
药物化学---药物的化学结构与体内代谢转化
方浩
第一部分概述
对人体而言,绝大多数药物是一类生物异源物质(Xenobiotics)。当药物进入机体后,一方面药物对机体产生诸多生理药理作用,即治疗疾病;另一方面,机体也对药物产生作用,即对药物的吸收、分布,排泄和代谢。药物代谢既是药物在人体内发生的化学变化,也是人体对自身的一种保护机能。
药物代谢是指在酶的作用下将药物(通常是非极性分子)转变成极性分子,再通过人体的正常系统排出体外。药物代谢多使有效药物转变为低效或无效的代谢物,或由无效结构转变成有效结构。在这过程中,也有可能将药物转变成毒副作用较高的产物。因此,研究药物在体内代谢过程中发生的化学变化,更能阐明药理作用的特点、作用时程、结构转变以及产生毒性的原因。
药物代谢在创新药物发现和临床药物合理应用中具有重要的地位。通过对近十年来许多创新药物在临床失败的案例,科学家们发现与药物代谢有关的问题是创新药物临床研究失败的重要原因。因此当前进行创新药物研究的过程中,应当在候选药物研究阶段就重视考察其药物代谢的相关问题,并将候选药物的代谢问题作为评判其成药性的重要研究内容。在药理学和生物药剂学课程中,对于药物在体内发生的药物代谢转化反应和代谢产物讲述内容较少。因此我们将在药物化学的讲述中,重点从药物代谢酶角度入手,讨论药物在体内发生的生物转化,以帮助大家更好的认识药物在体内所反应的代谢反应以及其与药物发现和临床合理应用的关系。
药物的代谢通常分为两相:即第Ⅰ相生物转化(PhaseⅠ)和第Ⅱ相生物转化(PhaseⅡ)。第Ⅰ相主要是官能团化反应,包括对药物分子的氧化、还原、水解和羟化等,在药物分子中引入或使药物分子暴露出极性基团,如羟基、羧基、巯基和氨基等。第Ⅱ相又称为结合反应(Conjugation),将第Ⅰ相中药物产生的极性基团与体内的内源性成分,如葡萄糖醛酸、硫酸、甘氨酸或谷胱甘肽,经共价键结合,生成极性大、易溶于水和易排出体外的结合物。但是也有药物经第Ⅰ相反应后,无需进行第Ⅱ相的结合反应,即可排出体外。
第二部分基本概念、基本知识及重点、难点
一、药物代谢的酶(Enzymes for Drug Metabolism)
第Ⅰ相生物转化是官能团化反应,是在体内多种酶系的催化下,对药物分子引入新的官能团或改变原有的官能团的过程。参与药物体内生物转化的酶类主要是氧化—还原酶和水解酶。本节主要介绍细胞色素P-450酶系、还原酶系、过氧化物酶和其它单加氧酶、水解酶。
(一)细胞色素P-450酶系
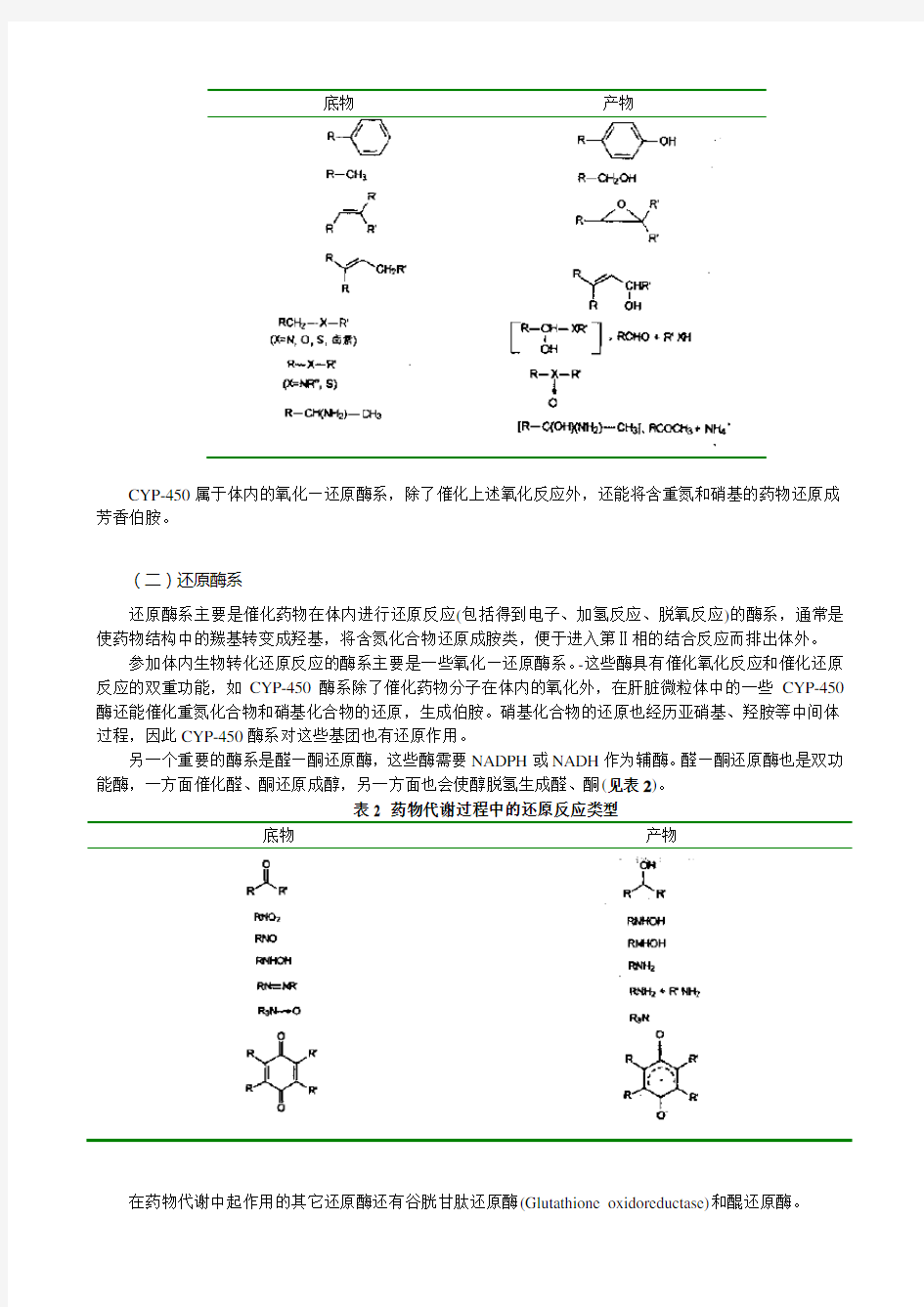
CYP-450(Cytochrome P-450 enzyme system,CYP-450)是一组酶的总称,由许多同功酶和亚型酶组成,是主要的药物代谢酶系,在药物和其它化学物质的代谢、去毒性中起着非常重要的作用。CYP-450存在于肝脏及其它肝脏外组织的内质网中,是一组由铁原卟啉偶联单加氧酶(Heme-coupled monooxygenases)、需要NADPH和分子氧共同参与、主要催化药物生物转化中氧化反应(包括失去电子、脱氢反应和氧化反应)的酶系。它主要是通过“活化”分子氧,使其中一个氧原子和有机物分子结合,同时将另一个氧原子还原成水,从而在有机药物的分子中引入氧。CYP-450催化的反应类型有烷烃和芳香化合物的氧化反应,烯烃、多核芳烃及卤代苯的环氧化反应,仲胺、叔胺及醚的脱烷基反应,胺类化合物的脱胺反应,将胺转化为N-氧化物、羟胺及亚硝基化合物以及卤代烃的脱卤反应。CYP-450还催化有机硫代磷酸酯的氧化裂解,氧化硫醚成亚砜等的反应(见表1)。
表1 CYP-450催化的一些药物代谢的氧化反应类型
底物产物
CYP-450属于体内的氧化—还原酶系,除了催化上述氧化反应外,还能将含重氮和硝基的药物还原成芳香伯胺。
(二)还原酶系
还原酶系主要是催化药物在体内进行还原反应(包括得到电子、加氢反应、脱氧反应)的酶系,通常是使药物结构中的羰基转变成羟基,将含氮化合物还原成胺类,便于进入第Ⅱ相的结合反应而排出体外。
参加体内生物转化还原反应的酶系主要是一些氧化—还原酶系。-这些酶具有催化氧化反应和催化还原反应的双重功能,如CYP-450酶系除了催化药物分子在体内的氧化外,在肝脏微粒体中的一些CYP-450酶还能催化重氮化合物和硝基化合物的还原,生成伯胺。硝基化合物的还原也经历亚硝基、羟胺等中间体过程,因此CYP-450酶系对这些基团也有还原作用。
另一个重要的酶系是醛—酮还原酶,这些酶需要NADPH或NADH作为辅酶。醛—酮还原酶也是双功能酶,一方面催化醛、酮还原成醇,另一方面也会使醇脱氢生成醛、酮(见表2)。
表2 药物代谢过程中的还原反应类型
底物产物
在药物代谢中起作用的其它还原酶还有谷胱甘肽还原酶(Glutathione oxidoreductase)和醌还原酶。
(三)过氧化物酶和其它单加氧酶
过氧化物酶属于血红素蛋白,是和CYP-450单加氧酶最为类似的一种酶。这类酶以过氧化物作为氧的来源,在酶的作用下进行电子转移,通常是对杂原子进行氧化(如N-脱烃基化反应)和1,4—二氢吡啶的芳构化。其它的过氧化酶还有前列腺素—内过氧化物合成酶、过氧化氢酶及髓过氧物酶。
单加氧酶中除了CYP-450酶系外,还有黄素单加氧酶(Flavin monooxygenase,FMO)和多巴胺β-羟化酶(Dopamine β-hydroxylase)。FMO和CYP-450酶系一起共同催化药物分子在体内的氧化,但FMO通常催化含N和S杂原子的氧化,而不发生杂原子的脱烷基化反应,如将叔胺、肼类化合物氧化成N-氧化物,仲胺氧化成羟基胺,羟胺氧化成硝基化合物,硫醇氧化成二硫醚,二硫醚氧化生成S—氧化物,硫醚氧化成亚砜和砜(见表3)。
表3 黄素单加氧酶催化药物代谢的氧化反应
底物产物
(四)水解酶
水解酶主要参与羧酸酯和酰胺类药物的代谢,这些非特定的水解酶大多存在于血浆、肝、肾和肠中。因此,大部分酯和酰胺类药物在这些部位发生水解。哺乳类动物的组织中也含有这些水解酶,使药物发生水解代谢。但是肝脏、消化道及血液具有更大的水解能力。
酯水解酶包括酯酶,胆碱酯酶及许多丝氨酸内肽酯酶。其它如芳磺酸酯酶、芳基磷酸二酯酶、β—葡萄糖苷酸酶和环氧化物酶(Epoxide hydrolase)等和酯水解酶的作用相似。
通常酰胺类化合物比酯类化合物稳定而难水解,水解速度较慢,因此大部分酰胺类药物是以原型从尿中排出。
二、第Ⅰ相的生物转化PhaseⅠBiotransformation
药物的第Ⅰ相生物转化是指体内各种酶对药物分子进行的官能团化反应,主要发生在药物分子的官能团上,或分子结构中活性较高、位阻较小的部位,包括引入新的官能团及改变原有的官能团。本节讲授的主要内容包括氧化反应、还原反应、脱卤素反应和水解反应。
(一)氧化反应
药物代谢中的氧化反应包括失去电子、氧化反应、脱氢反应等,是在CYP-450酶系、单加氧酶、过氧化酶等酶的催化下进行的反应。我们结合有关药物在体内的生物转化,探讨一下这些氧化反应的有关规律。
1.芳环及碳—碳不饱和键的氧化
(1)含芳环药物氧化代谢反应
下面是双氯酚酸钠在体内的代谢转化,我们可以看到其代谢产物主要以两个芳环引入酚羟基为主。
根据有关的研究发现,含芳环药物在体内被氧化为酚羟基的原理是CYP-450酶系首先将其氧化为环氧化合物。由于环氧化合物比较活泼,在质子的催化下会发生重排生成酚,或被环氧化物酶水解生成二羟基化合物。
芳环药物的氧化代谢以生成酚的代谢产物为主,一般遵照芳环亲电取代反应的原理,供电子取代基能
使反应容易进行,生成酚羟基的位置在取代基的对位或邻位;吸电子取代基则削弱反应的进行程度,生成酚羟基的位置在取代基的间位。
如果药物分子中含有二个芳环时,一般只有一个芳环发生氧化代谢。若二个芳环上取代基不同时,一般的是电子云较丰富的芳环易被氧化。如抗精神病药氯丙嗪(Chlorpromazine)易氧化生成7-羟基化合物,而含氯原子的苯环则不易被氧化。
(2)含烯烃和炔烃药物的代谢
由于烯烃化合物比芳香烃的π键活性较大,因此烯烃化合物也会被代谢生成环氧化合物。这些环氧化合物比较稳定,常常可以被分离出及确定其性质。例如抗癫痫药物卡马西平(Carbamazepine)在体内代谢生成10,11—环氧化合物,是卡马西平产生抗癫痫作用的活性成分。该环氧化合物会经进一步代谢,被环氧化物酶水解产生卡马西平的二羟基化合物,并随尿液排出体外。
烯烃类药物经代谢生成环氧化合物后,可以被转化为二羟基化合物,或将体内生物大分子如蛋白质、核酸等烷基化而产生毒性,导致组织坏死和致癌作用。例如黄曲霉素B1(Aflatoxin)经代谢后生成环氧化合物,该环氧化合物会进一步与DNA作用生成共价键化合物,是该化合物致癌的分子机理。
炔烃类反应活性比烯烃大,被酶催化氧化速度也比烯烃快。根据酶进攻炔键碳原子的不同,生成的产物也不同。若酶和氧连接在炔键的碳原子是端基碳原子,则随后发生氢原子的迁移,形成烯酮中间体,该烯酮可能被水解生成羧酸,也可能和蛋白质进行亲核性烷基化反应;若酶和氧连接在非端基炔键碳原子上,则炔烃化合物和酶中卟啉上的吡咯氮原子发生N-烷基化反应。这种反应使酶不可逆的去活化。如甾体化合物炔雌醇就会发生这类酶去活化反应。
2.饱和碳原子的氧化
(1)含脂环和非脂环结构药物的氧化
烷烃类药物经CYP-450酶系氧化后先生成含自由基的中间体,再经转化生成羟基化合物,酶在催化时具有区域选择性,这种选择性取决于被氧化碳原子附近的取代情况。生成的羟基化合物有可能一步脱水生成烯烃化合物。
长碳链的烷烃常在碳链末端甲基上氧化生成羟基,羟基化合物可被脱氢酶进一步氧化生成羧基称为ω—氧化;氧化还会发生在碳链末端倒数第二位碳原子上,称ω—1氧化。烷烃化合物除了ω—和ω—1氧化外,还会在有支链的碳康子上发生氧化,主要生成羟基化合物。如布洛芬(Ibuprofen)的氧化,其氧化主要发生在异丙基侧链上,有的氧化发生生末端甲基(ω—氧化),有的代谢产物属于ω—1氧化。部分ω-氧化产物进一步氧化为羧酸。
饱和的脂环容易发生氧化生成羟基化合物,如四氢萘的氧化主要是发生在脂肪环上,而不是在芳香环上。
取代的环己基药物在氧化代谢时,一般是环己基的C3或C4上氧化生成羟基化合物,并有顺、反式立体异构体。如降血糖格列本脲(Glibenclamide),其反式-4羟基代谢产物是主要代谢产物,顺式-3羟基代谢产物仅占15% 。
(2)和sp2碳原子相邻碳原子的氧化
当烷基碳原子和sp2碳原子相邻时,如羰基的 -碳原子、芳环的苄位碳原子及双键的α碳原子,由于受到sp2碳原子的作用使其反应活性增强,在CYP-450酶系的催化下,易发生氧化生成羟基化合物。
处于芳环和芳杂环的苄位,以及烯丙位的碳原子易被氧化生成苄醇或烯丙醇。对于伯醇会进一步氧化生成羧酸;仲醇会进一步氧化生成酮。如降酯药物吉非罗齐(Gemfibrozil)的代谢,先生成苄醇,最后形成羧酸。
(3)其它结构类型碳原子的氧化
非芳香杂环化合物通常是在和杂原子相邻的碳原子上发生氧化,先生成羟基化合物,再进一步氧化可
生成羰基化合物。如止吐药阿瑞匹坦(Aprepitant)发生N-脱烷基化后的产物,将进一步被氧化代谢为酰胺类化合物。
烷基氰化合物在CYP-450的作用下,经氧化代谢后会生成具有毒性的氰化物。氧化部位在氰基的α-缺电子的碳原子上,形成不稳定的氰醇,该化合物不稳定易分解生成氰化物和醛。
3.含氮化合物的氧化
含氮药物的氧化代谢主要发生在两个部位:一是在和氮原子相连接的碳原子上发生N-脱烷基化和脱氮反应,另一是发生N-氧化反应。
(1)N-脱烷基化和脱胺反应
N-脱烷基和氧化脱胺是一个氧化过程的两个不同方面,本质上都是碳-氮键的断裂,条件是与氮原子相连的烷基碳原子上应有氢原子(即α-氢原子)。该反应是在CYP-450酶的作用下,α-氢原子被氧化成羟基,生成的α-羟基胺是不稳定的中间体,会发生自动裂解。
胺类药物的N-脱烷基代谢是这类药物的主要的和重要的代谢途径之一。叔胺和仲胺氧化代谢后产生两种以上产物,而伯胺代谢后,只有一种产物。
胺类化合物氧化N-脱烷基化的基团通常是甲基、乙基、丙基、异丙基、丁基、烯丙基和苄基,以及其它α氢原子的基团。取代基的体积越小,越容易脱去。对于叔胺和仲胺化合物,叔胺的脱烷基化反应速度比仲胺快,这与它们之间的脂溶性有关。如利多卡因(Lidocaine)的代谢,脱第一个乙基比脱第二个乙基容易。
N-脱烷基后代谢产物极性加大,亲水性增加,因此扩散通过细胞膜的速度降低,和受体的作用减小,药物的生物活性下降。利多卡因进人中枢神经系统后产生的代谢产物由于难以扩散通过血—脑屏障,而产生中枢神经系统的毒副作用。
胺类药物代谢脱N-烷基化后,通常会产生活性更强的药物,如三环类抗郁抑药物丙米嗪(Imipramine)经脱N-甲基代谢生成地昔帕明(Desipramine),它也具有抗郁抑活性;或产生毒副作用,如上述的利多卡因的代谢以及N-异丙甲氧明经脱N-烷基后生成甲氧明(Methoxamine),会引起血压升高,临床上用于升高血压。
(2)N-氧化反应
一般来说,胺类药物在体内经氧化代谢生成稳定的N-氧化物主要是叔胺和含氮芳杂环,而伯胺和仲胺类药物的这种代谢通常比较少。伯胺和仲胺结构中如果无 -氢原子,则氧化代谢生成羟基胺、亚硝基或硝基化合物。酰胺类化合物的氧化代谢也与之相似。
参与N-氧化的酶类有黄素单加氧酶,CYP-450酶系及单胺氧化酶(MAO)。胺类的N-氧化反应是可逆反应,在CYP-450酶系和其它还原酶的作用下,氧化生成的N-氧化物又能脱氧还原成胺。
叔胺经N-氧化后,生成化学性质较稳定的N-氧化物,不再进一步发生氧化反应,如抗高血压胍乙啶(Guanethidine),在环上的叔胺氮原子氧化生成N-氧化物。
4.含氧化合物的氧化
主要是醚类药物在微粒体混合功能酶的催化下,发生O-去烃反应(O-脱烷基化)。
反应机制与N-去烃相同:氧化部位在氧原子的 -碳上,首先进行羟化,然后C-O键断裂,脱去烃基,生成醇或酚以及羰基化合物。O-去烃反应在药物中常见于芳醚类。例如:可待因在体内发生O-脱甲基反应生成吗啡。普萘洛尔在体内发生O去烷基反应而得到萘酚。
影响O-去烃反应速度的因素:烷基链的长度:链越长,烷基链越不容易脱去;烷基链的分支:分支越多,烷基链越不易脱除。药物分子如有多个醚基,发生O-脱烷基的位置与空间效应、电子效应、环上取代基等因素有关。
5.含硫化合物的氧化
含硫原子药物的氧化代谢反应主要有:S-去烃,氧化脱硫,S-氧化
(1)S-去烃:芳香族甲硫醚或脂肪族硫醚在CYP-450酶系催化下发生S-去烃反应,生成巯基和羰基化合物。反应机理同N-去烃、O-去烃相同。例如6-甲巯嘌呤的氧化脱甲基生成巯嘌呤。
(2)氧化脱硫:药物的氧化脱硫是指分子中的C=S、P=S双键被氧化生成C=O、P=O双键。
反应原理:C=S双键被单加氧酶催化氧化生成不稳定的硫单氧化物,进一步转化成硫双氧化物,很容易脱硫生成羰基化合物。例如:抗肿瘤药物塞替哌在体内经过脱硫反应,得到替哌(Tepa)而发挥生物烷化剂的作用。因此塞替哌属于前药。
(3)S-氧化反应:硫醚类药物可被氧化去烃,还可发生S-氧化反应。
6.醇和醛的氧化
含醇羟基药物在醇脱氢酶LAD作用下,脱氢氧化生成羰基化合物。脱氢酶LAD专一地需要辅酶ⅠNAD+作为受氢体。伯醇很容易氧化生成醛,体内醛不稳定,在体内进一步被氧化成羧酸。醇脱氢酶LAD 是双功能酶,既能氧化伯醇成醛,又能催化醛成醇。
酒精中毒主要就是因为乙醇氧化生成的乙醛在体内蓄积,乙醛和体内的生物大分子酶或蛋白质发生加成反应,导致细胞毒性。乙醇的氧化是体内代谢的主要途径。乙醇氧化先生成乙醛,进一步生成乙酸,乙酸最终被排除体外。
(二)还原反应
1.羰基的还原
(1)酮羰基在体内经酮还原酶的作用还原成仲醇。例如,美沙酮在体内被还原为美沙醇。
(2)酶催化还原的立体专一性;不对称酮羰基还原产生手性羟基,主要为S-构型
降血糖药醋磺己脲经代谢后生成S-(-)为主。
(3)酶催化还原在药物立体异构体间具立体选择性;
抗凝血药华法林有一手性中心,还原反应主要是R-(+)-异构体。
2.硝基的还原
芳香族硝基在代谢还原过程中,在CYP450酶系消化道细菌消极还原酶的催化下,还原生成芳香氨基。例如,氯霉素在体内经代谢,分子结构中的硝基被还原为芳伯胺。
还原是个多步骤过程,一般要经历亚硝基、羟胺等中间步骤。由于羟胺类化合物有毒,因此长期接触硝基苯化合物所引起的中毒,主要是由体内代谢还原的羟胺所致。另外,由于硝基还原成亚硝基是一个厌氧过程,氧气的存在会抑制不良反应。
3.偶氮基的还原
偶氮基的还原在很多方面与硝基还原相似,偶氮键先被还原为氢化偶氮键,最后断裂形成两个氨基。
通常,参与偶氮基团还原的代谢酶包括:CYP-450酶系、NADPH-CYP-450还原酶和消化道细菌还原酶。含有偶氮基的芳香族药物,一般被还原的通式为:
Ar—N=N-Ar’→Ar—NH2 + Ar’-NH2
由于偶氮键断裂还原也是一个厌氧过程,因此氧气的存在会抑制还原反应。
(三)脱卤素反应
卤代烃的体内代谢途径:一部分和谷胱甘肽或硫醚氨酸形成结合物排出体外,其余的在体内经过氧化脱卤素和还原脱卤素反应进行代谢。
例如:氯霉素在体内经氧化脱氯,结构中的二氯乙酰基被氧化为酰氯,易与体内蛋白质发生偶联,导致产生毒性。
(四)水解反应
水解反应是脂类和酰胺类药物在体内代谢的主要途径。主要由体内的酯酶和酰胺酶完成。能够通过水
解反应被代谢的药物结构类型包括:羧酸酯、硝酸酯、磺酸酯、磺胺类
影响水解反应的因素:
1.立体位阻
通常立体位阻越大,药物在体内的水解速度慢,甚至不水解。例如利多卡因结构中,苯环结构中存在的2,6-二甲基对邻近的酰胺键产生极大的立体位阻,提高了该药抗酶水解的能力。
2.酰胺较酯稳定,水解速度慢。例如:普鲁卡因属于羧酸酯结构,而普鲁卡因胺是酰胺结构。前者在体内基本水解,而后者在体内代谢后仍有60%以原形的形式存在。
3.水解选择性:母核结构选择性——如对脂肪族酯与芳香族酯的选择性水解
人体肝脏酶体内水解——水解酯环羧酸酯
人体肝脏酶体外水解——水解芳香羧酸酯
三第II相的生物转化(Phase II Biotransformation)
第Ⅱ相生物转化(结合反应)是在酶的催化下,将内源性的小分子如葡萄糖醛酸、硫酸、氨基酸、谷胱甘肽等结合到药物分子中或第Ⅰ相的药物代谢产物中。
第Ⅱ相生物转化的作用是导致药物去活化,另外还可以进一步生成水溶性的代谢物以利于药物从体内排出。药物的Ⅱ相生物转化(结合反应)的过程一般包括以下两步:首先内源性的小分子经活化成为其活化形式,第二步是药物或Ⅰ相药物代谢产物与内源性小分子活化形式结合。
药物Ⅱ相生物转化的主要类型有葡萄糖醛酸的结合、硫酸酯化结合、氨基酸结合、谷胱甘肽结合和乙酰化结合。下面分别概述如下:
(一)葡萄糖醛酸的结合——生成葡萄糖苷酸
葡萄糖醛酸的结合是药物代谢中最常见的一类结合反应。
葡萄糖苷酸的生成:
1.形成活化型尿苷-5-二磷酸-α-D-葡萄糖醛酸(UDPGA)
葡萄糖醛酸以α-糖苷键与尿苷二磷酸相连,形成活化型的尿苷二磷酸葡萄糖醛酸(UDPGA)
2.药物与活化型尿苷二磷酸葡萄糖醛酸(UDPGA)缩合生成葡萄糖苷酸。葡萄糖醛酸以β-糖苷键和药物相连。
葡萄糖醛酸的结合反应类型:
O-葡萄糖醛酸苷化——具有羟基和羧基的药物可发生
N-葡萄糖醛酸苷化——具氨基、酰胺基、磺酰胺基的药物可发生
S-葡萄糖醛酸苷化——具硫醇、硫代羧酸的药物可发生
C-葡萄糖醛酸苷化——具活泼C的药物(1,3二羰基结构的)的药物可发生
(二)硫酸酯化结合
在磺基转移酶的催化下,具有羟基、氨基、羟胺基的药物能和体内的活化型硫酸化剂提供的硫酸基酯化,形成硫酸化结合产物。
硫酸酯化反应类型:
O-硫酸酯化——具有羟基、羟胺基的药物
N-硫酸酯化——具有氨基的药物
(三)氨基酸结合
在辅酶A参与下,形成活化型酸,再与氨基酸(甘氨酸最常见)结合,形成氨基酸结合产物。参加氨基酸结合反应的药物类型主要是取代的苯甲酸,如水杨酸,生成物为酰化甘氨酸。
(四)谷胱甘肽结合
谷胱甘肽是由甘氨酸、谷氨酸和半胱氨酸组成的三肽化合物,具有硫醇基。硫醇基有较强的亲核作用,能和体内代谢产生的亲电性物质结合,从而使蛋白质的亲核中心免遭亲电性物质的毒害。
谷胱甘肽结合反应的类型:1、亲核取代反应(SN2)2、Micheal加成3、还原反应
(五)乙酰化结合
含伯胺基、氨基酸、磺酰胺,肼,酰肼等基团的药物在体内酰基转移酶的催化下,以乙酰辅酶A为辅酶,进行氨基乙酰化的反应。
(六)甲基化结合
甲基化反应在药物代谢反应中较为少见。甲基化结合实在甲基转移酶的作用下,以S-腺苷-L-甲硫氨酸(SAM)为辅酶进行的反应。
甲基化结合的作用:调节生物活性胺、蛋白质、核酸的生物活性——降低外来的内源性物质的生物活性;叔胺特殊:叔胺化合物甲基化后生成季铵盐,利于排泄
四药物代谢反应在新药研究中的重要作用
(一)寻找和发现新药
1.寻找和发现新的先导化合物
百浪多息最初发现时,在体外无效。经研究发现,其在体转变为磺胺而发挥药效,从而开启了磺胺类药物大发展的时代。因此从代谢产物中寻找新的先导化合物是开发新药的一个重要途径。许多药物上市后通过研究其代谢产物,发现某些代谢产物的活性比原型药物更高,从而促进了新药的诞生。例如保泰松作为非甾体抗炎药,其代谢产物羟布宗的活性比保泰松更高,已成为临床应用的有效药物之一。
2.先导化合物的结构修饰
利用药物代谢知识进行先导化合物的结构修饰,是进行新药发现的重要途径。例如:根据乙酰胆碱在体内易被乙酰胆碱酯酶水解的有关性质,以乙酰胆碱为基本骨架,通过引入甲基并将酯键改为氨基甲酸酯形式得到氯贝胆碱。
另一方面,利用酯和酰胺体内水解的性质,可以对活性分子进行修饰得到酯类或酰胺类的前药。这类药物在体外不具有生物活性,但在体内经代谢可转化为有生物活性的物质,从而起到治疗作用。有关前药的概念在过去教材中已多次出现,这里就不展开论述。
3.对新药研究的指导作用
根据药物代谢反应的一般规律,在新药研究开发的早期阶段,可以初步估计可能的代谢产物结构类型。然后应用动物模型进行药物的早期代谢产物的监测。对于创新药物而言,一般应用LC-MS进行代谢产物的监测和分析。针对液相给出的峰,通过LC-MS可以初步确定代谢产物结构类型。
根据可能出现的代谢产物结构类型,可以制备少量可能存在的代谢产物,以便进一步对相关代谢产物进行确证用监测。
(二)优化药物的动力学性质
1.缩短药物的作用时间
在某些药物结构中引入一些易代谢的基团,可以缩短药物的体内作用时间,避免药物的副作用。
2.延长药物的作用时间
根据药物的代谢稳定性以及其可能的代谢位点,通过结构改造限制化合物与代谢酶结合、降低代谢位点的反应性等结构修饰策略进一步提高化合物的代谢稳定性。常用的方法有:
(1)引入不易代谢的原子或基团
丁螺环酮(buspirone)是一新型的抗焦虑药物,由于其嘧啶环的5位极易被CYP3A4羟基化而缩短了其体内半衰期。将此位点用氟原子取代,阻断了该位点的代谢,从而显著提高其代谢稳定性,半衰期提高了近10倍。
(2)增加易代谢片段的位阻
CYP2D6的去甲基化反应是影响许多药物代谢稳定的重要因素之一。将环丙亚甲基替换美多洛尔的甲基得到了倍他洛尔,其在人体内的半衰期从前者的3.5~6小时延长至16~22小时。
(三)指导合理设计药物剂型
1.指导设计适当的药物剂型
药物必须制成制剂才能用于临床,如片剂、胶囊剂、针剂、口服液等都属药物的不同剂型。
(1)考虑药物体内的稳定性(理化性质)
(2)考虑药物的首过效应的大小
首过效应:口服药物在其到达作用部位之前,要经过胃肠道的消化酶和胃肠道壁及肝脏中的药物代谢酶的作用,即“首过效应”首过效应大的药物往往丧失活性。
如硝酸甘油:抗心绞痛药物,舌下给药,直接通过口腔黏膜进入血液,而不是口服通过胃肠道,继而
由肝门静脉进入血液。原因:首过效应大。
2.提高生物利用度
体内易代谢的药物,其生物利用度较低。
那么改变药物中易代谢的基团,将提高药物的生物利用度,其原理和延长体内药物的作用时间相同。
(四) 临床合理用药与避免可能的毒副作用
1.与细胞色素P450酶(cytochrome P450,CYP450)有关用药安全问题
CYP450作为体内的主要Ⅰ相代谢酶,是一组结构和功能相关的超家族基因编码的同工酶,主要分布在肝脏、小肠和肾脏,参与多种内、外源性化合物的代谢转化。在CYP450超家族中,CYP1A2、CYP2A6、CYP2C9、CYP2C19、CYP2D6和CYP3A4与目前市场95%以上药物代谢及药物间相互作用有关。药物对CYP450的抑制和诱导是导致药物相互作用的重要因素,当一些药物对CYP450产生诱导或抑制,使酶活性发生改变,结果使其自身或其他药物的代谢改变,影响其疗效或毒性。
药物代谢性相互作用中,70%由酶的抑制引起,酶的诱导所致的相互作用约占23%,其他则占7%。例如,特非那定需要CYP3A4同工酶的参与完成其体内代谢,然而红霉素却抑制了该酶的活性,从而使特非那定的血浓度增大到非正常水平,导致心脏QT间期延长,引发尖端扭转型心律失常。而诱导细胞色素P450酶的药物较少引起用药安全问题,但可降低药物的疗效。
2. 新药研究中针对CYP450的早期评价方法
近年来,通过建立的各种CYP450毒理学筛选方法评价先导分子或候选药物的类药性,设计出无CYP450诱导或抑制活性新化合物,已成为药物研究和开发过程中必要手段之一。
人肝细胞原代培养系统已成为体外药物代谢研究应用最为广泛的模型,该模型能全面地反映药物进入人体内的真实状态,其代谢处理系统与人体内极其相似,因此一直被认为是体外评价药物的“金标准”。其优点包括含有肝脏表达的所有参与药物代谢的CYP450酶,因此既可以用于研究任何目标CYP450酶,又可以用于评价某一药物是否为代谢依赖性的抑制剂,即某一药物经代谢生成的代谢产物可以抑制另外一个酶的活性;另外,肝微粒体还具有易于制备、可长期保存等特点。
用于评价CYP450各种亚型诱导活性的筛选方法主要有利用竞争结合测定、共转染报道基因系统、人源性PXR转基因小鼠等评价化合物对CYP3A的诱导活性;利用报告基因稳定转染细胞系评价对CYP1A 诱导能力。
CYP450的抑制效应可以利用不同的体外系统进行评价,包括cDNA表达的CYP450、人肝脏微粒体和肝细胞培养系统以及转基因细胞。每个系统都有其优点和局限性。由于肝脏微粒体是包括各种CYP450的混合体,因此,新化学实体的CYP450抑制效应多利用微粒体系统进行评价。
当发现先导化合物具有CYP抑制或诱导活性时,可以通过结构修饰来降低这种活性。例如咪唑类p38激酶抑制剂8-55虽然具有良好的体外抗肿瘤活性,但它对CYP3A4、2D6、2C9和1A22等都具有不同程度的抑制活性,限制了其进一步的应用。人们通过引入N-甲基哌啶基降低8-55的亲脂性,结构优化后的8-56对CYP的抑制活性大幅下降。
药物化学---药物的化学结构与体内代谢转化 方浩 第一部分概述 对人体而言,绝大多数药物是一类生物异源物质(Xenobiotics)。当药物进入机体后,一方面药物对机 体产生诸多生理药理作用,即治疗疾病;另一方面,机体也对药物产生作用,即对药物的吸收、分布,排泄和代谢。药物代谢既是药物在人体内发生的化学变化,也是人体对自身的一种保护机能。 药物代谢是指在酶的作用下将药物(通常是非极性分子)转变成极性分子,再通过人体的正常系统排出体外。药物代谢多使有效药物转变为低效或无效的代谢物,或由无效结构转变成有效结构。在这过程中,也有可能将药物转变成毒副作用较高的产物。因此,研究药物在体内代谢过程中发生的化学变化,更能阐明药理作用的特点、作用时程、结构转变以及产生毒性的原因。 药物代谢在创新药物发现和临床药物合理应用中具有重要的地位。通过对近十年来许多创新药物在临床失败的案例,科学家们发现与药物代谢有关的问题是创新药物临床研究失败的重要原因。因此当前进行创新药物研究的过程中,应当在候选药物研究阶段就重视考察其药物代谢的相关问题,并将候选药物的代谢问题作为评判其成药性的重要研究内容。在药理学和生物药剂学课程中,对于药物在体内发生的药物代谢转化反应和代谢产物讲述内容较少。因此我们将在药物化学的讲述中,重点从药物代谢酶角度入手,讨论药物在体内发生的生物转化,以帮助大家更好的认识药物在体内所反应的代谢反应以及其与药物发现和临床合理应用的关系。 药物的代谢通常分为两相:即第I相生物转化(Phase I )和第n相生物转化(Phase n )。第I相主要是官 能团化反应,包括对药物分子的氧化、还原、水解和羟化等,在药物分子中引入或使药物分子暴露出极性基团,如羟基、羧基、巯基和氨基等。第n相又称为结合反应(Conjugaten),将第I相中药物产生的极性 基团与体内的内源性成分,如葡萄糖醛酸、硫酸、甘氨酸或谷胱甘肽,经共价键结合,生成极性大、易溶于水和易排出体外的结合物。但是也有药物经第I相反应后,无需进行第n相的结合反应,即可排出体外。 第二部分基本概念、基本知识及重点、难点 一、药物代谢的酶(En zymes for Drug Metabolism) 第I相生物转化是官能团化反应,是在体内多种酶系的催化下,对药物分子引入新的官能团或改变原有的官能团的过程。参与药物体内生物转化的酶类主要是氧化一还原酶和水解酶。本节主要介绍细胞色素P-450酶系、还原酶系、过氧化物酶和其它单加氧酶、水解酶。 (一)细胞色素P-450酶系 CYP-450(Cytochrome P-450 enzyme system , CYP-450)是一组酶的总称,由许多同功酶和亚型酶组成, 是主要的药物代谢酶系,在药物和其它化学物质的代谢、去毒性中起着非常重要的作用。CYP-450存在于 肝脏及其它肝脏外组织的内质网中,是一组由铁原卟啉偶联单加氧酶(Heme-coupled monooxygenases)、需要NADPH和分子氧共同参与、主要催化药物生物转化中氧化反应(包括失去电子、脱氢反应和氧化反应)的酶系。它主要是通过“活化”分子氧,使其中一个氧原子和有机物分子结合,同时将另一个氧原子还原成水,从而在有机药物的分子中引入氧。CYP-450催化的反应类型有烷烃和芳香化合物的氧化反应,烯烃、多核芳烃及卤代苯的环氧化反应,仲胺、叔胺及醚的脱烷基反应,胺类化合物的脱胺反应,将胺转化为N-氧化物、羟胺及亚硝基化合物以及卤代烃的脱卤反应。CYP-450还催化有机硫代磷酸酯的氧化裂解,氧化 硫醚成亚砜等的反应(见表1)。 表1 CYP-450催化的一些药物代谢的氧化反应类型 底物产物
抗生素 1.抗生素按化学结构可分为哪几大类?各举一例药物。 (1)β-内酰胺抗生素:青霉素,氨苄西林,阿莫西林(2)四环素类抗生素:四环素(3)氨基糖苷类抗生素:阿米卡星,庆大霉素(4)大环内酯类抗生素:红霉素,罗红霉素,阿奇霉素(5)其他:氯霉素 2.简述青霉素对酸、碱、酶的不稳定性,试以反应式表示。 (1)在强酸条件下或氯化高汞的作用下,β-内酰胺环发生裂解,生成青霉酸,青霉酸与水生成青霉醛酸,青霉醛酸不稳定,释放出二氧化碳,生成青霉醛。另一途径为青霉酸脱二氧化碳生成青霉噻唑酸,在分解为D-青霉胺和青霉醛。在弱酸(pH=4)的室温条件下,侧链上羰基氧原子上的孤对电子作为亲核试剂进攻β-内酰胺环,再经重排生成青霉二酸,青霉二酸可进一步分解生成青霉胺和青霉醛。(2)在碱性条件下,碱性基团向β-内酰胺环进攻,生成青霉酸,青霉酸加热时易失去二氧化碳,生成青霉噻唑酸,遇氯化高汞青霉噻唑酸进一步分解生成青霉胺和青霉醛。(3)在β-内酰胺酶的作用下,酶中亲核性基团向β-内酰胺环进攻,生成青霉酸(青霉酸加热时易失去二氧化碳,生成青霉噻唑酸,遇氯化高汞青霉噻唑酸进一步分解生成青霉胺和青霉醛)。 3.简述寻找耐酸、耐酶、广谱青霉素的研究方法。 (1)耐酸青霉素的设计原理:天然青霉素V的6位酰胺侧链上连有吸电子基,可阻碍电子转移,避免分子内重排,增加了对酸的稳定性。为寻找耐酸青霉素提供了基本思想,即在6位酰胺基的α位引入O、N、X等电负性原子,从而合成了一系列耐酸的青霉素。(2)耐酶青霉素的设计原理:通过改变6位侧链,引入立体障碍大的基团,可以阻止青霉素和β-内酰胺酶的活性中心作用,同时可以限制侧链和酰胺C=O之间的单键旋转,迫使青霉素分子变成一种与酶活性中心不易适应的构型,降低了青霉素与酶活性中心作用的适应性,从而保护了分子中的β-内酰胺环。(3)广谱青霉素的设计原理:对G+菌的作用低于青霉素G,但对G-菌却显示较强的抑制作用。分析原因是由于其侧链为亲水性。受之启发,合成一系列含有NH2,COOH,SO3H的侧链的半合成青霉素。 4.为什么青霉素G不能口服?而青霉素V却可以口服?为什么青霉素G的钠盐或钾盐必须做成粉针剂型? 青霉素G在酸性条件下不稳定,易发生重排而失活。因此不能口服,通常将其做成钠盐或钾盐注射使用,但其钠盐或钾盐的水溶性碱性较强,β-内酰胺环会发生开环生成青霉酸,失去抗菌活性。因此青霉素的钠盐或钾盐必须做成粉针剂使用。 青霉素V:具有耐酸性,不易被胃酸破坏。 5.奥格门汀是由哪两种药物组成?说明两者合用起增效作用的原理。 临床上使用克拉维酸和阿莫西林组成复方制剂称为奥格门汀,可使阿莫西林增效130倍,用于治疗耐阿莫西林细菌所引起的感染。阿莫西林为半合成的光谱青霉素,通过抑制细菌细胞壁的合成而发挥抗菌作用,但会被细菌所产生的β-内酰胺酶水解而失活。克拉维酸是有效的β-内酰胺酶抑制剂,可与多数β-内酰胺酶牢固结合,可使阿莫西林免受β-内酰胺酶的钝化,用于治疗耐阿莫西林细菌所引起的感染。 6.简述天然四环素类抗生素的不稳定性,并说明四环素类抗生素不能和牛奶等富含金属离子的食物一起使用的原因。 (1)天然四环素具有易产生耐药性,化学结构在酸、碱条件下不稳定等缺点。不稳定部位为C-6位的羟基和C-4位的二甲胺基。在酸性条件下,C-6上的羟基和C-5α上氢发生消除反应,生成无活性橙黄色脱水物。在pH2-6条件下,C-4二甲胺基很易发生可逆反应的差向异构化,生成差向异构体。4位差向异构化产物在酸性条件也还会进一步脱水生成脱水差向异构化产物。在碱性条件下,C-6上的羟基形成氧负离子,向C-11发生分子内亲核进攻,经电子转移,C环破裂,生成无活性的具有内酯结构的异构体。(2)分子中含有许多羟基、烯醇羟基及羰基,在近中性条件下能与多种金属离子形成不溶性螯合物。四环素类抗生素能和钙离子形成黄色的络合物沉积在骨骼和牙齿上,小儿服用后会发生牙齿变黄色,孕妇服用后其产儿可能发生牙齿变色、骨骼生长抑制。因此小儿和孕妇应慎用或禁用。 7.试从红霉素的不稳定性说明半合成红霉素药物的设计原理,并举出两例药物。 结构存在多个羟基以及在其9位上有一个羰基,因此在酸性条件下不稳定,易发生分子内的脱水环合。在酸性液中,C-6上的羟基与C-9的羰基形成半缩酮的羟基,再与C-8上氢消去一分子水,生成8,9-脱水-6,9-半缩酮衍生物。然后C-12上的羟基与C-8-C-9双键加成,进行分子内环合,生成6,9,-9,12-螺旋酮。(2)①早期对红霉素的结构修饰主要是将红霉素制成各种酯类和盐类的前体药物目的是增加红霉素的稳定性和水溶性。红霉素乳糖醛酸盐、琥乙红霉素②后期主要是针对红霉素酸降解的机制对大环内酯进行改造。在红霉素在酸降解反应中,参与反应的基团有C-9酮,C-6羟基,C-12羟基和C-8氢,因此结构修饰主要在这些部位进行。罗红霉素、克拉霉素、阿齐霉素 8.氨基糖苷类抗生素有哪些共性?为什么氨基糖甙类抗生素易产生耐药性? (1)共同特点:①结构:含氨基糖,碱性多元醇②抗菌谱:广谱,对G-菌的作用强于G+③作用机制相似:抑制核糖体蛋白质的合成④副作用相同,听觉毒性,肾毒性⑤易产生耐药性。(2)一些耐药菌会产生氨基糖苷钝化酶,使氨基糖苷类抗生素灭活。包括氨基糖苷磷酸转移酶、氨基糖苷乙酰转移酶、氨基糖苷腺苷转移酶。这些酶的作用均使卡那霉素失去活性。 9.为什么红霉素口服后生物利用度极低? 水溶性小,只能口服,但在胃酸中不稳定,易分解迅速失去活性。 合成抗菌药
《药物化学》课程标准 一、前言 (一)课程的性质 该课程是兽药生产与营销专业的专业基础课,目标是让学生现代药物化学基本理论和技能,对常用药物的结构类型、药物合成、理化性质、构效关系及其应用有一个较系统的认识,并了解现代药物化学的发展,为以后在制药实践中合成并合理使用常用药物打下坚实的基础。它是以有机化学和药理学等课程的学习为基础,也是进一步学习药物制剂技术等课程的基础。 (二)设计思路 按教学大纲规定,认真备课,重视启发式教学,课堂教学多采用多媒体教学。通过阅读教材有关内容,结合观看有关教学VCD、多媒体课件等,培养学生的自学能力,以增加学生的感性认识,启迪学生的科学思维。注意运用理论知识指导学习,通过理论的学习加深对实践的理解。 二、课程目标 通过本课程的学习,要求学生掌握常用药物或代表药物的化学结构、化学名、理化性质、合成制备、构效关系;能够熟练、安全地合成药物;熟悉药物发展史和设计思想,研究构效关系和合理设计药物。 通过本课程的实验,学生能根据所学合成原理进行原料药中间体的合成、化学药物的合成、抗生素的合成;能对合成的粗品进行纯化;能鉴别药物中的杂质。 通过理论与实践一体化的教学方式,让学生在完成具体项目的过程中完成相应工作任务,并构建相关理论知识,发展职业能力,使学生获得的知识,技能真正满足化学制药、药物制剂、药品检验不同岗位发展的需求。为学生今后的专业学习和职业生涯发展、在兽药企业工作中奠定坚实的专业信念、知识与技能的基础。 职业能力培养目标: 1.能根据所学的合成原理进行原料药中间体的合成操作。 2.能进行化学药物的合成操作。 3.能进行抗生素的合成操作。 4.能对合成的粗品进行纯化。 5.能鉴别药物中杂质。 6.能按照药物的理化性质判断其储存条件。 7.能熟练对常用药物或代表药物进行鉴别操作。 8.能按照药物的性质给出调剂的要求。
药物的化学结构与药效的关系 A型题(最佳选择题) (1题-20题) 1.下列对生物电子等排原理叙述错误的是 A以生物电子等排体的相互替换,对药物进行结构的改造,以提高药物的疗效。 B以生物电子等排体的相互替换,对药物进行结构的改造,以降低药物的毒副作用。 C凡具有相似的物理性质和化学性质,又能产生相似生物活性的基团或分子都称为生物电子等排体。 D生物电子等排体可以以任何形式相互替换,来提高药物的疗效,降低毒副作用。 E 在药物结构中可以通过基团的倒转、极性相似、范德华半径相似等进行电子等排体的相互替换,找到疗效更高,毒性更小的新药。 2.下列对前药原理的作用叙述错误的是 A 前药原理可以改善药物在体内的吸收; B 前药原理可以缩短药物在体内的作用时间; C前药原理可以提高药物的稳定性; D前药原理可以消除药物的苦味; E前药原理可以改善药物的溶解度; 3.药物分子中引入烃基、卤素原子、硫醚键等,可使药物的 A 脂溶性降低; B 脂溶性增高; C 脂溶性不变; D 水溶性增高; E 水溶性不变; 4.药物分子中引入羟基、羧基、脂氨基等,可使药物的 A 水溶性降低; B 脂溶性增高; C 脂溶性不变; D 水溶性增高; E 水溶性不变; 5.一般来说,酸性药物在体内随介质pH增大 A解离度增大,体内吸收率降低; B解离度增大,体内吸收率升高;
C解离度减小,体内吸收率降低; D解离度减小,体内吸收率升高; E解离度不变,体内吸收率不变; 6.一般来说,碱性药物在体内随介质pH增大 A解离度增大,体内吸收率降低; B解离度增大,体内吸收率升高; C解离度减小,体内吸收率降低; D解离度减小,体内吸收率升高; E解离度不变,体内吸收率不变; 7.药物的基本结构是指 A具有相同药理作用的药物的化学结构; B 具有相同化学结构的药物; C 具有相同药理作用的药物的化学结构中相同部分; D 具有相同理化性质的药物的化学结构中相同部分; E 具有相同化学组成药物的化学结构; 8.在药物的基本结构中引入烃基对药物的性质影响叙述错误的是 A 可以改变药物的溶解度; B 可以改变药物的解离度; C 可以改变药物的分配系数; D 可以改变药物分子结构中的空间位阻; E 可以增加位阻从而降低药物的稳定性; 9.在药物的基本结构中引入羟基对药物的性质影响叙述错误的是 A 可以增加药物的水溶性; B 可以增强药物与受体的结合力; C 取代在脂肪链上,使药物的活性和毒性均下降; D取代在芳环上,使药物的活性和毒性均下降; E可以改变药物生物活性; 10.在药物的基本结构中引入羧基对药物的性质影响叙述错误的是
1.地西泮 2.苯妥因钠 3.普罗加比 4.盐酸氯丙嗪 5.氟奋乃静 6.氯普噻吨 7.舒必利 8.吗啡 9.哌替啶 10.咖啡因 11.硫酸阿托品11.麻黄碱 12.苯海拉明 13.马来酸氯苯那敏 14.阿斯咪唑 15.普鲁卡因 16.利多卡因 17.硝苯地平 18.利血平 19.卡托普利20.奎尼丁 21.普萘洛尔 22.美托洛尔 23.(双)氢氯噻嗪 24.甲苯磺丁脲 25.雷尼替丁 26.奥美拉唑 27.昂丹司琼 28.甲氧普胺 29.阿司匹林 30.贝诺酯 31.对乙酰氨基酚 32.吲哚美辛 33.环磷酰胺 34. 5-氟尿嘧啶 35. 紫杉醇 36. 顺铂 37. 青霉素钾 38. 苯唑西林 39.氨苄西林 40. 苯唑西林 42. 头孢氨苄 43. 磺胺嘧啶 44. 甲氧苄啶 45. 诺氟沙星(氟哌酸) 46. 利福平 47.异烟肼 48. 硝酸益康唑 49. 三氮唑核苷 50. 奎宁 51. 青蒿素 52.红霉素 53.链霉素 54.四环素 55.氯霉素 56雄甾烷-3-酮 57. 雌激素 雄激素 氢化可的松 地塞米松 维生素C 吡罗昔康:第一个临床使用的1,2苯并 噻嗪类解热镇痛药 氯氮平:第一个上市的非经典抗精神病 药 哌替啶:苯基哌啶类的第一个合成镇痛 药 洛伐他汀:第一个投放市场的
HMG-CoA还原酶抑制剂 氯沙坦:第一个上市的血管紧张素Ⅱ受体拮抗剂 苯唑西林:第一个耐酸耐酶青霉素,口服、注射均可 克拉维酸:第一个β-内酰胺酶抑制剂阿奇霉素:第一个环内含氮的15元大环内酯抗生素 链霉素:第一个用于抗结核病的药物齐多夫定:美国FDA批准的第一个用于艾滋病及其相关症状治疗的药物 沙奎那韦:第一个批准上市治疗艾滋病的蛋白酶抑制剂 金霉素:第一个四环素类抗生素 碘苷:第一个用于临床的抗病毒核苷类药物 阿昔洛韦:第一个上市的开环鸟苷类似物广谱抗病毒药 氨苄霉素:第一个使用的广谱口服抗生素 酮康唑:第一个口服有效的咪唑类广谱抗真菌药物 帕瑞昔布:全球第一种注射用选择性COX-2 抑制剂 药物化学各类药物分类总结 镇静催眠药7 p; P c6 Z% S# m, 巴比妥类:苯巴比妥、硫喷妥钠 苯二氮卓类:地西泮、奥沙西泮2 K$ 氨基甲酸酯类:甲丙氨酯5 o/ @$ {7 其他类:水合氯醛 抗癫痫药, b) F3 w% k7 `) Q 巴比妥类、 苯并二氮卓类:地西泮. w9 a+ G, L 乙内酰脲类:苯妥英钠* j/ Z3 `; ^6 H0 二苯并氮杂卓类:卡马西平$ e0 h* 脂肪羧酸类:丙戊酸钠 磺酰胺类 抗精神失常药/ {; i' J1 m) Y# N0 吩噻嗪类:氯丙嗪8 K) m+ U9 y% G9 丁酰苯类:氟哌啶醇 二苯并氮卓类:氯氮平) \3 j: b6 p* k* 噻吨类:氯普噻吨 抗抑郁药" |! ~/ k, Y; v* J1 s6 Y1 P7 E 去甲肾上腺素重摄取抑制剂;5-羟色胺重摄取抑制剂;盐酸阿米替林单胺氧化酶抑制剂;非典型抗抑郁 解热镇痛药 水杨酸类:阿司匹林 乙酰苯胺类:对乙酰氨基酚 吡唑酮类 非甾类抗炎药(了解)# ~; ?3 x1 @' f } 水杨酸类:贝诺酯阿司匹林与对乙 酰氨基酚成酯形成的前药,特别适 合于儿童 吡唑酮类 芳基烷酸类:吲哚美辛、双氯芬酸 钠、布洛芬、萘普生 N-芳基邻氨基苯甲酸类:灭酸类 1,2-苯并噻嗪类:美洛昔康 其他类 镇痛药Z$ C ~( x3 B0 V3 i 天然生物碱:盐酸吗啡 半合成镇痛药:磷酸可待因 合成镇痛药:盐酸哌替啶、美沙酮 内源性多肽 胆碱受体激动剂9 v6 Z' [9 {1 U5 a8 S M胆碱受体激动剂:毛果芸香碱 胆碱酯酶复活剂:碘解磷定 胆碱受体拮抗剂 乙酰胆碱酯酶抑制剂:新斯的明 M胆碱受体拮抗剂+ X K; F2 E' r$ z 茄科生物碱:对中枢作用:东莨菪 碱>阿托品>樟柳碱>山莨菪碱 全合成M胆碱受体拮抗剂:硫酸 阿托品、氯琥珀胆碱 肾上腺素能受体激动剂# D$ |+ B0 k, ]3 t8 苯乙胺类:肾上腺素、多巴胺、克 仑特罗、特布他林 苯异丙胺类:麻黄碱、甲氧明 肾上腺素:对α和β受体都有激动 作用。临床用于急性心力衰竭、支 气管哮喘及心搏骤停的抢救。 盐酸多巴胺:多巴胺受体激动剂, 抗休克药。 重酒石酸去甲肾上腺素:主要兴奋 α受体。主要升压,静滴用于休克, 口服用于消化道出血。2 k% E* s/ M" 盐酸异丙肾上腺素:兴奋β受体。 用于支气管哮喘、过敏性哮喘、慢性肺 气肿及低血压等。& s9 L+ T3 Y, D& y 盐酸麻黄碱:α和β受体均有激动作用。 盐酸甲氧明:激动α受体,用于外伤和 周围循环不全时低血压急救 肾上腺素能受体拮抗剂 α受体阻断剂:盐酸哌唑嗪 β受体阻断剂:普萘洛尔、阿替洛尔 降血脂药- C: o A( g6 ~" U! g$ F 分类(掌握)' c! o3 E1 Y* L4 U 苯氧乙酸类:氯贝丁酯、吉非贝齐 烟酸类 羟甲戊二酰辅酶A还原酶抑制剂:洛 伐他丁 其他 抗心绞痛药7 b3 H; f' m+ ^. G! Q6 |6 ` 硝酸酯和亚硝酸酯类:硝酸异山梨酯 钙拮抗剂: a.二氢吡啶类:硝苯地平、尼索地平 b.苯烷基胺类:维拉帕米,左旋体室上 性心动过速的首选药物,右旋体治疗心 绞痛 c.苯噻氮卓类:地尔硫卓 d.二苯哌嗪类:氟桂利嗪、桂利嗪,直 接扩张血管平滑肌 β受体阻断剂I) Q$ {- S% [: R5 O& h. K% h: 抗高血压药% k8 l9 c" P8 u# A3 M ⑴作用于自主神经系统4 ?" q. H/ [; a.外周抗去甲肾上腺素能神经末梢药: 利血平胍乙啶2 ^2 h( N7 c2 c7 Q b.中枢性交感神经抑制药:可乐定 甲基多巴; [( X. L% l% Q2 ] c.直接扩血管药:肼屈嗪硝普钠 d.神经节阻断药:咪噻吩9 ?) y4 e P9 e.肾上腺素α1受体阻断药:哌唑嗪。 ⑵作用于RAS系统 a.血管紧张素转换酶抑制药:卡托普利。 b.血管紧张素Ⅱ受体拮抗剂:氯沙坦。 c.肾素抑制剂:肽类。 ⑶作用于离子通道 钙拮抗药:硝苯地平尼群地平4 K6 钾通道开放剂 抗心律失常药- ^+ S6 D e( W" Y (1)Ⅰ类:钠通道阻滞药- {) [" T1 A' Y! {" ⅠA类:适度阻滞心肌细胞钠通道 奎尼丁; D' S: t# ]: G) d" X8 q
药物—特殊化学品:用来预防、治疗、诊断疾病;为了调节人体生理机能、提高生活质量、保持身体健康 药物化学就是一门发现与发明科学、合成化学药物、阐明药物化学性质、研究药物分子与机体细胞(生物大分子)直接相互作用规律得综合性学科。 先导化合物:具有特定生理活性得化合物,可作为结构修饰与结构改造得模型,从而获得预期药理作用得药物。发现途径与方法:从天然产物得到;以现有药物作为先导化合物;用活性内源性物质作;利用组合化学与高通量筛选得到;利用计算机进行靶向筛选得到。优化方法:采用生物电子等排体进行替换、前药设计、软药设计、定量构效关系研究。 新化学实体(NCE)在以前文献中为未报道过,并且能以安全、有效得方式治疗疾病得新化合物。 新药发现:靶分子得确定与选择,靶分子得优化,先导化合物得发现,先导化合物得优化。ADM E:吸收、分布、代谢、排泄。化学物既定理化性质。 脂水分配系数:药物在正辛醇中与水中分配达到平衡时得浓度比值。P=Co/Cw。 亲水:扩散至血液体液亲脂:通过生物膜 立体化学作用:几何异构,光学异构,构象异构
优势构象:分子势能最低得构象。未必未药效构象,与受体作用实际构象。 药效构象:药物与受体作用就是所采取得实际构象。 构象等效性:药物分子得基本结构不同,但可能会以相同得作用机制引起相同得药理或毒理作用,这就是由于它们具有共同得构象,即构象等效性。 代谢拮抗:设计与生物体内基本代谢物结构有某种相似度得化合物,使与基本代谢物竞争性或干扰基本代谢物被利用,或掺入生物大分子中形成伪生物大分子,导致致死合成,影响细胞生长 计算机辅助药物设计(CADD)利用计算机得快速计算功能,全方位得逻辑制断功能,一目了然得图形显示功能,将量子化学、分子力学、药物化学、生命科学、计算机图形学与信息科学等学科交叉,从药物分子得作用机制入手进行药物设计。 生物靶点:能够与药物分子结合并产生药理效应得生物大分子受体、酶、离子通道、核酸 生物电子等排体:具有相似得物理及化学性质得基团或取代基产生得大致相似、相关或相反得生物活性得一种物质。 如果药物经过化学结构修饰后得到得化合物,在体外没有或很少有活性在生物体或人体内通过酶得作用又转化为原来得药物而发挥药效时,称原来得药物为母体药物,修饰后得到得化合物为前体药物,简称
第一章药物的化学结构与药效的关系 本章提示: 大多数药物的作用依赖于药物分子的化学结构,因此药物的药效和药物的理化性质,如疏水性、酸碱性、药物的解离度等有关;与药物结构的立体构型、空间构型、电子云密度等有关。此外还与药物与生物分子的作用强弱有关。 第一节影响药物药效的因素和药效团 药物从给药到产生药效是一个非常复杂的过程,包括吸收、分布、代谢、组织结合,以及在作用部位产生作用等等。在这一过程中影响药物产生药效的主要因素有两个方面: 1.药物到达作用部位的浓度。对于静脉注射给药时,由于药物直接进入血液,不存在药物被吸收的问题。而对于其它途径给药时都有经给药部位吸收进入血液的问题。进入血液后的药物,随着血液流经各器官或组织,使药物分布于器官或组织之间,这需要药物穿透细胞膜等生物膜,最后到达作用部位。而药物只有到达作用部位,才能产生药效。在这一系列的过程中,药物的理化性质产生主要的影响。此外药物随血液流经肝脏时会产生代谢,改变药物的结构和疗效,流经肾脏时产生排泄,减少了药物在体内的数量。这些也与药物结构中的取代基的化学反应性有一定的联系。 2.药物与受体的作用。药物到达作用部位后,与受体形成复合物,产生生理和生化的变化,达到调节机体功能或治疗疾病的目的。药物与受体的作用一方面依赖于药物特定的化学结构,以及该结构与受体的空间互补性,另一方面还取决于药物和受体的结合方式,如化学的方式通过共价键结合形成不可逆复合物,或以物理的方式,通过离子键、氢键、离子偶极、范德华力和疏水性等结合形成可逆的复合物。 这二个影响因素都与药物的化学结构有密切的关系,是药物结构-药效关系(构-效关系)研究的主要内容。 但对于药物的作用方式来讲,又有两种不同类型。一类是药物的药效作用主要受药物的理化性质影响而与药物的化学结构类型关系较少,如全身麻醉药,尽管这些药物的化学结构类型有多种,但其麻醉作用与药物的脂水分配系数有关,这类药物称为结构非特异性药物;另一类药物的作用依赖于药物分子特异的化学结构,该化学结构与受体相互作用后才能产生影响,因此化学结构的变化会直接影响其药效,这类药物称为结构特异性药物。而大多数药物属于结构特异性药物。 结构特异性药物中,能被受体所识别和结合的三维结构要素的组合又称为药效团。这样受体必须首先要识别所趋近的分子是否具有结合所需的性质,然后与其结合。药效团又可分为两种类型:一类具有相同药理作用的类似物,它们具有某种基本结构;另一类则可能是一组化学结构完全不同的分子,但可以与同一受体以相同的机理键合,产生同样的药理作用。受体与药物的结合实际上是与药物结构中药效团的结合,这与药物结构上官能团的静电性、疏水性及基团的大小有关。 第二节药物理化性质和药效的关系 在药物作用的过程中,药物的理化性质对药物的吸收、转运都产生重要的影响,而且对于结构非特异性药物,药物的理化性质直接影响药物的活性。药物的理化性质主要有药物的溶解度、分配系数和解离度。
脂类代 一、问答题 1、为什么摄入糖量过多容易长胖? 答:因为脂肪酸合成的起始原料乙酰CoA主要来自糖酵解产物丙酮酸,摄入糖量过多则糖酵解产生的丙酮酸也多,进而导致合成脂肪酸的起始原料乙酰CoA也多,原料多合成的脂肪酸自然就多了,所以摄入糖量过多容易长胖。 2、比较脂肪酸β—氧化和脂肪酸的合成有哪些不同点? 答:①细胞中发生部位不同:合成发生在细胞质,氧化发生在线粒体; ②酰基载体不同:合成所需载体为ACP—SH,氧化所需载体为乙酰CoA;③二碳片段的加入与裂解方式:合成是以丙二酰ACP加入二碳片段,氧化的裂解方式是乙酰CoA;④电子供体或受体:合成的供体是NADPH,氧化的受体是FAD、FAD+;⑤酶系不同:合成需7种酶,氧化需4种酶;⑥原料转运方式:合成是柠檬酸转运系统,氧化是肉碱穿梭系统;⑦能量变化:合成耗能,氧化产能。 3、试计算1mol甘油彻底氧化成CO2和H2O可净生成多少molATP。答:甘油氧化产生的乙酰CoA进入三羧酸循环彻底氧化。经过4次脱氢反应生成3molNADH+H+、1molFADH2、以及2molCO2,并发生一次底物水平磷酸化,生成1molGTP。依据生物氧化时每1molNADH+H+和1molFADH2 分别生成2.5mol、1.5mol的ATP,因
此,1mol甘油彻底氧化成CO2和H2O生成ATP摩尔数为6×2.5+1×1.5+3-1=18.5。 4、1mol硬脂酸(即18碳饱和脂肪酸)彻底氧化成CO2和H2O时净生成的ATP的摩尔数。 答:1mol硬脂酸彻底氧化需经8次循环,产生9个乙酰CoA,每摩尔乙酰CoA进入三羧酸循环产生10molATP,这样共产生90molATP。8molFADH2进入电子传递链产生12molATP,8molNADH进入电子传递链共产生20molATP。脂肪酸的活化需消耗2个高能磷酸键,这样彻底氧化1mol硬脂酸净得120molATP。 5、胆固醇在体可转变成哪些重要物质?合成胆固醇的基本原料和关键酶各是什么? 答:转变成胆汁酸、甾类激素、维生素D; 基本原料:二甲基丙烯焦磷酸酯(DPP)、异戊烯醇焦磷酸酯 关键酶:羟甲基戊二酸单酰CoA还原酶(HMGCoA还原酶) 6、为什么在长期饥饿或糖尿病状态下,血液中酮体浓度会升高?答:由于糖供应不足或利用率降低,机体需动员大量的脂肪酸供能,同时生成大量的乙酰CoA。此时草酰乙酸进入糖异生途径,又得不到及时的回补而浓度降低,因此不能与乙酰CoA缩合成柠檬酸。在这种情况下,大量积累的乙酰CoA衍生为丙酮、乙酰乙酸、β—羟丁酸。
异戊巴比妥 5-乙基-5-(3-甲基丁基)-2,4,6-(1H ,3H ,5H )嘧啶三酮 地西泮 1-甲基-5-苯基-7-氯-1,3-二氢-2H-1,4-苯并二氮杂卓-2-酮 N N O Cl 124 5 7 唑吡坦 Zolpidem N N O N 1 3 6 苯妥英钠 5,5-二苯基-2,4-咪唑烷二酮钠盐 N H N O ONa 1 5 卡马西平 酰胺咪嗪 N O NH 2 卤加比 Progabide OH F N Cl NH 2 O 盐酸氯丙嗪 N ,N-二甲基-2-氯-10H-吩噻嗪-10-丙胺 盐酸盐 . HCl N S Cl N 25 10 氟哌啶醇 氯氮平 N N N N H Cl 盐酸丙咪嗪 N ,N-二甲基-10,11-二氢-5H-二苯并[b ,f]氮杂卓-5-丙胺 盐酸盐 N N HCl 氟西汀 O H N F F F HCl * 吗啡 Morphine 17-甲基-4, 5a-环氧-7, 8-二脱氢 吗啡喃 -3, 6a-二醇盐酸盐 三水合物
O OH N HO 13 4 5 67 8 9101112 1314 1516 17. HCl . 3H 2O 盐酸哌替啶 1-甲基-4-苯基-4-哌啶甲酸乙酯盐酸盐 N O O . HCl 盐酸美沙酮 N O . HCl 喷他佐辛 N HO H 咖啡因 Caffeine 1,3,7-三甲基-3,7-二氢-1H - 嘌呤 -2,6-二酮一 水合物 N N N N O O . H 2O 137 吡拉西坦 2-(2-氧代-吡咯烷-1-基)乙酰胺 NH 2N O O 氯贝胆碱 Bethanechol Chloride O O H 2N N +(CH 3)3 Cl -CH 3 毛果芸香碱 N N O H 3C CH 3 O 溴新斯的明 Neostigmine Bromide N +(CH 3)3 Br - O N O H 3C CH 3 多奈哌齐 硫酸阿托品 Atropine Sulphate . H 2SO 4 . H 2O N O OH O CH 3 2 溴丙胺太林 Br - O O O N H 3C CH 3CH 3 CH 3 CH 3 + 哌仑西平
药物化学—--药物的化学结构与体内代谢转化 方浩 第一部分概述 对人体而言,绝大多数药物是一类生物异源物质(Xenobiotics)。当药物进入机体后,一方面药物对机体产生诸多生理药理作用,即治疗疾病;另一方面,机体也对药物产生作用,即对药物的吸收、分布,排泄和代谢.药物代谢既是药物在人体内发生的化学变化,也是人体对自身的一种保护机能。 药物代谢是指在酶的作用下将药物(通常是非极性分子)转变成极性分子,再通过人体的正常系统排出体外。药物代谢多使有效药物转变为低效或无效的代谢物,或由无效结构转变成有效结构.在这过程中,也有可能将药物转变成毒副作用较高的产物.因此,研究药物在体内代谢过程中发生的化学变化,更能阐明药理作用的特点、作用时程、结构转变以及产生毒性的原因。 药物代谢在创新药物发现和临床药物合理应用中具有重要的地位.通过对近十年来许多创新药物在临床失败的案例,科学家们发现与药物代谢有关的问题是创新药物临床研究失败的重要原因。因此当前进行创新药物研究的过程中,应当在候选药物研究阶段就重视考察其药物代谢的相关问题,并将候选药物的代谢问题作为评判其成药性的重要研究内容。在药理学和生物药剂学课程中,对于药物在体内发生的药物代谢转化反应和代谢产物讲述内容较少。因此我们将在药物化学的讲述中,重点从药物代谢酶角度入手,讨论药物在体内发生的生物转化,以帮助大家更好的认识药物在体内所反应的代谢反应以及其与药物发现和临床合理应用的关系。 药物的代谢通常分为两相:即第Ⅰ相生物转化(PhaseⅠ)和第Ⅱ相生物转化(PhaseⅡ)。第Ⅰ相主要是官能团化反应,包括对药物分子的氧化、还原、水解和羟化等,在药物分子中引入或使药物分子暴露出极性基团,如羟基、羧基、巯基和氨基等。第Ⅱ相又称为结合反应(Conjugation),将第Ⅰ相中药物产生的极性基团与体内的内源性成分,如葡萄糖醛酸、硫酸、甘氨酸或谷胱甘肽,经共价键结合,生成极性大、易溶于水和易排出体外的结合物.但是也有药物经第Ⅰ相反应后,无需进行第Ⅱ相的结合反应,即可排出体外。 第二部分基本概念、基本知识及重点、难点 一、药物代谢的酶(Enzymes forDrug Metabolism) 第Ⅰ相生物转化是官能团化反应,是在体内多种酶系的催化下,对药物分子引入新的官能团或改变原有的官能团的过程.参与药物体内生物转化的酶类主要是氧化-还原酶和水解酶。本节主要介绍细胞色素P—450酶系、还原酶系、过氧化物酶和其它单加氧酶、水解酶。 (一)细胞色素P-450酶系 CYP—450(Cytochrome P-450enzyme system,CYP—450)是一组酶的总称,由许多同功酶和亚型酶组成,是主要的药物代谢酶系,在药物和其它化学物质的代谢、去毒性中起着非常重要的作用。CYP-450存在于肝脏及其它肝脏外组织的内质网中,是一组由铁原卟啉偶联单加氧酶(Heme—coupled monooxygenases)、需要NADPH和分子氧共同参与、主要催化药物生物转化中氧化反应(包括失去电子、脱氢反应和氧化反应)的酶系。它主要是通过“活化”分子氧,使其中一个氧原子和有机物分子结合,同时将另一个氧原子还原成水,从而在有机药物的分子中引入氧。CYP-450催化的反应类型有烷烃和芳香化合物的氧化反应,烯烃、多核芳烃及卤代苯的环氧化反应,仲胺、叔胺及醚的脱烷基反应,胺类化合物的脱胺反应,将胺转化为N-氧化物、羟胺及亚硝基化合物以及卤代烃的脱卤反应。CYP-450还催化有机硫代磷酸酯的氧化裂解,氧化硫醚成亚砜等的反应(见表1)。 表1CYP-450催化的一些药物代谢的氧化反应类型
掌握内容: 必需脂酸的概念及种类: 人体需要但又不能合成,必须从食物中获取的脂酸。人体必需的脂酸是亚油酸,亚麻酸,花生四烯酸。 脂肪动员: 概念及过程:储存于脂肪细胞中的甘油三酯,在三种脂肪酶的作用下逐步水解为游离脂酸和甘油,释放入血供其他组织氧化利用的过程,称脂肪动员。甘油三酯脂肪酶是脂肪动员的限速酶。(过程PPT29、30) 激素敏感性脂肪酶的定义和作用: 甘油三酯脂肪酶是脂肪动员的限速酶,其活性受多种激素调节故称激素敏感性脂肪酶 脂解激素:增加脂肪动员限速酶活性,促进脂肪动员活性的激素。(肾上腺素、去甲状腺激素、胰高血糖素、促肾上腺皮质激素、促甲状腺激素 抗脂解激素:抑制脂肪动员,(胰岛素,前列腺素E2,烟酸) 甘油的代谢甘油的主要去路: *经糖异生转变为葡萄糖 *氧化分解为水、二氧化碳、提供能量 *参与TG和磷脂的合成 甘油→3-磷酸甘油→磷酸二羟丙酮→氧化分解,供能 ↓↓
合成磷脂和TG 糖异生 脂酸的氧化分解 概念:脂酸在胞液中活化成脂酰辅酶A,在肉碱的帮助下进入线粒体基质进行β--氧化,每次β--氧化可产生1MOL乙酰辅酶A和比原来少两个碳原子的脂酰辅酶A,偶数碳脂酸最终产生乙酰辅酶A,奇数碳脂酸除乙酰辅酶A外还有1MOL 丙酰辅酶A. 部位:肝、肌肉(脑和成熟红细胞不行) 反应阶段:1)脂酸的活化(胞液) 2)脂酰辅酶A进入线粒体 3)脂酰COA的β--氧化(线粒体) 过程及酶;
有关能量的计算:脂酰COA+7FAD+7NAD++7COA-SH+7H2O→8乙酰COA+7FADH2+7(NADH+H+) 1)软脂酸(16C饱和脂酸的)活化—2ATP 2)7次β--氧化4*7ATP 3)8乙酰COA进入TCA循环彻底氧化10*8ATP 净生成106ATP 脂酰辅酶Aβ--氧化小结 部位:线粒体 四部连续反应:脱氢、加水、再脱氢、硫解
药物化学相关药物母核大全 杂环化合物 环数名称别名及其他信息结构式衍生物 单环 三元环 吖丙啶C2H5N 环氧乙烷C2H4O环氧丙烷环硫乙烷C2H4S 四元环吖丁啶C3H7N 恶丁烷C3H6O 噻丁环C3H6S 五元环含 一 个 杂 原 子 呋喃 含氧五元杂环化合物, 又称氧(杂)茂。 四氢呋喃 呋喃甲醛吡咯 含有一个氮杂原子的五元杂 环化合物, 又称氮(杂)茂。 还原成 二氢和 四氢吡咯噻吩 含有一个硫杂原子的五元杂 环化合物。 四氢噻吩 含 两 个 杂 原 子 吡唑 1,2-二氮唑; 邻二氮杂茂。 咪唑 1,3-二氮杂环戊二烯; 1,3-二氮唑,间二氮茂。 恶唑 含有一个氧和一个氮杂原子 的五元杂环化合物;环中的氧 和氮原子分别占1,3两位,
又称氮代呋喃 异恶唑氧和氮原子分别占1,2位,则称为异恶唑 噻唑噻唑含有一个硫和一个氮杂 原子的五元杂环化合物,分子式C3H3NS。唑字由外文字尾azole译音而来,意为含氮的五元杂环,除吡咯外都称为某唑。硫和氮占1,3两位的称为噻唑。 异噻唑硫和氮占1,2两位的,称为异噻唑。 六元环含 一 个 杂 原 子 吡啶 是含有一个氮杂原子的六元杂 环化合物。可以看做苯分子中的 一个(CH)被N取代的化合物, 故又称氮苯。 六氢吡啶 烟酸 烟酸胺 异烟肼吡喃 含有一个氧杂原子的六元杂 环化合物。 噻喃C5H6S 含 两 个 杂 原 子 哒嗪 1、2位含两个氮杂原子的六元 杂环化合物, 又称邻二氮苯。 嘧啶 1、3两位的称为嘧啶,由2个 氮原子取代苯分子间位上的2个 碳形成,是一种二嗪。
吡嗪占1、4两位的称为吡嗪 哌嗪对二氮己环 七元环及 以上 杂??指环庚三烯正离子 稠环 五元及六元稠杂环 吲哚 吲哚是吡咯与苯并联的化合 物。 苯并咪唑间(二)氮茚。 咔唑9H-咔唑。 喹啉吡啶与苯并联的化合物。 异喹啉 蝶啶 吡嗪和嘧啶并联而成的二杂环 化合物 7H-嘌呤
第二章药物代谢 本章提示: 药物代谢是在体内酶的作用下使药物的化学结构发生变化,大多使有效药物转变为低效或无效的代谢物,有时也会产生活性代谢物;也有可能转变成毒副作用较高的产物。而前药设计则是通过代谢转变产生有效药物。执业药师应熟悉药物在体内代谢的化学变化类型,以及药物的化学结构变化后产生生物活性的变化。 药物进入机体后,一方面药物对机体产生诸多生理药理作用,即对疾病治疗作用;另一方面对机体来讲药物是一种外来的化学物质,机体组织将对药物进行作用设法将其排出体外,这就是药物的代谢。药物代谢是指在酶的作用下将药物(通常是非极性分子)转变成极性分子,再通过人体的正常系统排泄至体外的过程;是药物在人体内发生的化学变化,也是人体对自身的一种保护机能。因此研究药物在体内代谢过程中发生的化学变化,更能阐明药理作用的特点,作用时程,结构的转变以及产生毒副作用的原因。 药物的代谢通常分为二相:第Ⅰ相生物转化(Phase Ⅰ),也称为药物的官能团化反应,是体内的酶对药物分子进行的氧化、还原、水解、羟基化等反应,在药物分子中引入或使药物分子暴露出极性基团,如羟基、羧基、巯基、氨基等。第Ⅱ相生物结合(Phase Ⅱ),是将第Ⅰ相中药物产生的极性基团与体内的内源性成分,如葡萄糖醛酸、硫酸、甘氨酸或谷胱甘肽,经共价键结合,生成极性大、易溶于水和易排出体外的轭合物。但是也有药物经第Ⅰ相反应后,无需进行第Ⅱ相的结合反应,即排出体外。其中第Ⅰ相生物转化反应对药物在体内的活性影响最大。 由于催化反应时酶对底物化学结构有一定的要求,因此不同化学结构的药物,其代谢的情况也不一样。 第一节药物的官能团化反应(第Ⅰ相生物转化) 一、含芳环药物的代谢 含芳环的药物主要发生氧化代谢,是在体内肝脏CYP 450酶系催化下,首先将芳香化合物氧化成环氧化合物,然后在质子的催化下会发生重排生成酚,或被环氧化物水解酶水解生成二羟基化合物。生成的环氧化合物还会在谷胱甘肽S-转移酶的作用下和谷胱甘肽生成硫醚;促进代谢产物的排泄。但是环氧化物若和体内生物大分子如DNA或RNA中的亲核基团反应,生成共价键的结合物,而使生物大分子失去活性,则产生毒性。 含芳环药物的氧化代谢是以生成酚的代谢产物为主,芳环上的供电子取代基能使反应容易进行,生成酚羟基的位置在取代基的对位或邻位;吸电子取代基则削弱反应的进行程度,生成酚羟基的位置在取代基的间位。和一般芳环的取代反应一样,芳环的氧化代谢部位也受到立体位阻的影响,通常发生在立体位阻较小的部位。如果药物分子中含有二个芳环时,一般只有一个芳环发生氧化代谢。如苯妥英(Phenytoin)在体内代谢后生成羟基苯妥英失去生物活性。 H H
脂类代谢 一、问答题 1、为什么摄入糖量过多容易长胖? 答:因为脂肪酸合成的起始原料乙酰CoA主要来自糖酵解产物丙酮酸,摄入糖量过多则糖酵解产生的丙酮酸也多,进而导致合成脂肪酸的起始原料乙酰CoA也多,原料多合成的脂肪酸自然就多了,所以摄入糖量过多容易长胖。 2、比较脂肪酸β—氧化与脂肪酸的合成有哪些不同点? 答:①细胞中发生部位不同:合成发生在细胞质,氧化发生在线粒体;②酰基载体不同:合成所需载体为ACP—SH,氧化所需载体为乙酰CoA; ③二碳片段的加入与裂解方式:合成就是以丙二酰ACP加入二碳片段,氧化的裂解方式就是乙酰CoA;④电子供体或受体:合成的供体就是NADPH,氧化的受体就是FAD、FAD+;⑤酶系不同:合成需7种酶,氧化需4种酶;⑥原料转运方式:合成就是柠檬酸转运系统,氧化就是肉碱穿梭系统;⑦能量变化:合成耗能,氧化产能。 3、试计算1mol甘油彻底氧化成CO2与H2O可净生成多少molATP。答:甘油氧化产生的乙酰CoA进入三羧酸循环彻底氧化。经过4次脱氢反应生成3molNADH+H+、1molFADH2、以及2molCO2,并发生一次底物水平磷酸化,生成1molGTP。依据生物氧化时每1molNADH+H+与1molFADH2 分别生成2、5mol、1、5mol的ATP,
因此,1mol甘油彻底氧化成CO2与H2O生成ATP摩尔数为6×2、5+1×1、5+3-1=18、5。 4、1mol硬脂酸(即18碳饱与脂肪酸)彻底氧化成CO2与H2O时净生成的ATP的摩尔数。 答:1mol硬脂酸彻底氧化需经8次循环,产生9个乙酰CoA,每摩尔乙酰CoA进入三羧酸循环产生10molATP,这样共产生90molATP。8molFADH2进入电子传递链产生12molATP,8molNADH进入电子传递链共产生20molATP。脂肪酸的活化需消耗2个高能磷酸键,这样彻底氧化1mol硬脂酸净得120molATP。 5、胆固醇在体内可转变成哪些重要物质?合成胆固醇的基本原料与关键酶各就是什么? 答:转变成胆汁酸、甾类激素、维生素D; 基本原料:二甲基丙烯焦磷酸酯(DPP)、异戊烯醇焦磷酸酯 关键酶:羟甲基戊二酸单酰CoA还原酶(HMGCoA还原酶) 6、为什么在长期饥饿或糖尿病状态下,血液中酮体浓度会升高?答:由于糖供应不足或利用率降低,机体需动员大量的脂肪酸供能,同时生成大量的乙酰CoA。此时草酰乙酸进入糖异生途径,又得不到及时的回补而浓度降低,因此不能与乙酰CoA缩合成柠檬酸。在这种情况下,大量积累的乙酰CoA衍生为丙酮、乙酰乙酸、β—羟丁酸。