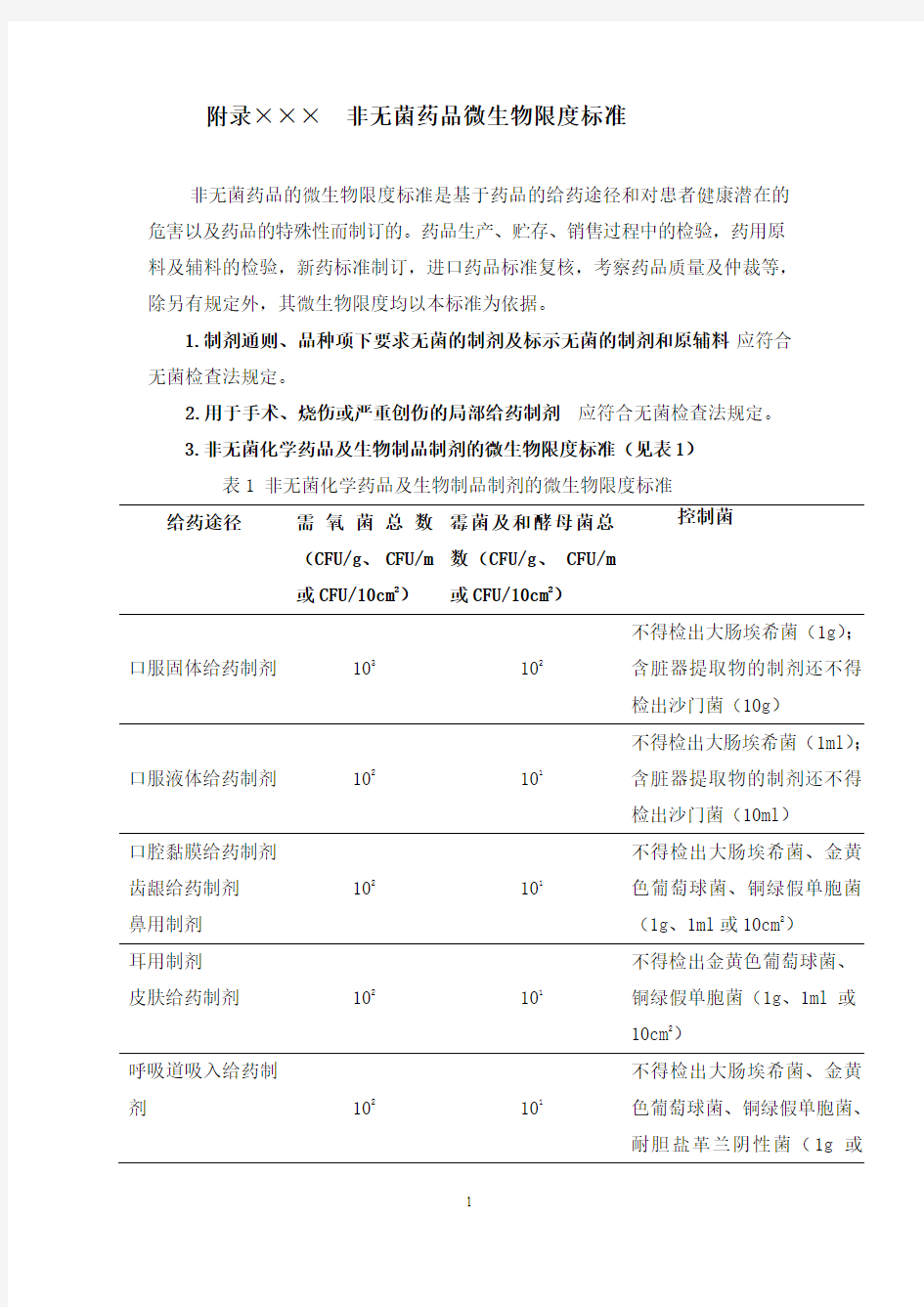

附录×××非无菌药品微生物限度标准
非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。药品生产、贮存、销售过程中的检验,药用原料及辅料的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的制剂及标示无菌的制剂和原辅料应符合无菌检查法规定。
2.用于手术、烧伤或严重创伤的局部给药制剂应符合无菌检查法规定。
3.非无菌化学药品及生物制品制剂的微生物限度标准(见表1)
表1 非无菌化学药品及生物制品制剂的微生物限度标准
给药途径需氧菌总数
(CFU/g、 CFU/m
或CFU/10cm2)霉菌及和酵母菌总
数(CFU/g、 CFU/m
或CFU/10cm2)
控制菌
口服固体给药制剂103102不得检出大肠埃希菌(1g);含脏器提取物的制剂还不得检出沙门菌(10g)
口服液体给药制剂102101不得检出大肠埃希菌(1ml);含脏器提取物的制剂还不得检出沙门菌(10ml)
口腔黏膜给药制剂
齿龈给药制剂鼻用制剂102101
不得检出大肠埃希菌、金黄
色葡萄球菌、铜绿假单胞菌
(1g、1ml或10cm2)
耳用制剂
皮肤给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)
呼吸道吸入给药制
剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌、耐胆盐革兰阴性菌(1g或
1ml)
阴道、尿道给药制
剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌(1g、1ml或10cm2)
直肠给药
固体制剂液体制剂103
102
102
102
不得检出金黄色葡萄球菌、
铜绿假单胞菌(1g或1ml)
其他局部给药制剂102102不得检出金黄色葡萄球菌、
铜绿假单胞菌(1g、1ml或
10cm2)
4. 非无菌的药用原料及辅料微生物限度标准(见表2)
表2 非无菌药用原料及辅料微生物限度标准
需氧菌总数(CFU/g 或 CFU/m)霉菌和酵母菌总
数(CFU/g或
CFU/mL)
控制菌
药用原料及辅料103102* 注*:限度未做统一规定。
5.有兼用途径的制剂应符合各给药途径的标准。
6.霉变、长螨者以不合格论。
非无菌药品的需氧菌总数、霉菌和酵母菌总数照“非无菌产品微生物限度检查:微生物计数法”(附录×××)检查;非无菌药品的控制菌照“非无菌产品微生物限度检查:控制菌检查法”(附录×××)检查。各品种项下规定的微生物限度标准解释如下:
101cfu:可接受的最大菌数为20;
102cfu:可接受的最大菌数为200;
103cfu:可接受的最大菌数为2000:依此类推。
本限度标准所列的控制菌对于控制某些药品的微生物质量可能并不全面,因此,对于原料、辅料及某些特定的制剂,根据原辅料及其制剂的特性和用途、制剂的生产工艺等因素,可能还需检查其他具有潜在危害的微生物。
除了本限度标准所列的控制菌外,药品中若检出其他可能具有潜在危害性的微生物,应从以下方面进行评估:
药品的给药途径:给药途径不同,其危害不同(眼、鼻或呼吸道给药);
药品的特性:药品是否促进微生物生长,或者药品是否有足够的抑制微生物生长能力;
药品的使用方法;
用药对象:用药对象不同,如新生儿、婴幼儿及体弱者,风险可能不同;
患者使用免疫抑制剂和甾体类固醇激素等药品的情况;
存在疾病、伤残和器官损伤,等等。
当进行上述相关因素的风险评估时,评估人员应经过微生物学和微生物数据分析等方面的专业知识培训。评估原辅料微生物质量时,应考虑相应制剂的生产工艺、现有的检测技术及原辅料符合该标准的必要性。
1.目的:建立非无菌药品微生物限度检查检验标准操作规程,规范检验操作,确保检验结果准确。 2.适用范围:适用于本公司所有采用非无菌药品微生物限度检查法测定的供试品。 3.责任者:QC检验员、QC经理。 4.正文: 4.1非无菌产品微生物限度检查:微生物计数法 4.1.1简述 微生物计数法系用于能在有氧条件下生长的嗜温细菌和真菌的计数。 当本法用于检查非无菌制剂及其原、辅料等是否符合规定的微生物限度标准时,应按下述规定进行检验,包括样品的取样量和结果的判断等。除另有规定外,本法不适用于活菌制剂的检查。 本检查法可采用替代的微生物检查法,包括自动检测方法,但必须证明替代方法等效于药典规定的检查方法。 微生物计数试验应在受控洁净环境下的局部洁净度不低于B级的单向流空气区域内进行。检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。单向流空气区域、工作台面及环境应定期进行监测。 如供试品有抗菌活性,应尽可能去除或中和。供试品检查时,若使用了中和剂或灭活剂,应确认其有效性及对微生物无毒性。 供试液制备时如果使用了表面活性剂,应确认其对微生物无毒性以及与所使用中和剂或灭活剂的相容性。 4.1.2计数方法
计数方法包括平皿法、薄膜过滤法和最可能数法(Most-Probable-NumberMethod,简称MPN 法)。MPN法用于微生物计数时精确度较差,但对于某些微生物污染量很小的供试品,MPN法可能是更适合的方法。 供试品检查时,应根据供试品理化特性和微生物限度标准等因素选择计数方法,所选的方法必须具备检测充足样品量的能力,以保证所获得的试验结果能够判断供试品是否符合规定。所选方法的适用性须经确认。 4.1.3计数培养基适用性检查和供试品计数方法适用性试验 供试品微生物计数中所使用的培养基应进行适用性检查。 供试品的微生物计数方法应进行方法适用性试验,以确认所采用的方法适合于该产品的微生物计数。 若检验程序或产品发生变化可能影响检验结果时,计数方法应重新进行适用性试验。 表1试验菌液的制备和使用
附录3: 生物制品 第一章范围 第一条生物制品的制备方法是控制产品质量的关键因素。采用下列制备方法的生物制品属本附录适用的范围: (一)微生物和细胞培养,包括DNA重组或杂交瘤技术; (二)生物组织提取; (三)通过胚胎或动物体内的活生物体繁殖。 第二条本附录所指生物制品包括:细菌类疫苗(含类毒素)、病毒类疫苗、抗毒素及抗血清、血液制品、细胞因子、生长因子、酶、按药品管理的体内及体外诊断制品,以及其它生物活性制剂,如毒素、抗原、变态反应原、单克隆抗体、抗原抗体复合物、免疫调节剂及微生态制剂等。 第三条生物制品的生产和质量控制应当符合本附录要求和国家相关规定。 第二章原则 第四条生物制品具有以下特殊性,应当对生物制品的生产过程和中间产品的检验进行特殊控制: (一)生物制品的生产涉及生物过程和生物材料,如细胞培养、活生物体材料提取等。这些生产过程存在固有的可变性,因而其副产物的范围和特性也存在可变性,甚至培养过程中所用的物料也是污染微生物生长的良好培养基。
(二)生物制品质量控制所使用的生物学分析技术通常比理化测定具有更大的可变性。 (三)为提高产品效价(免疫原性)或维持生物活性,常需在成品中加入佐剂或保护剂,致使部分检验项目不能在制成成品后进行。 第三章人员 第五条从事生物制品生产、质量保证、质量控制及其他相关人员(包括清洁、维修人员)均应根据其生产的制品和所从事的生产操作进行专业知识和安全防护要求的培训。 第六条生产管理负责人、质量管理负责人和质量受权人应当具有相应的专业知识(微生物学、生物学、免疫学、生物化学、生物制品学等),并能够在生产、质量管理中履行职责。 第七条应当对所生产品种的生物安全进行评估,根据评估结果,对生产、维修、检验、动物饲养的操作人员、管理人员接种相应的疫苗,并定期体检。 第八条患有传染病、皮肤病以及皮肤有伤口者、对产品质量和安全性有潜在不利影响的人员,均不得进入生产区进行操作或质量检验。 未经批准的人员不得进入生产操作区。 第九条从事卡介苗或结核菌素生产的人员应当定期进行肺部X 光透视或其它相关项目健康状况检查。 第十条生产期间,未采用规定的去污染措施,员工不得从接触活有机体或动物体的区域穿越到生产其它产品或处理不同有机体的区域中去。
微生物限度检查若干问题 微生物限度检查若干问题 微生物限度检查是对非规定灭菌制剂及其原、辅料受到微生物污染程度的一种检查方法,是药品微生物学检验的重要容之一。中国药典2000年版二部附录ⅪJ“微生物限度检查法”、《中国药品检验标准操作规》(2000年版)及参考书《药品微生物学检验手册》均做了详尽的阐述。下面对几个比较重要的问题,谈一点工作学习体会,仅供参考。 一、实验室要求 开展微生物限度检查工作,首先要按照《药品检验所实验室质量管理规》及《药品生产质量管理规》的要求,建立一个布局合理,使用方便,操作安全的实验室,并且配有完善的实验设施和管理制度。微生物限度检查、无菌检查、抗生素微生物检定实验室以及接种室(接种对照菌及菌种传代)均应严格分开。 (一)无菌室: 1结构与要求: 最好设在二楼以上(防潮、防霉、采光好),面积不超过10平方米,高度不超过24米,由2个缓冲间和操作间组成。缓冲间与操作间之间应有样品传递窗,出入操作间和缓冲间的门不应直对。无菌室应六面光滑平整,能耐受清洗消毒,墙壁与地面、墙壁与天花板连接处应呈凹弧形,无缝隙、无死角。 无菌室光照应不低于300勒克斯,缓冲间和操作间均应装有紫外线杀
菌灯2-25w/m3作空气消毒用。紫外线波长200-300nm者''具有杀菌作用,其中以265-266nm最强,这与DNA的吸收光谱围一致。其杀菌机理可能是在DNA中引起胸腺嘧啶双聚体形成,从而干扰DNA复制,导致细菌变异或死亡。紫外线杀菌灯1m以距离杀菌效果最佳''每次开灯照射时间为 20min。其缺点:穿透力弱,一纸可以挡住,不能穿透固体物,对人的皮肤眼睛有损伤,所以紫外线灯开关应装在无菌室外便于开关。 2温度、湿度:温度应控制在18-26℃''相对湿度40-60''操作间或净化工作台的洁净空气应保持对环境形成正比''不低于49Pa。 3操作间:应安装空气除菌过滤层流装置,操作间不应安装下水道,洁净度不低于1万级,净化工作台局部应为100级。操作间应准备电子称(感量01g''最大称量300g''乙醇灯''火柴''2碘酊及75乙醇棉球,大、小橡皮乳头,记号笔,灭菌的剪刀,镊子、注射器等。 4缓冲间:应有洗手盒、消毒液、无菌衣、帽、口罩,拖鞋等,不应放置培养箱和其他杂物。 (二)洁净级别及检查方法: 通常采用尘粒数及浮游菌数或沉降菌数测定法。 98年国家药监局颁布药品生产质量管理规''把药品生产洁净区空气洁 净度划分为四个级别: 空气洁净度级别表 ──────────────────────────── 尘埃数/立方米活微生物数个 洁净级别─────────────────────
GMP(2010年版)附录1 无菌药品考试题姓名:日期:分数: 一、填空题(每空1分,共45分) 1.无菌药品的生产须满足其质量和预定用途的要求,应当、和的污染。、及其是达到上述目标的关键因素。 2.无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当并监测压差。 3.洁净区的设计必须符合相应的要求,包括达到和的标准。 4.无菌药品生产所需的洁净区可分为那4个级别::A级区单向流系统在其工作区域必须均匀送风,风速为。 5.培养基模拟灌装试验通常应当按照生产工艺,每次至少。 6.评估无菌生产的微生物状况的监测方法有、和等。 7.无菌生产的隔离操作器所处的环境至少应为洁净区。 8.凡在洁净区工作的人员(包括清洁工和设备维修工)应当定期,使无菌药品的操作符合要求。培训的内容应当包括和方面的基础知识。未受培训的外部人员(如外部施工人员或维修人员)在生产期间需进入洁净区时,应当对他们进行特别详细的是。 9.从事无菌药品生产的员工应当随时报告任何可能的异常情况,包括污染的类型和程度。 10.工作服及其质量应当与及相适应,其式样和穿着方式应当能够满足保护产品和人员的要求。 11.A/B级洁净区的着装要求:应当用将所有头发以及胡须等相关部位全部遮盖,应当塞进衣领内,应当戴以防散发飞沫,必要时戴防护目镜。
12.个人外衣不得带入通向B级或C级洁净区的更衣室。每位员工每次进入A/B 级洁净区,应当。 13.无菌生产的洁净区内禁止设置水池和地漏。 14.气锁间两侧的门同时打开。 15.关键设备,如灭菌柜、空气净化系统和工艺用水系统等,应当经过,并进行计划性维护,经方可使用。 16.无菌原料药精制、无菌药品配制、直接接触药品的包装材料和器具等最终清洗、A/B级洁净区内消毒剂和清洁剂配制的用水应当符合的质量标准。 17.当无菌生产正在进行时,应当特别注意减少洁净区内的各种活动。应当减少人员,避免剧烈活动散发过多的和。 18.洁净区内应当避免使用的容器和物料;在无菌生产的过程中,不得使用此类容器和物料。 19.为确认A级洁净区的级别,悬浮粒子每个采样点的采样量不得少于。 20.在,连续或有规律地出现少量≥5.0 μm的悬浮粒子时,应当进行调查。 21.应当对进行动态监测,评估无菌生产的微生物状况。监测方法有、和等。动态取样应当避免对洁净区造成不良影响。 22.处于未完全密封(1)状态下产品的操作和转运,如产品灌装(或灌封)、分装、压塞、轧盖(2)等应当选择的操作环境。 23.A/B级洁净区应当使用消毒剂和清洁剂。 二、名词解释:(每题3分,共15分) 无菌药品: 单向流:
标准操作规程 目的:建立一个药品微生物限度检验标准操作规程。 范围:适用于本企业生产的所有品种,本企业所有洁净生产区域,QC微生物限度检查室,洁净工作室等。 责任者:QC主任、化验员。 规程: 本规程引至《中国药典》2000年版。 1. 概述:微生物限度检查系指非规定灭菌制剂及其原辅料受到微生物污染程度的一种检查方法,包括染菌量及控制菌的检查。我公司QC设无菌操作室,用于微生物限度检查。无菌操作室的管理及使用制度见本文附录一。 2. 抽样:供试品应按批号随即抽样,一般抽样量为检验用量(2个以上最小包装单位)的3倍。抽样时,凡发现有异常可疑的样品,应缺陷选用疑问的样品,但因机构损伤明显破裂的包装不得作为样品,凡已能从药品、瓶口(外盖内侧及瓶口周围)外观看出长螨、长霉、虫蛀及变质的药品,可直接判为不合格,无需要再抽样检验。 3. 供试品的保存:供试品在检验之前,应保存在阴凉干燥处,以防供试品中的污染菌因保藏条件所引起致死、损伤或繁殖。供试品在检验之前,应该保持原有包装状态,严禁开启,包装已开启的样品不得作为供试品。 4. 检查: 4.1. 使用设备:电热恒温培养箱、电热恒温水温箱、试管、刻度吸管、量筒、三角瓶、培养皿、试管架、注射器、针头、注射器盒、研钵、75%酒精棉球、紫外灯(365nm波长)。 4.2. 检查的全过程应严格遵守无菌操作,严防再污染。使用设备、仪器、人员及无菌操作室常用的消毒、灭菌方法见本文附录二。除另有规定外,供试品制备成供试液后,均在均匀状态取样。制成供试液后,应该在60分钟内注皿操作完毕。 标准操作规程
4.3. 培养:除另有规定外,本检查法中细菌培养温度为30-35℃,霉菌、酵母菌培养温度为25-28℃,控制菌培养温度为36±1℃,检验结果的报告以1g、1ml或10cm2为单位。 4.4. 复检 4.4.1. 菌数测定不合格者应复检,控制菌检查以一次检出为准,不再复试,但应保留检出菌株一个月备查。 4.4.2. 以复试项下不合格项目为准,作单项复试。复试需另取同批号样品,测定2次。 4.4.3. 复试报告,以3次测定结果的算术平均值报告。 4.5. 检验报告 4.5.1. 检验报告以1g、1ml或10cm2为单位。 4.5.2. 测定菌数报告,以每次测定结果全部平均值报告。 4.5.3. 控制菌按检验结果报告,如未检出控制菌时,报告为“按规定抽样检验结果未检出XX菌”,如抽样中任意一样品要检出控制菌时,报告为“按规定抽样,检出XX菌,不符合药品微生物限度标准。” 4.6. 培养基、试药及稀释剂的相关内容见本文件附录三。 4.7. 供试液的制备:按供试液的理化特性与生物学特性可采取适宜的方法制成供试液。 4.7.1. 供试品的取样及注意事项 供试品取样应有一定数量,以使检验结果具有代表性,正常的供试品一般每批应随机抽取两瓶或两盒以上的包装单位,检验时每次应分取两瓶(盒)以上的样品共10g或10ml。 供试品在检验前,应严格保持包装的原有状态,不得启开,并放在阴凉干燥处,防止微生物再繁殖,以免影响检验结果,凡已将原包装启开,则无代表性应另取样。 供试品稀释后须在1-2小时内操作完毕,防止微生物繁殖或死亡。 供试品稀释成供试液后,应在均匀状态下取样,凡因抑菌或不溶于水的剂型,其供试品应作特殊处理后进行检验。 4.7.2. 液体供试品:取供试品10ml,加入稀释剂90ml中,混匀,作为供试液。油剂可加适量 聚山梨酯80,混匀,吸取相当于10g或10ml供试品,标准操作规程 再稀释成100ml作为供试液;合剂(系指含王桨或蜂蜜者,下同)可用供试品作为供试液。 4.7.3. 固体、半固体或黏稠液供试品:称取供试品10g,置0.9%无菌氯化钠溶液100ml中,用匀浆仪或其他适宜的方法混匀后作为供试液。在制备过程中,必要时可加适量聚山梨酯80,并适当加温,但不应超过45℃。 (1)非水溶性供试品:取供试品5g(5ml)加入含溶化的无菌司盘80 5g、单硬脂酸甘油酯3g、聚山梨酯80 10g混合物的烧杯中,用无菌玻棒搅拌成团后,慢慢加入45℃左右的0.9%无菌氯化钠溶
无菌药品 第一章范围 第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药。 第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。 第二章原则 第三条无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染。生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验(包括无菌检查)。 第四条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。 第五条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。 第六条物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。 第七条应当根据产品特性、工艺和设备等因素,确定无菌药品生产用洁净区的级别。每一步生产操作的环境都应当达到适当的动态洁净度标准,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。 第三章洁净度级别及监测 第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。 第九条无菌药品生产所需的洁净区可分为以下4个级别: A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。应当有数据证明单向流的状态并经过验证。 在密闭的隔离操作器或手套箱内,可使用较低的风速。
2017年质量月GMP知识竞赛题库(生产部) 无菌药品 一、判断题 1、无菌药品在B级洁净区要采用与A级洁净区相同的监测系统。 (×)(无菌药品,在B级洁净区可采用与A级洁净区相似的监测系统) 2 、在灌装/分装时,由于产品本身产生粒子或液滴,允许灌装点≥5.0μm的悬浮粒子出现不符合标准的情况(√ ) 3、无菌药品生产,一般情况下,洗手设施应安装在更衣的任何阶段. (×)(无菌药品生产,一般情况下,洗手设施只能安装在更衣的第一阶段。) 4、无菌药品生产设备及辅助装置的设计和安装,应当尽可能便于在洁净区进行操作、保养和维修。 (×)(无菌药品生产设备及辅助装置的设计和安装,应当尽可能便于在洁净区外进行操作、保养和维修。) 5、在无菌生产的过程中,应当尽量避免使用易脱落纤维的容器和物料。 (×)(在无菌生产的过程中,不得使用易脱落纤维的容器和物料。) 6、流通蒸汽处理不属于最终灭菌. (√ ) 7、非最终灭菌产品直接接触药品的包装材料、器具灭菌后处于密闭容器内的转运和存放可以在C级区操作(×)(应在B级区) 8、非最终灭菌产品的轧盖操作可选择在C级或D级背景下的A级送风环境中进行,但A级送风环境至少符合A级区的静态要求。(√ ) 9、可以使用化学或生物指示剂监控灭菌工艺来替代物理测试。(×)(不能使用) 10、最终灭菌的产品可以用过滤除菌工艺替代最终灭菌工艺。(×)(不可以) 11、企业的厂房、设施、设备和检验仪器应当经过确认,应当采用经过验证的生产工艺、操作规程和检验方法进行生产、操作和检验,并保持持续的验证状态。(√ ) 12、制药用水应当适合其用途,并符合《中华人民共和国药典》的质量标准及相关要求。制药用水至少应当采用纯化水。(×) 13、每批药品的检验记录应当包括中间产品、待包装产品和成品的质量检验记录,可追溯该批药品所有相关的质量检验情况。(√ )
附录1: 无菌药品 第一章范围 第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药。 第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。 第二章原则 第三条无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染。生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验(包括无菌检查)。 第四条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。 第五条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。 第六条物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。 第七条应当根据产品特性、工艺和设备等因素,确定无菌药品生产用洁净区的级别。每一步生产操作的环境都应当达到适当的动态洁净
度标准,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。 第三章洁净度级别及监测 第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。 第九条无菌药品生产所需的洁净区可分为以下4个级别: A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。应当有数据证明单向流的状态并经过验证。 在密闭的隔离操作器或手套箱内,可使用较低的风速。 B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。 C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。 以上各级别空气悬浮粒子的标准规定如下表: 注: (1)为确认A级洁净区的级别,每个采样点的采样量不得少于1立方米。A级洁净区空气悬浮粒子的级别为ISO 4.8,以≥5.0μm的悬浮粒子为限度标准。B级洁净区(静态)的空气悬浮粒子的级别为ISO 5,同时包括表中两种粒径的悬浮粒子。对
药品微生物限度标准 王知坚
?我国药品微生物限度标准的历史沿革?药品微生物限度检查在制剂通则中的修订内容 ?药品微生物限度标准的修订内容 ?限度标准的注意事项 ?国外药典微生物限度标准的收载情况
历史沿革
?检查法的历史沿革 –国务院1973年121号文件标志着我国药品微生 物限度检查工作正式启动 –在1974年颁布了74版《卫生部药品卫生学检查法》,是首次颁布与药品微生物限度检查有关 的检查方法 –先后于84年和90年两次修订,颁布新的检查法–1995年版《中国药典》首次在附录中收载微生物限度检查法 –2000年版、2005年版和2010年版《中国药 典》先后收载该检查法,并进行了不同程度的 修订和完善
?限度标准的变迁 –1986年版《卫生部部颁药品微生物限度标准》?国内首次颁布与药品微生物质量有关的限度标准 ?限度标准的分类依据为剂型,不同剂型制订不同的 限度标准值;控制菌检查的分类依据为给药途径, 不同途径的制剂有不同的检查内容 –1989年版《卫生部部颁药品卫生补充规定》?是对86版限度标准的修订和补充 ?分类方式上与86版相同
–《中国药典》2000年版 ?首次将微生物限度标准收载入国家药典 ?在编制体例上仍延用部颁标准的做法,按剂型制订限度标准,按给药途径和剂型特点制订控制菌标准–《中国药典》2005年版 ?首次确定按给药途径来制订不同产品的限度标准和控制菌标准 ?首次在制剂通则中对某一类制剂制订较为特殊的微生物控制要求,如眼用液体制剂需要达到近似无菌的要求 ?在标准的具体规定上,体现了当时对微生物质量的研究水平,如含生药原粉的制剂要求开展大肠菌群检查,用于深部组织的制剂要求不得检出梭菌。
药品微生物限度检验方法标准操作规 程
标准操作规程 目的:建立一个药品微生物限度检验标准操作规程。 范围:适用于本企业生产的所有品种,本企业所有洁净生产区域,QC微生物限度检查室,洁净工作室等。 责任者:QC主任、化验员。 规程: 本规程引至《中国药典》。 1. 概述:微生物限度检查系指非规定灭菌制剂及其原辅料受到微生物污染程度的一种检查方法,包括染菌量及控制菌的检查。我公司QC设无菌操作室,用于微生物限度检查。无菌操作室的管理及使用制度见本文附录一。 2. 抽样:供试品应按批号随即抽样,一般抽样量为检验用量(2个以上最小包装单位)的3倍。抽样时,凡发现有异常可疑的样品,应缺陷选用疑问的样品,但因机构损伤明显破裂的包装不得作为样品,凡已能从药品、瓶口(外盖内侧及瓶口周围)外观看出长螨、长霉、虫蛀及变质的药
品,可直接判为不合格,无需要再抽样检验。 3. 供试品的保存:供试品在检验之前,应保存在阴凉干燥处,以防供试品中的污染菌因保藏条件所引起致死、损伤或繁殖。供试品在检验之前,应该保持原有包装状态,严禁开启,包装已开启的样品不得作为供试品。 4. 检查: 4.1. 使用设备:电热恒温培养箱、电热恒温水温箱、试管、刻度吸管、量筒、三角瓶、培养皿、试管架、注射器、针头、注射器盒、研钵、75%酒精棉球、紫外灯(365nm波长)。 4.2. 检查的全过程应严格遵守无菌操作,严防再污染。使用设备、仪器、人员及无菌操作室常见的消毒、灭菌方法见本文附录二。除另有规定外,供试品制备成供试液后,均在均匀状态取样。制成供试液后,应该在60分钟内注皿操作完毕。 标准操作规程 4.3. 培养:除另有规定外,本检查法中细菌培养温度为30-35℃,霉菌、酵母菌培养温度为25-28℃,控制菌培养温度为36±1℃,检验结果的报告以1g、1ml或10cm2为单位。 4.4. 复检
Appendix XVI D. Microbiological Quality of Non- sterile Pharmaceutical Preparations and Substances 非无菌制剂的微生物限度 (Ph. Eur. general text 5.1.4) The presence of certain micro-organisms in non-sterile preparations may have the potential to reduce or even inactivate the therapeutic activity of the product and has a potential to adversely affect the health of the patient. Manufacturers therefore have to ensure a low bioburden of finished dosage forms by implementing current guidelines on Good Manufacturing Practice during the manufacture, storage and distribution of pharmaceutical preparations. 非无菌制剂中存在一定量的微生物可能导致药物的治疗活性降低或消失,并对患者健康有着潜在的危害。因此生产商应在药品生产、储存、运输过程中实施GMP管理,使制剂的微生物负荷保持在低水平。 Microbial examination of non-sterile products is performed according to the methods given in general chapters 2.6.12 and 2.6.13. Acceptance criteria for non-sterile pharmaceutical products based upon the total aerobic microbial count (TAMC) and the total combined yeasts/moulds count (TYMC) are given in Tables 5.1.4.-1 and 5.1.4.-2. Acceptance criteria are based on individual results or on the average of replicate counts when replicate counts are performed (e.g. direct plating methods). 非无菌制剂的微生物检测按照通则2.6.12和2.6.13的方法执行。非无菌制剂的微生物限度基于表5.1.4-1和5.1.4-2的需氧微生物总数(TAMC)及酵母菌/霉菌总数(TYMC)。接受标准适用于单个检出结果或者做重复样品时结果的平均数(例如,直接平板法)。 When an acceptance criterion for microbiological quality is prescribed it is interpreted as follows: 给出的微生物质量接受标准解读如下: — 101 CFU: maximum acceptable count = 20; — 101 CFU: 最大可接受数= 20; — 102 CFU: maximum acceptable count = 200; — 102 CFU: 最大可接受数= 200; — 103 CFU: maximum acceptable count = 2000; — 103 CFU: 最大可接受数= 2000; Table 5.1.4.-1 includes a list of specified micro-organisms for which acceptance criteria are set. The list is not necessarily exhaustive and for a given preparation it may be necessary to test for other micro-organisms depending on the nature of the starting materials and the manufacturing process. 表5.1.4-1给出了各种剂型的具体微生物接受标准。此表格并不完全,根据起始物料和生产工艺的性质可能需要其他的微生物检查。 If it has been shown that none of the prescribed tests will allow valid enumeration of micro-organisms at the level prescribed, a validated method with a limit of detection as close as possible to the indicated acceptance criterion is used.
1. 目的:建立非无菌药品微生物限度检查检验标准操作规程,规范检验操作,确保检验结果准确。 2. 适用范围:适用于本公司所有采用非无菌药品微生物限度检查法测定的供试品。 3. 责任者:QC检验员、QC经理。 4. 正文: 4.1 非无菌产品微生物限度检查:微生物计数法 4.1.1 简述 微生物计数法系用于能在有氧条件下生长的嗜温细菌和真菌的计数。 当本法用于检查非无菌制剂及其原、辅料等是否符合规定的微生物限度标准时,应按下述规定进行检验,包括样品的取样量和结果的判断等。除另有规定外,本法不
适用于活菌制剂的检查。 本检查法可采用替代的微生物检查法,包括自动检测方法,但必须证明替代方法等效于药典规定的检查方法。 微生物计数试验应在受控洁净环境下的局部洁净度不低于B 级的单向流空气区域内进行。检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。单向流空气区域、工作台面及环境应定期进行监测。 如供试品有抗菌活性,应尽可能去除或中和。供试品检查时, 若使用了中和剂或灭活剂,应确认其有效性及对微生物无毒性。 供试液制备时如果使用了表面活性剂,应确认其对微生物无毒性以及与所使用中和剂或灭活剂的相容性。 4.1.2 计数方法 计数方法包括平皿法、薄膜过滤法和最可能数法(Most-Probable-NumberMethod,简称MPN 法)。MPN 法用于微生物计数时精确度较差,但对于某些微生物污染量很小的供试品,MPN 法可能是更适合的方法。 供试品检查时, 应根据供试品理化特性和微生物限度标准等因素选择计数方法,所选的方法必须具备检测充足样品量的能力,以保证所获得的试验结果能够判断供试品是否符合规定。所选方法的适用性须经确认。 4.1.3 计数培养基适用性检查和供试品计数方法适用性试验 供试品微生物计数中所使用的培养基应进行适用性检查。 供试品的微生物计数方法应进行方法适用性试验,以确认所采用的方法适合于该产品的微生物计数。 若检验程序或产品发生变化可能影响检验结果时,计数方法应重新进行适用性试
附录×××非无菌药品微生物限度检查指导原则 为更好应用非无菌产品微生物限度检查:微生物计数法(附录×××)、 非无菌产品微生物限度检查:控制菌检查法(附录×××)及非无菌药品 微生物限度标准(附录×××),特制定本指导原则。 非无菌药品中污染的某些微生物可能导致药物活性降低,甚至使药品丧失疗效,从而对患者健康造成潜在的危害。因此,在药品生产、贮藏和流通各个环节中,药品生产企业应严格遵循GMP的指导原则,以降低产品受微生物污染程度。非无菌产品微生物计数法、控制菌检查法及药品微生物限度标准可用于判断非规定无菌制剂及原料、辅料是否符合药典的规定,也可用于指导制剂、原料、辅料的微生物质量标准的制定,及指导生产过程中间产品微生物质量的监控。本指导原则将对标准和方法中的特定内容及标准的应用做进一步的说明。 1.非无菌药品微生物限度检查中,受控的洁净环境是指不低于GMP现行版要求的D级洁净环境。 2. 非无菌药品微生物限度检查过程中,如使用表面活性剂、灭活剂及中和剂,在确定其能否适用于所检样品及其用量时,除应证明该试剂对所检样品的处理有效外,还须确认该试剂不影响样品中可能污染的微生物的检出(即无毒性),因此无毒性确认试验的菌株不能仅局限于验证试验菌株,而应当包括产品中可能污染的微生物。 3.供试液制备方法、抑菌成分的消除方法及需氧菌总数、霉菌和酵母菌总数计数方法应尽量选择微生物计数方法中操作简便、快速的方法,同时,所选用的方法应避免损伤供试品中污染的微生物。对于抑菌作用较强的供试品,在供试品溶液性状允许的情况下,应尽量选用薄膜过滤法进行试验。 4.对照培养基系指按培养基处方特别制备、质量优良的培养基,用于培养基适用性检查,以保证药品微生物检验用培养基的质量。对照培养基由中国食品药品检定研究院研制及分发。 5.进行微生物计数方法适用性试验时,若因没有适宜的方法消除供试品中的抑菌作用而导致微生物回收的失败,应采用能使微生物生长的更高稀释级供试液进行方法适用性试验。此时更高稀释级供试液的确认要从低往高的稀释级进行,
附录1:无菌药品 第一章范围 第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括注射剂、眼用制剂、无菌软膏剂、无菌混悬剂等。 第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。 第三条悬浮粒子、浮游菌、沉降菌和表面微生物等测试方法应按照相关标准执行。 第二章原则 第四条无菌药品的生产须满足其质量和预定用途的要求,应最大限度降低微生物、各种微粒和热原的污染。生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验。 第五条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。 第六条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,如采用机械连续传输物料时,应采用正压气流保护并监测压差。物料准备、产品配制和灌装或分装等操作必须在洁净区内分区(室)进行。 第七条应按所需环境的特点确定无菌药品洁净生产区的级别。每一步生产操作的环境都应达到适当的动态洁净度标准,以尽可能降低产品或所处理的物料被微粒或微生物污染的风险。 第三章洁净度级别及监测 第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。 第九条无菌药品生产所需的洁净区可分为以下4个级别: A级
高风险操作区,如:灌装区、放置胶塞桶、敞口安瓿瓶、敞口西林瓶的区域及无菌装配或连接操作的区域。通常用层流操作台(罩)来维持该区的环境状态。层流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。应有数据证明层流的状态并须验证。 在密闭的隔离操作器或手套箱内,可使用单向流或较低的风速。 B级 指无菌配制和灌装等高风险操作A级区所处的背景区域。 C级和D级 指生产无菌药品过程中重要程度较低的洁净操作区。 以上各级别空气悬浮粒子的标准规定如下表: 注: (1) 为了确定A级区的级别,每个采样点的采样量不得少于1m3。A级区空气尘埃粒子的级别为ISO 4.8,以≥0.5μm的尘粒为限度标准。B级区(静态)的空气尘埃粒子的级别为ISO 5,同时包括表中两种粒径的尘粒。对于C级区(静态和动态)而言,空气尘埃粒子的级别分别为ISO 7和ISO 8。对于D级区(静态)空气尘埃粒子的级别为ISO 8。测试方法可参照ISO14644-1。 (2) 在确认级别时,应使用采样管较短的便携式尘埃粒子计数器,以避免在远程采样系统长的采样管中≥5.0μm尘粒的沉降。在单向流系统中,应采用等动力学的取样头。 (3) 可在常规操作、培养基模拟灌装过程中进行测试,证明达到了动态的级别,但培养基模拟试验要求在“最差状况”下进行动态测试。 第十条应对洁净区的悬浮粒子进行动态监测。
1、目得:建立非无菌药品微生物限度检查检验标准操作规程,规范检验操作,确保检验结果准确。 2、适用范围:适用于本公司所有采用非无菌药品微生物限度检查法测定得供试品. 3、责任者:QC检验员、QC经理。 4、正文: 4、1 非无菌产品微生物限度检查:微生物计数法 4.1.1简述 微生物计数法系用于能在有氧条件下生长得嗜温细菌与真菌得计数。
当本法用于检查非无菌制剂及其原、辅料等就是否符合规定得微生物限度标准时,应按下述规定进行检验,包括样品得取样量与结果得判断等。除另有规定外,本法不适用于活菌制剂得检查。 本检查法可采用替代得微生物检查法,包括自动检测方法,但必须证明替代方法等效于药典规定得检查方法。 微生物计数试验应在受控洁净环境下得局部洁净度不低于B 级得单向流空气区域内进行。检验全过程必须严格遵守无菌操作,防止再污染,防止污染得措施不得影响供试品中微生物得检出。单向流空气区域、工作台面及环境应定期进行监测。 如供试品有抗菌活性,应尽可能去除或中与。供试品检查时,若使用了中与剂或灭活剂,应确认其有效性及对微生物无毒性。 供试液制备时如果使用了表面活性剂,应确认其对微生物无毒性以及与所使用中与剂或灭活剂得相容性。 4.1。2计数方法 计数方法包括平皿法、薄膜过滤法与最可能数法(Most—Probable—Num berMethod,简称MPN 法)。MPN 法用于微生物计数时精确度较差,但对于某些微生物污染量很小得供试品,MPN 法可能就是更适合得方法。 供试品检查时, 应根据供试品理化特性与微生物限度标准等因素选择计数方法,所选得方法必须具备检测充足样品量得能力,以保证所获得得试验结果能够判断供试品就是否符合规定。所选方法得适用性须经确认。 4.1。3 计数培养基适用性检查与供试品计数方法适用性试验 供试品微生物计数中所使用得培养基应进行适用性检查。 供试品得微生物计数方法应进行方法适用性试验,以确认所采用得方法适合于该
药品微生物限度检查结果影响因素分析及控制 方法 文件管理序列号:[K8UY-K9IO69-O6M243-OL889-F88688]
药品微生物限度检查结果影响因素分析及控制方法 blueski推荐?[2010-3-4] 出处:来自网上 作者:房宇辉董艳丽 ? 摘要:微生物限度检查法系指非规定灭菌制剂及其原、辅料受到微生物污染程度的一种检查方法,包括染菌量及控制菌的检查,是对生产单位所用的药品原料、器具设备、工艺流程、生产环境和操作者的卫生状况进行卫生评价的综合依据。由于微生物限度检查程序繁琐,操作严格,检查周期长,干扰因素多等原因都会造成检验结果误差,这样难......微生物限度检查法系指非规定灭菌制剂及其原、辅料受到微生物污染程度的检查方法,包括染菌量及控制菌的检查,是对生产单位所用的药品原料、器具设备、工艺流程、生产环境和操作者的卫生状况进行卫生评价的综合依据。由于微生物限度检查程序繁琐,操作严格,检查周期长,干扰因素多等原因都会造成检验结果误差,样难以对生产做出系统、科学的管理。现就微生物检查时,容易引起误差的几方面原因以及控制方法分析如下。 1 实验者主观因素及操作技能中的误差
1.1 无菌观念淡薄一些操作者认为,微生物限度范围定得宽,多几个、少几个菌对结果影响不大,因此操作马虎、随意,这样容易造成供试品的再次污染以及已污染菌的繁殖及死亡,从而不能真实的反映供试品的染菌情况。因此每一个检验者应明确的了解,在检验过程中的每一个环节,每个空间,每个操作,每个工具及材料,没灭菌者都是带菌者,都会对检验结果产生不确定影响,因而应牢固的建立无菌观念。 1.2 操作技能中误差一些操作者在操作不熟练,及实验条件控制不严格,细节不够注意,每次操作前的消毒处理必须彻底,不得留有死角,对实验所用器具根据材料的性质进行相应的灭菌;开启供试品包装的工具,如剪刀、砂轮等也应消毒,实验用的器具如吸管应清洗干净,灭菌,放液时不得接触液面,每ml样品必须放尽,否则影响细菌记数的准确性;这些细小环节都有可能污染供试品的可能,使平皿染菌数增多,导致实验误差,所以应该对实验每个环节都要考虑,分析,根据本单位具体情况建立个系统、科学的操作规范(SOP),正确熟练的掌握无菌技术及操作规范是准确完成检验结果、得出可靠结论的基本条件。 2 操作环境不符合要求带来的误差 按无菌要求,无菌室应由无菌操作间和两个缓冲间组成,操作间与缓冲间之间应有样品传递箱,操作间和缓冲间的门不应直对;操作间内不应安装下水道,无菌室的照明灯应嵌装在天花板内,室内六壁应光滑平整,能耐受清洗消毒;无菌室的温湿度和洁净级别应准确无误,并有相应的检测设备,由于条件限制,一些单位的无菌室在设计上不符合要求,
非无菌药品微生物限度检验原始记录 样品编号:检验开始时间:年月日样品名称:检验完成时间:年月日检测项目:检测依据: □水溶性供试品:用胰酪大豆胨液体培养基制成1:10供试液,必要时用胰酪大豆胨液做稀释液进一步10倍系列稀释。也可用原液作为供试液。 □水溶性非油脂类供试品:用胰酪大豆胨液体培养基制成1:10供试液,分散力较差的供试品可在稀释液中加入表面活性剂,必要时用胰酪大豆胨液做稀释液进一步10倍系列稀释。也可用原液作为供试液。 □油脂类供试品:取供试品,加入无菌十四烷酸异丙酯使溶解制成1:10供试液,必要时用十四烷酸异丙酯进一步10倍系列稀释。 取供试样品10g或10ml;膜剂为100cm2,根据试验性质不同,按照上述方法进行处理,制成10倍系列稀释液。 一、需氧菌总数、霉菌和酵母菌总数:吸取1ml样品均液于无菌平皿内,每个稀释度做2个平皿。同时,分别吸取1ml空白稀释液加入两个无菌平皿内作空白对照,胰酪大豆胨琼脂培养基平板在30~35℃培养3~5天,观察计算需氧菌总数;沙氏葡萄糖琼脂平板在20~25℃培养5~7天,观察计算霉菌和酵母菌总数。 计算及结果:需氧菌总数(CFU/g,ml,cm2): 霉菌和酵母菌总数(CFU/g,ml,cm2): 电子天平编号:使用状况试验前:试验后: 培养箱编号:使用状况试验前:试验后: 检测人:校核人:审核人:
样品编号: 二、 注:+生长或有可疑菌落;-未生长或无可疑菌落。 电子天平编号:使用状况试验前:试验后:培养箱编号:使用状况试验前:试验后:检测人:校核人:审核人:
抗生素残留量检查原始记录 样品编号:检验开始时间:年月日样品名称:检验完成时间:年月日检测项目:检测依据: 取直径8cm的培养皿,注入融化的抗生素II号培养基15~20ml,使在碟底内均匀摊布,放置水平台上使凝固,作为底层。取抗生素II号培养基15~20ml置于1支50℃水浴预热的试管中,加入0.5%~1.5%(ml/ml)的菌悬液300μm混匀,取适量注入已铺至底层的培养皿中,放置水平台上,冷却后,在每个培养皿上等距离均匀放置钢管,于钢管中依次滴加供试品溶液、阴性对照溶液(磷酸盐缓冲液)及对照品溶液,培养皿置于37℃培养18~22h,进行结果判定。 “+”表示有抑菌圈,“-”表示无抑菌圈,“A”表示大抑菌圈,“B”表示小抑菌圈 电子天平编号:使用状况试验前:试验后: 培养箱编号:使用状况试验前:试验后: 检测人:校核人:审核人: