下图为氨基酸分子的结构通式,下列叙述正确的是() A.结构④在生物体内约有20种 B.氨基酸脱水缩合产生水,水中的氢来自于②和③ C.结构④中含有的氨基或羧基全部都参与脱水缩合 D.生物体内n个氨基酸形成一条多肽链需要n种密码子 解析①为氨基,③为羧基,④为侧链基团(R基)。构成人体氨基酸的种类约有20种,A正确;脱水缩合形成水,水中氢来自①③,B错误;R基中的氨基或羧基不参与脱水缩合,C错误;生物体内n个氨基酸形成一条多肽链需要n个密码子而不是需要n种密码子,D错误。 答案 A 解答本类题目的关键是熟记氨基酸的结构通式,如下图所示
找出氨基酸的共同体,即图中“不变部分”(连接在同一碳原子上的—NH2、—COOH和—H),剩下的部分即为R基。倘若找不到上述“不变部分”,则不属于构成蛋白质的氨基酸。
蛋白质纯化与结晶的原理 获得蛋白质的晶体结构的第一个瓶颈,就是制备大量纯化的蛋白质(>10 mg),其浓度通常在10 mg/ml 以上,并以此为基础进行结晶条件的筛选。运用重组基因的技术,将特定基因以选殖(clone)的方式嵌入表现载体(expression vector)内,此一载体通常具有易于调控的特性。之后再将带有特定基因的载体送入可快速生长的菌体中,如大肠杆菌(Escherichia coli),在菌体快速生长的同时,也大量生产表现载体上的基因所解译出之蛋白质。一般而言纯度越高的蛋白质比较有机会形成晶体,因此纯化蛋白质的步骤就成为一个重要的决定因素。 在取得高纯度的蛋白质溶液后,接下来就是晶体的培养。蛋白质晶体与其他化合物晶体的形成类似,是在饱和溶液中慢慢产生的,每一种蛋白质养晶的条件皆有所差异,影响晶体形成的变量很多,包含化学上的变量,如酸碱度、沈淀剂种类、离子浓度、蛋白质浓度等;物理上的变数,如溶液达成过饱和状态的速率、温度等;及生化上的变数,如蛋白质所需的金属离子或抑制剂、蛋白质的聚合状态、等电点等,皆是养晶时的测试条件。截至目前为止,并无一套理论可以预测结晶的条件,所以必须不断测试各种养晶溶液的组合后,才可能得到一颗完美的单一晶体(图一) 。 蛋白质晶体的培养,通常是利用气相扩散法(Vapor Diffusion Method) 的原理来达成;也就是将含有高浓度的蛋白质(10-50 mg/ml)溶液加入适当的溶剂,慢慢降低蛋白质的溶解度,使其接近自发性的沈淀状态时,蛋白质分子将在整齐的堆栈下形成晶体。举例来说,我们将蛋白质溶于低浓度(~1.0 M) 的硫酸铵溶液中,将它放置于一密闭含有高浓度(~2.0 M)硫酸铵溶液的容器中,由气相平衡,可以缓慢提高蛋白质溶液中硫酸铵的浓度,进而达成结晶的目的(图二)。 蛋白质晶体在外观上与其他晶体并无明显不同之处,但在晶体的内部,却有很大的差异。一般而言,蛋白质晶体除了蛋白质分子外,其他的空间则充满约40 %至60 %之间的水溶液,其液态的成分不仅使晶体易碎,也容易使蛋白质分子在晶格排列上有不规则的情形出现,造成晶体处理时的困难及绕射数据上的搜集不易等缺点。但也由于高含水量的特性,让蛋白质分子在晶体内与水溶液中的状态,极为相似。所以由晶体所解出的蛋白质结构,基本上可视为自然状态下的结构。 蛋白质结构解析的方法简介 到目前为止,蛋白质结构解析的方法主要是两种,x射线衍射和NMR。近年来还出现了一种新的方法,叫做Electron Microscopy。其中X射线的方法产生的更早,也更加的成熟,解析的数量也更多,我们知道,第一个解析的蛋白的结构,就是用x晶体衍射的方法解析的。而NMR方法则是在90年代才成熟并发展起来的。这两种方法各有优点和缺点。 首先来说一下,这两种方法的一般的步骤和各自的优点和缺点。电子显微镜(electron microsco py)作为一种新型的技术,目前的应用还是非常少,并且比较狭窄,我可能等到最后在给它作些
蛋白质凭借游离的氨基和羧基而具有两性特征,在等电点易生成沉淀。不同的蛋白质等电点不同,该特性常用作蛋白质的分离提纯。生成的沉淀按其有机结构和化学性质,通过pH的细微变化可复溶。蛋白质的两性特征使其成为很好的缓冲剂,并且由于其分子量大和离解度低,在维持蛋白质溶液形成的渗透压中也起着重要作用。这种缓冲和渗透作用对于维持内环境的稳定和平衡具有非常重要的意义。 在紫外线照射、加热煮沸以及用强酸、强碱、重金属盐或有机溶剂处理蛋白质时,可使其若干理化和生物学性质发生改变,这种现象称为蛋白质的变性。酶的灭活,食物蛋白经烹调加工有助于消化等,就是利用了这一特性。 (二)蛋白质的分类 简单的化学方法难于区分数量庞杂、特性各异的这类大分子化合物。通常按照其结构、形态和物理特性进行分类。不同分类间往往也有交错重迭的情况。一般可分为纤维蛋白、球状蛋白和结合蛋白三大类。 1.纤维蛋白包括胶原蛋白、弹性蛋白和角蛋白。 (1) 胶原蛋白胶原蛋白是软骨和结缔组织的主要蛋白质,一般占哺乳动物体蛋白总量的30%左右。胶原蛋白不溶于水,对动物消化酶有抗性,但在水或稀酸、稀碱中煮沸,易变成可溶的、易消化的白明胶。胶原蛋白含有大量的羟脯氨酸和少量羟赖氨酸,缺乏半胱氨酸、胱氨酸和色氨酸。 (2) 弹性蛋白弹性蛋白是弹性组织,如腱和动脉的蛋白质。弹性蛋白不能转变成白明胶。 (3) 角蛋白角蛋白是羽毛、毛发、爪、喙、蹄、角以及脑灰质、脊髓和视网膜神经的蛋白质。它们不易溶解和消化,含较多的胱氨酸(14-15%)。粉碎的羽毛和猪毛,在15-20磅蒸气压力下加热处理一小时,其消化率可提高到70-80%,胱氨酸含量则减少5-6%。 2.球状蛋白 (1) 清蛋白主要有卵清蛋白、血清清蛋白、豆清蛋白、乳清蛋白等,溶于水,加热凝固。 (2) 球蛋白球蛋白可用5-10%的NaCl溶液从动、植物组织中提取;其不溶或微溶于水,可溶于中性盐的稀溶液中,加热凝固。血清球蛋白、血浆纤维蛋白原、肌浆蛋白、豌豆的豆球蛋白等都属于此类蛋白。 (3) 谷蛋白麦谷蛋白、玉米谷蛋白、大米的米精蛋白属此类蛋白。不溶于水或中性溶液,而溶于稀酸或稀碱。 (4) 醇溶蛋白玉米醇溶蛋白、小麦和黑麦的麦醇溶蛋白、大麦的大麦醇溶蛋白属此类蛋白。不溶于水、无水乙醇或中性溶液,而溶于70-80%的乙醇。 (5) 组蛋白属碱性蛋白,溶于水。组蛋白含碱性氨基酸特别多。大多数组蛋白在活细胞中与核酸结合,如血红蛋白的珠蛋白和鲭鱼精子中的鲭组蛋白。 (6) 鱼精蛋白鱼精蛋白是低分子蛋白,含碱性氨基酸多,溶于水。例如鲑鱼精子中的鲑精蛋白、鲟鱼的鲟精蛋白、鲱鱼的鲱精蛋白等。鱼精蛋白在鱼的精子细胞中与核酸结合。 球蛋白比纤维蛋白易于消化,从营养学的角度看,氨基酸含量和比例也较纤维蛋白更理想。 3. 结合蛋白 结合蛋白是蛋白部分再结合一个非氨基酸的基团(辅基)。如核蛋白(脱氧核糖核蛋白、核糖体),磷蛋白(酪蛋白、胃蛋白酶),金属蛋白(细胞色素氧化酶、铜蓝蛋白、黄嘌呤氧化酶),脂蛋白(卵黄球蛋白、血中β1-脂蛋白),色蛋白(血红蛋白、细胞色素C、黄素蛋白、视网膜中与视紫质结合的水溶性蛋白)及糖蛋白(γ球蛋白、半乳糖蛋白、甘露糖蛋白、氨基糖蛋白)。
获得蛋白质的晶体结构的第一个瓶颈,就是制备大量纯化的蛋白质(>10mg),其浓度通常在10mg/ml 以上,并以此为基础进行结晶条件的筛选。运用重组基因的技术,将特定基因以选殖(clone)的方式嵌入表现载(expressionvector)内,此一载体通常具有易于调控的特性。之后再将带有特定基因的载体送入可快速生长的菌体中,如大肠杆菌(Escherichia coli),在菌体快速生长的同时,也大量生产表现载体上的基因所解译出之蛋白质。一般而言纯度越高的蛋白质比较有机会形成晶体,因此纯化蛋白质的步骤就成为一个重要的决定因素。 在取得高纯度的蛋白质溶液后,接下来就是晶体的培养。蛋白质晶体与其他化合物晶体的形成类似,是在饱和溶液中慢慢产生的,每一种蛋白质养晶的条件皆有所差异,影响晶体形成的变量很多,包含化学上的变量,如酸碱度、沈淀剂种类、离子浓度、蛋白质浓度等;物理上的变数,如溶液达成过饱和状态的速率、温度等;及生化上的变数,如蛋白质所需的金属离子或抑制剂、蛋白质的聚合状态、等电点等,皆是养晶时的测试条件。截至目前为止,并无一套理论可以预测结晶的条件,所以必须不断测试各种养晶溶液的组合后,才可能得到一颗完美的单一晶体。 蛋白质晶体的培养,通常是利用气相扩散法(Vapor Diffusion Method)的原理来达成;也就是将含有高浓度的蛋白质(10-50 mg/ml)溶液加入适当的溶剂,慢慢降低蛋白质的溶解度,使其接近自发性的沈淀状态时,蛋白质分子将在整齐的堆栈下形成晶体。举例来说,我们将蛋白质溶于低浓度(~1.0M)的硫酸铵溶液中,将它放置于一密闭含有高浓度(~2.0M)硫酸铵溶液的容器中,由气相平衡,可以缓慢提高蛋白质溶液中硫酸铵的浓度,进而达成结晶的目的。 蛋白质晶体在外观上与其他晶体并无明显不同之处,但在晶体的内部,却有很大的差异。一般而言,蛋白质晶体除了蛋白质分子外,其他的空间则充满约40%至60%之间的水溶液,其液态的成分不仅使晶体易碎,也容易使蛋白质分子在晶格排列上有不规则的情形出现,造成晶体处理时的困难及绕射数据上的搜集不易等缺点。但也由于高含水量的特性,让蛋白质分子在晶体内与水溶液中的状态,极为相似。所以由晶体所解出的蛋白质结构,基本上可视为自然状态下的结构。
生物芯片北京国家工程研究中心 湖南中药现代化药物筛选分中心 暨湖南涵春生物有限公司 常用数据库名录 1、蛋白质数据库 PPI - JCB 蛋白质与蛋白质相互作用网络 ?Swiss-Prot - 蛋白质序列注释数据库 ?Kabat - 免疫蛋白质序列数据库 ?PMD - 蛋白质突变数据库 ?InterPro - 蛋白质结构域和功能位点 ?PROSITE - 蛋白质位点和模型 ?BLOCKS - 生物序列分析数据库 ?Pfam - 蛋白质家族数据库 [镜像: St. Louis (USA), Sanger Institute, UK, Karolinska Institutet (Sweden)] ?PRINTS - 蛋白质 Motif 数据库 ?ProDom - 蛋白质结构域数据库 (自动产生) ?PROTOMAP - Swiss-Prot蛋白质自动分类系统 ?SBASE - SBASE 结构域预测数据库 ?SMART - 模式结构研究工具 ?STRING - 相互作用的蛋白质和基因的研究工具
?TIGRFAMs - TIGR 蛋白质家族数据库 ?BIND - 生物分子相互作用数据库 ?DIP - 蛋白质相互作用数据库 ?MINT - 分子相互作用数据库 ?HPRD - 人类蛋白质查询数据库 ?IntAct - EBI 蛋白质相互作用数据库 ?GRID - 相互作用综合数据库 ?PPI - JCB 蛋白质与蛋白质相互作用网络 2、蛋白质三级结构数据库 ?PDB - 蛋白质数据银行 ?BioMagResBank - 蛋白质、氨基酸和核苷酸的核磁共振数据库?SWISS-MODEL Repository - 自动产生蛋白质模型的数据库 ?ModBase - 蛋白质结构模型数据库 ?CATH - 蛋白质结构分类数据库 ?SCOP - 蛋白质结构分类 [镜像: USA | Israel | Singapore | Australia] ?Molecules To Go - PDB数据库查询 ?BMM Domain Server - 生物分子模型数据库 ?ReLiBase - 受体/配体复合物数据库 [镜像: USA] ?TOPS - 蛋白质拓扑图 ?CCDC - 剑桥晶体数据中心 (剑桥结构数据库 (CSD))
生物信息研究中常用蛋白质数据库简述 内蒙古工业大学理学院呼和浩特孙利霞 2010.1.5 摘要:在后基因组时代生物信息学的研究当中,离不开各种生物信息学数据库。尤其在蛋白质从序列到功能的研究当中,目前各种行之有效的方法都是基于各种层次和结构的蛋白质数据库。随着计算机技术及网络技术的发展,目前的蛋白质数据库不论是所包含数据量还是功能都日新月异,新的数据库层出不穷。一个新手面对如此浩瀚的数据量往往无从下手。本文粗浅地为目前蛋白质数据库的使用勾画出一个轮廓,作为自己蛋白质研究入门的一个引导。 关键词:蛋白质;数据库 0 引言 随着科技的发展,个人的知识往往赶不上快速膨胀的信息量,人们为了解决这个问题,便创建了形形色色的数据库。蛋白质数据库是指:在蛋白质研究领域根据实际需要,对蛋白质序列、蛋白质结构以及文献等数据进行分析、整理、归纳、注释,构建出具有特殊生物学意义和专门用途的数据库。蛋白质数据库总体上可分为两大类:蛋白质序列数据库和蛋白质结构数据库,蛋白质序列数据库来自序列测定,结构数据库来自X-衍射和核磁共振结构测定(详见图1)。这些数据库是分子生物信息学的基本数据资源。上世纪90年代,我国从事蛋白质研究的学者使用的蛋白质数据库储存介质还是国外实验室发布的激光光盘[1]。信息的传播储存甚为不便。随着蛋白质研究的发展飞快,同时伴随着计算机和因特网发展,蛋白质数据库的储存传播方式也发生的巨大的变化。进入21世纪后,我们所用的各种蛋白质数据库都发展成为存储在网络服务器上,基于“服务器—客户机”的访问查询方式。伴随着计算机及物理测试技术的发展数据库的容量和功能成数量级膨胀。但是面对如此浩瀚的数据,新手往往感到无从下手,在需要时找不到自己需要的合适数据库。 本文从目前蛋白质数据库建立的的逻辑层次出发,系统地简绍了常用蛋白质数据的概况,它们的查询方法以及它们相互之间的联系。同时尽量不涉及数据库建设和维护方面的计算机和网络这些数据库底层的技术,为蛋白质研究的入门者及对蛋白质感兴趣的人员的一个引导。
1.蛋白质的二级结构主要有哪些类型,其特点如何? 答:α-右手螺旋,β-折叠,无规卷曲,U型回折(β-转角) <1>α-右手螺旋 α-螺旋为右手螺旋,每一圈含有3.6个aa残基(或肽平面),每一圈高5.4?,即每一个aa 残基上升1.5?,旋转了100度,直径为5 ?,2个二面角(ф,ψ)=(-570,-480)。维持α-右手螺旋的力量是螺旋内氢键,它产生于一个肽平面的C=O与相邻一圈的在空间上邻近的另一个肽平面的N-H之间,它的方向平行于螺旋轴,每个氢键串起的长度为3.6个肽平面或3.6个aa残基,被氢键串起来的这个环上含有13个原子,故α-右手螺旋也被称为 3.613螺旋。Pro破坏α-螺旋。 <2>β-折叠 肽链在空间的走向为锯齿折叠状,二面角(ф,ψ)=(-119℃,+113℃)。维持β-折叠的力量是折叠间的氢键,它产生于一个肽平面的C=O与相邻肽链的在空间上邻近的另一个肽平面的N-H之间,两条肽链上的肽平面互相平行,有平行式和反平行式两种, <3>U型回折:也叫β-转角,肽链在某处回折1800所形成的结构。这个结构包括的长度为 4个aa残基,其中的第三个为Gly,稳定该结构的力量是第一和第四个aa残基之间形成的氢键。 <4>无规卷曲:无固定的走向,但也不是任意变动的,它的2个二面角(ф,ψ)有个变化 范围。论 述 04 蛋 白 质 简述蛋白质一级结构的分析方法。 第一步:前期准备,第二步:肽链的端点测定,第三步:每条肽链aa顺序的测定,第四步:二硫键位置的确定。 <1>第一步:前期准备 分离纯化蛋白质:纯度要达到97%以上。 蛋白质分子量的测定:用于判断分子的大小,估计肽链的数目,有渗透压法、凝胶电泳法(聚丙烯酰胺、SDS)、凝胶过滤法、超离心法等 aa组成的测定:用于最后核对,氨基酸自动分析仪。 肽链拆分:非共价键的如氢键、离子键、疏水键、范德华力4种,可用尿素或盐酸胍等有机溶液来拆分。共价键的仅二硫键1种,可用巯基乙醇、碘代乙酸、过甲酸来拆分。 <2>第二步:肽链的端点测定 N端测定:Sanger法,DNFB→DNP-肽→水解→乙醚萃取→层析鉴定。 Edman法,PITC→PTC-肽→PTH-aa→层析鉴定。 C端测定:肼解法。 <3>第三步:每条肽链aa顺序的测定 事先要将蛋白质打断成多肽甚至寡肽,再上机分析,而且要2套以上,便于以后拼接。 常用的工具酶和特异性试剂有: 胰蛋白酶:-(Arg、Lys)↓-。产物为C端Arg、Lys的肽链。 糜蛋白酶:表示为-(Trp、Tyr、Phe)↓-。 CNBr:-Met↓-。 <4>第四步:二硫键位置的确定 包括链内和链间二硫键的位置,用对角线电泳来测,这项工作在AA序测定完毕后进行。在肽链未拆分的情况下用胃蛋白酶水解之,可以得到被二硫键连着的多肽产物。先进行第一向电泳,将产物分开。再用巯基乙醇处理,将二硫键打断。最后进行第二向电泳,条件与第一向电泳完全相同。选取偏离对角线的样品(多肽或寡肽),它们就是含二硫键的片段,上机测aa顺序,根据已测出的蛋白质的aa顺序,把这些片段进行定位,就能找到二硫键的位置。
蛋白质晶体结构分析及其发展 范海福 中国科学院,物理研究所,北京,100080 物质的各种宏观性质源出于本身的微观结构。探索物质结构与性质之间的关系,是凝聚态物理、结构化学、材料科学、分子生物等许多学科的一个重要研究内容。晶体结构分析,是在原子的层次上测定固态物质微观结构的主要手段,它与上述众多学科有着密切的联系。就其本身而言,晶体结构分析是物理学中的一个小分支。这主要研究如何利用晶态物质对X-射线、电子、以及中子的衍射效应来测定物质的微观结构。晶体结构分析服务于许多不同的学科,因而许多学科的发展都对晶体结构分析产生深刻的影响。另一方面,晶体结构分析有自己独立的体系,它本身的发展又对所服务的学科起着促进作用。 晶体结构分析是伦琴发现X-射线以后创站的最重要学科之一。它奠基于物理学的几项重要进展。其中包括1895年W. C. Roentgen发现X-射线,1912年M. von Laue发现晶体对X-射线的衍射,1927年C. J. Davisson和G. P. Thomson发现晶体对电子的衍射,以及1931年E. Ruska建造第一台电子显微镜。上述几项重大的物理学进展使人类掌握了在原子层次上研究物质内部结构的手段,它们分别获得1901、1914、1937和1986年的诺贝尔物理学奖。其中,1901年伦琴获得的诺贝尔奖还是历史上第一个诺贝尔物理奖。通过研究物质内部结构与性质的关系,晶体结构分析有力地促进了各相关学科的发展。晶体结构分析的发展,是一个不断完善自身和不断扩大应用的过程。诺贝尔将的年谱记录了晶体结构分析历史上的重大事件并展示了它与其他学科相互作用所产生的丰硕成果。 晶体结构分析的方法主要有两大类。这就是以X-射线衍射为代表的衍射分析方法和以电子显微术为代表的显微成像方法。电了显微成像也可以认为是两上相继的电子衍射过程。因此,可以说衍射分析是晶体结构分析的核心。用衍射分析方法测定晶体结构的理论依据,在于晶体结构同它的衍射效应之间存在着互为Fourier变换的关系。这里说的衍射效应,是指从晶体向各个方向发出的衍射的振幅和相位。从衍射实验可以记录下各个方向上衍射波的振幅。但是在目前以及可见的将来,还不容易找到有普遍意义的实用方法来记录由晶体发出的衍射波的相位。因此要想从衍射效应的Fourier变换解出晶体结构,必须先设法找回"丢失了的"相位。这就是晶体学中的"相位问题",它一直是研究晶体结构分析方法的关键问题。 紧接着Laue发现X-射线衍射,Bragg父子(W. H. Bragg和W. L. Bragg) 就迅速建立了用X-射线衍射方法测定晶体结构的实验手段和理论基础。这使人类得以定量地观测原子在晶体中的位置。为此他们两人同获1915年的诺贝尔物理学奖。晶体结构分析最初用于一些简单的无机化合物。对碱金属卤化物结构的研究导至W. L. Bragg提出原子半径的概念。不久Bragg又将晶体结构分析应用于研究硅酸盐以及金属和合金。硅酸盐晶体结构分析的工作为硅酸盐结构化学提供了最早的实验基础,而有关金属和合金的工作则作物理冶金、金属物理、以及相平衡图的研究推上了一个新的台阶,使有关工作深入到原子的层次。 晶体结构分析在研究无机化合物上取得成功,引起人们对有机物尤其是生命物质内部结构的兴趣。英国从二十年代中期就开始研究有机物晶体结构。但是过了十年多仍未见有重大的突破。原因是当时的分析技术和方法还很原始。于是迎来了三、四十年代晶体结构分析方法和技术大发展的时期。如前所述,晶体结构分析中所谓"相位问题"。早期的晶体结构分析用以解决相位问题的方法是所谓尝试法。其要点是:先根据已尼掌握的线索猜想出一个结构模型,再从这个模型计算出相应的一组理论衍射强度,然后同实验所犁衍射强度作比较并据此对模型进行修改。。上述步骤须经多次反复,直至理论和实验的衍射强度得以吻合。用这样的"方法"来测定晶体结构,说明科学试验却更像艺术创作。它显然适应不了测定复杂的晶
搞蛋白质的童鞋们,甭要只查NCBI了~蛋白质相关数据库启蒙~ ★ 小木虫(金币+1):奖励一下,谢谢提供资源 qinhy:恭喜,您的帖子被版主审核为资源贴了,别人回复您的帖子对资源进行评价后,您就可以获得金币了理由:资源贴2011-11-26 16:56 本来是带图的,可是弄过来就变成米图了,附件里面一个是PDF版、一个是WORD版均是带图的,童鞋们看带图的可能比较方便点哦~ 基于蛋白质序列的蛋白质相互作用位点预测(闲谈版) 这个不是论文不是论文啊~~这个是应某某的要求帮他找的,所以都是用现成的免费的网站数据库做的预测分析。无论文为依托,无原理为根据,纯粹就是流连各大网站作个的闲谈。 1、用这些网站先查查你要研究的蛋白质的底细。 这些网站的数据库大多数是实验或者一些相关文献报道的数据的组成。 ★String http://string.embl.de/ 输入你要搜寻的蛋白,它就把这个蛋白相关的数据反映给你,分confidence、evidence的数据可信度参考,同时还具有actions选项,反应它们之间可能是激活/抑制的关系。按按+、-号可以扩大缩小关联蛋白的数量范围。 往下拉一点点就是数据,哈哈,我们都要看数据吃饭啊~~ 分析的数据源自Neighborhood、Fusion、Occurrence、Coexpression、Experiments Database、Textminin及Homology,表示点得证明有数据,根据各项数据给出综合评分。评分越高相互存在关系可能性越高。点击下方各项图标等详细看到各项数据内容。 设条件确定筛选范围。 ★DIP https://www.doczj.com/doc/de10911074.html,/dip/Main.cgi 跟上面的大同小异的功能,装上它附带的软件可能操作性会好一点,不过我米有试过哦。倒是跟它有链接的几个数据库都很强大,大家可以点击看看。 ★BIND http://www.bind.ca 文献有介绍的网站,不过我不能理解为什么我注册就注不了……. 2、继续查,用这些网站将要研究的蛋白质的家庭背景,月收入也大起底。 这里的网站可能跟相互作用方面的关系不大,但是如果知道这些,可以对研究的蛋白有更深的了解。 ★PDB https://www.doczj.com/doc/de10911074.html,/pdb/home/home.do 要查3D结构就往这里查~通常说的PDB号为文献号末4位。 ★PIR https://www.doczj.com/doc/de10911074.html,/pirwww/index.shtml 在蛋白质方面如NCBI般强大的网站,去上面晃荡下吧,会有收获滴。 ★KEGG http://www.genome.jp/kegg/ 粉强大的一个网站,我只说说它的KEGG PA THW AY子项,能迅速掌握一个蛋白质的功能通路,对于小白的偶们来说,很有用,有木有。 3、正题正题,做完上面那些后,接着就是纯预测的成分。也因为如此,要找着这些网站是很悲催的一件事。就算你找着了,你不懂语言,不懂算法,到底结果的可靠性怎样,见人见智。 需要PDB号作分析: promate http://bioinfo.weizmann.ac.il/promate/
SWISS-MODEL 蛋白质结构预测 SWISS-MODEL是一项预测蛋白质三级结构的服务,它利用同源建模的方法实现对一段未知序列的三级结构的预测。该服务创建于1993年,开创了自动建模的先河,并且它是讫今为止应用最广泛的免费服务之一。 同源建模法预测蛋白质三级结构一般由四步完成: 1. 从待测蛋白质序列出发,搜索蛋白质结构数据库(如PDB,SWISS-PROT等),得到许多相似序列 (同源序列),选定其中一个(或几个)作为待测蛋白质序列的模板; 2. 待测蛋白质序列与选定的模板进行再次比对,插入各种可能的空位使两者的保守位置尽量对齐; 3. 建模:调整待测蛋白序列中主链各个原子的位置,产生与模板相同或相似的空间结构——待测蛋白 质空间结构模型; 4. 利用能量最小化原理,使待测蛋白质侧链基团处于能量最小的位置。 最后提供给用户的是经过如上四步(或重复其中某几步)后得到的蛋白质三级结构。 SWISS-MODEL工作模式 SWISS-MODEL服务器是以用户输入信息的最小化为目的设计的,即在最简单的情况下,用户仅提供一条目标蛋白的氨基酸序列。由于比较建模程序可以具有不同的复杂性,用户输入一些额外信息对建模程序的运行有时是有必要的,比如,选择不同的模板或者调整目标模板序列比对。该服务主要有以下三种方式: ?First Approach mode(简捷模式):这种模式提供一个简捷的用户介面:用户只需要输入一条氨基酸序列,服务器就会自动选择合适的模板。或者,用户也可以自己指定模板(最多5条),这些模板可以来自ExPDB 模板数据库(也可以是用户选择的含坐标参数的模板文件)。如果一条模板与提交的目标序列相似度大于25%,建模程序就会自动开始运行。但是,模板的可靠性会随着模板与目标序列之间的相似度的降低而降低,如果相似度不到50%往往就需要用手工来调整序列比对。这种模式只能进行大于25个残基的单链蛋白三维结构预测。 ?Alignment Interface(比对界面):这种模式要求用户提供两条已经比对好的序列,并指定哪一条是目标序列,哪一条是模板序列(模板序列应该对应于ExPDB模板数据库中一条已经知道其空间结构的蛋白序列)。服务器会依据用户提供的信息进行建模预测。 ?Project mode(工程模式):手工操作建模过程:该模式需要用户首先构建一个DeepView工程文件,这个工程文件包括模板的结构信息和目标序列与模板序列间的比对信息。这种模式让用户可以控制许多参数,例如:模板的选择,比对中的缺口位置等。此外,这个模式也可以用于“first approach mode简捷模式”输出结果的进一步加工完善。 此外,SWISS-MODEL还具有其他两种内容上的模式: ?Oligomer modeling(寡聚蛋白建模):对于具有四级结构的目标蛋白,SWISS-MODEL提供多聚模板的模式,用于多单体的蛋白质建模。这一模式弥补了简捷模式中只能提交单个目标序列,不能同时预测两条及以上目标序列的蛋白三维结构的不足。 ?GPCR mode(G蛋白偶联受体模式):是专门对7次跨膜G蛋白偶联受体的结构预测。
《蛋白质结构解析研究进展》 一、蛋白质结构分类 人类对于进化的认识及蛋白质结构相似性比较的研究使蛋白质结构分类成为可能,而且近年来取得的研究进展表明,大部分蛋白质可以成功的分入到适当数目的家族中。目前国际上流行的蛋白质结构分类数据库基本上采取两种不同的思路,一种是数据库中储存所有结构两两比较的结果;第二种思路是致力于构建非常正式的分类体系。由于所有分类方法反映了各研究小组在探究这个重要领域的不同角度,所以这些方法是同等有效的。目前,被广泛应用的四种分类标准是:手工构造的层次分类数据库SCOP,全自动分类的MMDB和FSSP,和半手工半自动的CATH。 蛋白质结构自动分类问题可以被纳入机器学习的范畴,通过提取分析蛋白质结构的关键特征,构造算法来学习蕴含于大量已知结构和分类的数据中的专家经验知识,来实现对未知蛋白质结构的分类预测。目前,对蛋白质结构的不同层次分类,结果比较好的机器学习方法是:神经网络多层感知器、支持向量机和隐马尔可夫模型。支持向量机应用于分类问题最终归结于求解一个最优化问题。上世纪90 年代中期,隐马尔可夫模型与其他机器学习技术结合,高效地用于多重比对、数据挖掘和分类、结构分析和模式发现。多层感知器即误差反向传播神经网络,它是在各种人工神经网络模型中,在机器学习中应用最多且最成功的采用BP学习算法的分类器。 二、蛋白质结构的确定 蛋白质三维空间结构测定方法主要包括X射线晶体学分析、核磁共振波谱学技术和三维电镜重构,这三种方法都可以完整独立地在原子分辨水平上测定出蛋白质的三维空间结构。蛋白质数据库PDB中80%的蛋白质结构是由X射线衍射分析得到的,约15%的蛋白质结构是由核磁共振波谱学这种新的结构测定方法得到。 1、X射线晶体学
蛋白质相互作用数据库和分析方法 1. 蛋白质相互作用的数据库 蛋白质相互作用数据库见下表所示: 数据库名 说明 网址 BIND 生物分子相互作用数据库 http://bind.ca/ DIP 蛋白质相互作用数据库 https://www.doczj.com/doc/de10911074.html,/ IntAct 蛋白质相互作用数据库 https://www.doczj.com/doc/de10911074.html,/intact/index.html InterDom 结构域相互作用数据库 https://www.doczj.com/doc/de10911074.html,.sg/ MINT 生物分子相互作用数据库 http://mint.bio.uniroma2.it/mint/ STRING 蛋白质相互作用网络数据库 http://string.embl.de/ HPRD 人类蛋白质参考数据库 https://www.doczj.com/doc/de10911074.html,/ HPID 人类蛋白质相互作用数据库 http://wilab.inha.ac.kr/hpid/ MPPI 脯乳动物相互作用数据库 http://fantom21.gsc.riken.go.jp/PPI/ biogrid 蛋白和遗传相互作用数据,主要来自于酵母、线虫、果蝇和人 https://www.doczj.com/doc/de10911074.html,/ PDZbase 包含PDZ 结构域的蛋白质相互作用数据库 https://www.doczj.com/doc/de10911074.html,/services/pdz/start Reactome 生物学通路的辅助知识库 https://www.doczj.com/doc/de10911074.html,/ 2. 蛋白质相互作用的预测方法 蛋白质相互作用的预测方法很非常多,以下作了简单的介绍 1) 系统发生谱 这个方法基于如下假定:功能相关的(functionally related)基因,在一组完全测序的基因组中预期同时存在或不存在,这种存在或不存在的模式(pattern)被称作系统发育谱;如果两个基因,它们的序列没有同源性,但它们的系统发育谱一致或相似.可以推断它们在功能上是相关的。
蛋白质预测分析网址集锦? 物理性质预测:? Compute PI/MW?? ?? SAPS?? 基于组成的蛋白质识别预测? AACompIdent???PROPSEARCH?? 二级结构和折叠类预测? nnpredict?? Predictprotein??? SSPRED?? 特殊结构或结构预测? COILS?? MacStripe?? 与核酸序列一样,蛋白质序列的检索往往是进行相关分析的第一步,由于数据库和网络技校术的发展,蛋白序列的检索是十分方便,将蛋白质序列数据库下载到本地检索和通过国际互联网进行检索均是可行的。? 由NCBI检索蛋白质序列? 可联网到:“”进行检索。? 利用SRS系统从EMBL检索蛋白质序列? 联网到:”,可利用EMBL的SRS系统进行蛋白质序列的检索。? 通过EMAIL进行序列检索?
当网络不是很畅通时或并不急于得到较多数量的蛋白质序列时,可采用EMAIL方式进行序列检索。? 蛋白质基本性质分析? 蛋白质序列的基本性质分析是蛋白质序列分析的基本方面,一般包括蛋白质的氨基酸组成,分子质量,等电点,亲水性,和疏水性、信号肽,跨膜区及结构功能域的分析等到。蛋白质的很多功能特征可直接由分析其序列而获得。例如,疏水性图谱可通知来预测跨膜螺旋。同时,也有很多短片段被细胞用来将目的蛋白质向特定细胞器进行转移的靶标(其中最典型的例子是在羧基端含有KDEL序列特征的蛋白质将被引向内质网。WEB中有很多此类资源用于帮助预测蛋白质的功能。? 疏水性分析? 位于ExPASy的ProtScale程序(?)可被用来计算蛋白质的疏水性图谱。该网站充许用户计算蛋白质的50余种不同属性,并为每一种氨基酸输出相应的分值。输入的数据可为蛋白质序列或SWISSPROT数据库的序列接受号。需要调整的只是计算窗口的大小(n)该参数用于估计每种氨基酸残基的平均显示尺度。? 进行蛋白质的亲/疏水性分析时,也可用一些windows下的软件如,bioedit,dnamana等。? 跨膜区分析? 有多种预测跨膜螺旋的方法,最简单的是直接,观察以20个氨基酸为单位的疏水性氨基酸残基的分布区域,但同时还有多种更加复杂的、精确的算法能够预测跨膜螺旋的具体位置和它们的膜向性。这些技术主要是基于对已知
蛋白质的分类 摘要:蛋白质的种类繁多,结构复杂,所以分类也就各异。 一、按来源分类 蛋白质按来源可以分为动物蛋白和植物蛋白,两者所含的氨基酸是不同的。动物性蛋白质主要为提取自牛奶的乳清蛋白,其所含必需氨基酸种类齐全,比例合理,但是含有胆固醇。植物性蛋白质主要来源于大豆的大豆蛋白,最多的优点就是不含胆固醇。 二、按组成成分分类 按照化学组成,蛋白质通常可以分为简单蛋白质、结合蛋白质和衍生蛋白质。简单蛋白质经水解得氨基酸和氨基酸衍生物;结合蛋白质经水解得氨基酸、非蛋白的辅基和其他(结合蛋白质的非氨基酸部分称为辅基);蛋白质经变性作用和改性修饰得到衍生蛋白质。 1—脂 如酪蛋 铜的有血蓝蛋白等。 ⑥黄素蛋白(flavoproteins):辅基为黄素腺嘌呤二核苷酸,如琥珀酸脱氢酶、D—氨基酸氧化酶等。 ⑦金属蛋白(metalioproteins):与金属直接结合的蛋白质,如铁蛋白含铁,乙醇脱氢酶含锌,黄嘌呤氧化酶含钼和铁等。 衍生蛋白质,天然蛋白质变性或者改性、修饰和分解产物。 ①一级衍生蛋白质:不溶于所有溶剂,如变性蛋白质。 ②二级衍生蛋白质:溶于水,受热不凝固,如胨、肽。 ③三级衍生蛋白质:功能改进,如磷酸化蛋白、乙酰化蛋白、琥珀酰胺蛋白。 三、按分子形状分类 根据分子形状的不同,可将蛋白质分为球状蛋白质和纤维状蛋白质两大类。以长轴和短轴之比为标准,球状蛋白
质小于5,纤维状蛋白质大于5。纤维状蛋白多为结构蛋白,是组织结构不可缺少的蛋白质,由长的氨基酸肽链连接成为纤维状或蜷曲成盘状结构,成为各种组织的支柱,如皮肤、肌腱、软骨及骨组织中的胶原蛋白;球状蛋白的形状近似于球形或椭圆形。许多具有生理活性的蛋白质,如酶、转运蛋白、蛋白类激素与免疫球蛋白、补体等均属于球蛋白。 四、按结构分类 蛋白质按其结构可分为:单体蛋白、寡聚蛋白、多聚蛋白。 单体蛋白:蛋白质由一条肽链构成,最高结构为三级结构。包括由二硫键连接的几条肽链形成的蛋白质,其最高结构也是三级。多数水解酶为单体蛋白。 寡聚蛋白:包含2个或2个以上三级结构的亚基。可以是相同亚基的聚合,也可以是不同亚基的聚合。 多聚蛋白:由数十个亚基以上,甚至数百个亚基聚合而成的超级多聚体蛋白。 五、按功能分类 1. 2. 3.
生物大分子结构与功能 1、什么叫晶体衍射的结构因子?结构因子F与晶体衍射的衍射点的强度I有什 么关系? 结构因子是定量表征原子排布以及原子种类对衍射强度影响规律的参数,即晶体结构对衍射强度的影响因子。 晶体X光衍射强度与几何结构因子的平方成正比 2、什么叫傅里叶变换?用公式说明为什么说蛋白质晶体的电子密度与晶体衍 射的结构因子互为傅里叶变换与反变换的关系? The Fourier transform is a mathematical operation that decomposes a function into its constituent frequencies, known as a frequency spectrum. The term "Fourier transform" refers to both the transform operation and to the complex-valued function it produces. 结构因数同电子密度分布函数之间存在着傅里叶变换的关系: 由傅里叶变换公式得: for every real number ξ. When the independent variable x represents time (with SI unit of seconds), the transform variable ξ represents frequency (in hertz). Under suitable conditions, ? can be reconstructed from by the inverse transform: for every real number x. 晶体对X射线的衍射是一种傅里叶变换,把正空间的电子密度变换为倒易空间的衍射强度。
蛋白质序列数据库 我们可以根据基因组序列预测新基因,预测编码区域,并推测其产物(即蛋白质)的序列。因此,随着基因组序列的不断增长,蛋白质序列也在不断增加。 PIR 历史上,蛋白质数据库的出现先于核酸数据库。在1960年左右,Dayhoff和其同事们搜集了当时所有已知的氨基酸序列,编著了《蛋白质序列与结构图册》。从这本图册中的数据,演化为后来的蛋白质信息资源数据库PIR(Protein Information Resource)。 PIR是由美国生物医学基金会NBRF(National Biomedical Research Foundation)于1984年建立的,其目的是帮助研究者鉴别和解释蛋白质序列信息,研究分子进化、功能基因组,进行生物信息学分析。它是一个全面的、经过注释的、非冗余的蛋白质序列数据库。所有序列数据都经过整理,超过99%的序列已按蛋白质家族分类,一半以上还按蛋白质超家族进行了分类。PIR提供一个蛋白质序列数据库、相关数据库和辅助工具的集成系统,用户可以迅速查找、比较蛋白质序列,得到与蛋白质相关的众多信息。目前,PIR已经成为一个集成的生物信息数据源,支持基因组研究和蛋白质组研究。至2004年,PIR 有近30万个蛋白质的登录数据项,包括来自不同生物体的蛋白质序列。 除了蛋白质序列数据之外,PIR还包含以下信息: (1)蛋白质名称、蛋白质的分类、蛋白质的来源; (2)关于原始数据的参考文献; (3)蛋白质功能和蛋白质的一般特征,包括基因表达、翻译后处理、活化等; (4)序列中相关的位点、功能区域。 对于数据库中的每一个登录项,有与其它数据库的交叉索引,包括到GenBank、EMBL、DDBJ、GDB、MELINE等数据库的索引。PIR中一个具体的登录项如图4.4所示。
蛋白质结构解析的方法对比综述 工程硕士李瑾 摘要:到目前为止,蛋白质结构解析的方法主要是两种,x射线衍射法和NMR 法,这两种方法各有优点和不足。 关键词:x射线衍射法 NMR法 到目前为止,蛋白质结构解析的方法主要是两种,x射线衍射法和NMR法。其中X射线的方法产生的更早,也更加的成熟,解析的数量也更多,第一个解析的蛋白的结构,就是用x晶体衍射的方法解析的。而NMR方法则是在90年代才成熟并发展起来的。这两种方 [1]法各有优点和不足。 首先是X射线晶体衍射法。该方法的前提是要得到蛋白质的晶体。通常是将表达目的蛋白的基因经PCR扩增后克隆到一种表达载体中,然后转入大肠杆菌中诱导表达,目的蛋白提纯之后摸索结晶条件,等拿到晶体之后,将晶体进行x射线衍射,收集衍射图谱,通过 [2]一系列的计算,得到蛋白质的原子结构。 x射线晶体衍射法的优点是:速度快,通常只要拿到晶体,最快当天就能得出结构,另外不受肽链大小限制,无论是多大分子量的蛋白质或者RNA、DNA,甚至是结合多种小分子的复合体,只要能够结晶就能够得到其原子结构。所以x射线方法解析蛋白的关键是摸索蛋白结晶的条件。该方法得到的是蛋白质分子在晶体状态下的空间结构,这种结构与蛋白质分子在生物细胞内的本来结构有较大的差别。晶体中的蛋白质分子相互间是有规律地、紧密地排列在一起的,运动性较差;而自然界的生物细胞中的蛋白质分子则是处于一种溶液状态,周围是水分子和其他的生物分子,具有很好的运动性。而且,有些蛋白质只能稳定地存
[2]在于溶液状态,无法结晶。 核磁共振NMR(nuclear magnetic resonance)现象很早就被科研人员观察到了,但将这种方法用来解析蛋白质结构,却是近一二十年的事情。NMR法具体原理是对水溶液中的蛋白质样品测定一系列不同的二维核磁共振图谱,然后根据已确定的蛋白质分子的一级结构,通过对各种二维核磁共振图谱的比较和解析,在图谱上找到各个序列号氨基酸上的各种氢原子所对应的峰。有了这些被指认的峰,就可以根据这些峰在核磁共振谱图上所呈现的相互之 [3]间的关系得到它们所对应的氢原子之间的距离。可以想象,正是因为蛋白质分子具有空间结构,在序列上相差甚远的两个氨基酸有可能在空间距离上是很近的,它们所含的氢原子所 [4] 对应的NMR峰之间就会有相关信号出现。通常,如果两个氢原子之间距离小于0.5纳米的话,它们之间就会有相关信号出现。一个由几十个氨基酸残基组成的蛋白质分子可以得到几百个甚至几千个这样与距离有关的信号,按照信号的强弱把它们转换成对应的氢原子之间的距离,然后运用计算机程序根据所得到的距离条件模拟出该蛋白质分子的空间结构。该结构既要满足从核磁共振图谱上得到的所有距离条件,还要满足化学上有关原子与原子结合的 [4]一些基本限制条件,如原子间的化学键长、键角和原子半径等。 NMR解析蛋白结构常规步骤如下:首先通过基因工程的方法,得到提纯的目的蛋白,在蛋白质稳定的条件下,将未聚合,而且折叠良好的蛋白样品(通常是 1mM,3mM,500ul,PH6,7的PBS)装入核磁管中,放入核磁谱仪中,然后由写好的程序控制谱仪,发出一系 1313列的电磁波,激发蛋白中的H、N、C原子,等电磁波发射完毕,再收集受激发的原子所