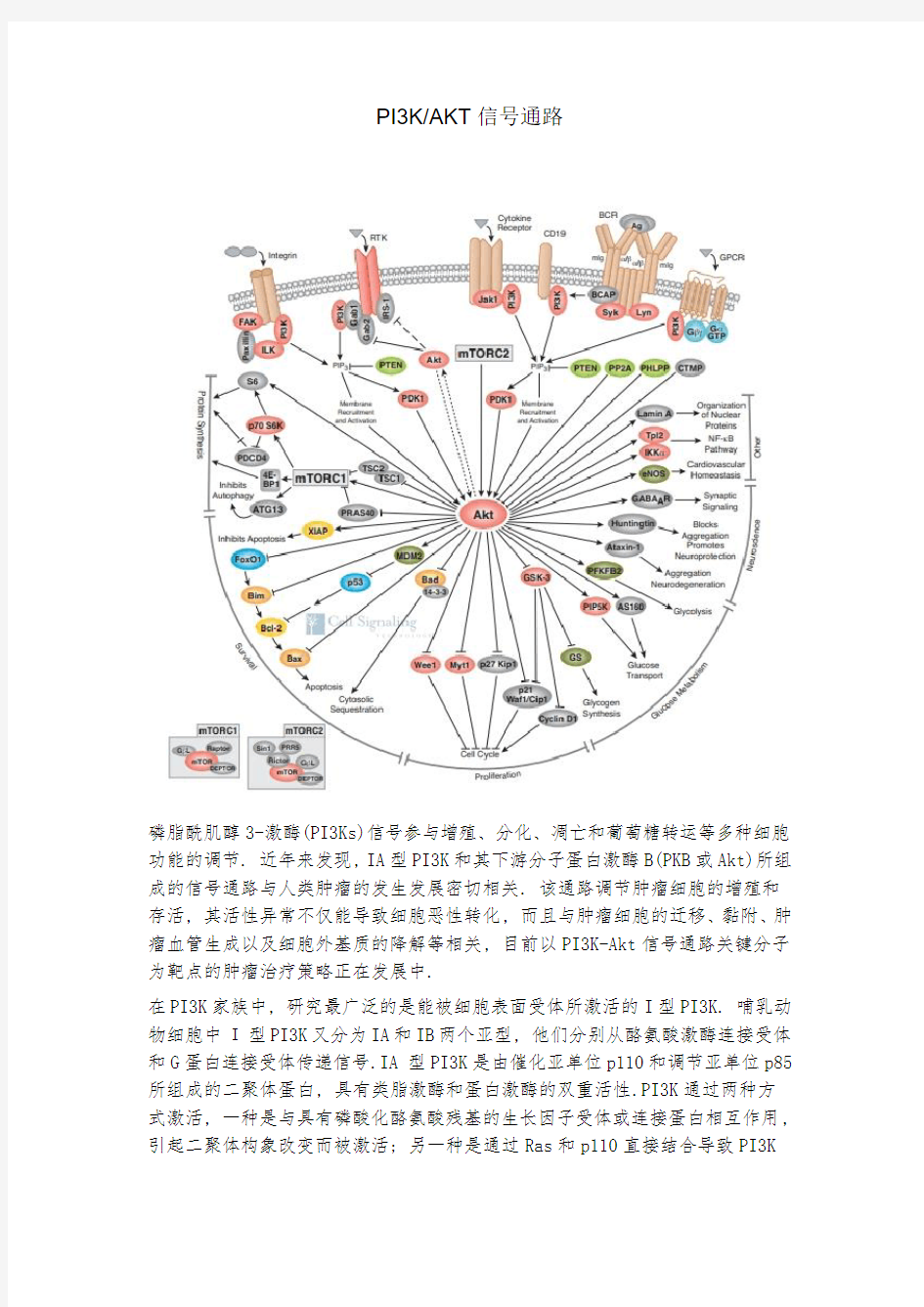

PI3K/AKT信号通路
磷脂酰肌醇3-激酶(PI3Ks)信号参与增殖、分化、凋亡和葡萄糖转运等多种细胞功能的调节. 近年来发现, IA型PI3K和其下游分子蛋白激酶B(PKB或Akt)所组成的信号通路与人类肿瘤的发生发展密切相关. 该通路调节肿瘤细胞的增殖和存活, 其活性异常不仅能导致细胞恶性转化, 而且与肿瘤细胞的迁移、黏附、肿瘤血管生成以及细胞外基质的降解等相关, 目前以PI3K-Akt信号通路关键分子为靶点的肿瘤治疗策略正在发展中.
在PI3K家族中, 研究最广泛的是能被细胞表面受体所激活的I型PI3K. 哺乳动物细胞中Ι型PI3K又分为IA和IB两个亚型, 他们分别从酪氨酸激酶连接受体和G蛋白连接受体传递信号.IA 型PI3K是由催化亚单位p110和调节亚单位p85所组成的二聚体蛋白, 具有类脂激酶和蛋白激酶的双重活性.PI3K通过两种方
式激活, 一种是与具有磷酸化酪氨酸残基的生长因子受体或连接蛋白相互作用, 引起二聚体构象改变而被激活; 另一种是通过Ras和p110直接结合导致PI3K
的活化. PI3K激活的结果是在质膜上产生第二信使PIP3, PIP3与细胞内含有PH 结构域的信号蛋白Akt和PDK1(phosphoinositidedependentkinase-1)结合, 促使PDK1磷酸化Akt蛋白的Ser308导致Akt的活化. Akt还能通过PDK2(如整合素连接激酶ILK)对其Thr473的磷酸化而被激活.活化的Akt通过磷酸化作用激活或抑制其下游靶蛋白Bad 、Caspase9、NF-κB、GSK-3、FKHR、 p21Cip1和
p27 Kip1等, 进而调节细胞的增殖、分化、凋亡以及迁移等.
PI3K-Akt信号通路的活性被类脂磷酸酶PTEN(phosphatase and tensin homolog deleted on chromosome ten)和SHIP(SH2-containing inositol 5-phosphatase)负调节, 他们分别从PIP3的3′和5′去除磷酸而将其转变成PI(4,5)P2和
PI(3,4)P2而降解. 迄今为止, 尚未发现下调Akt活性的特异磷酸酶, 但用磷酸酶抑制剂处理细胞后, 发现Akt的磷酸化和活性均有所增加. 最近发现Akt能被一种C末端调节蛋白(CTMP)所失活, CTMP能结合Akt并通过抑制Akt的磷酸化而阻断下游信号的传递, CTMP的过表达能够逆转v-Akt转化细胞的表型. 热休克蛋白90(HSP90)亦能结合Akt, 阻止Akt被PP2A磷酸酶的去磷酸化而失活, 因此具有保护Akt的作用.
本信号转导涉及的信号分子主要包括
Integrin,FAK,Paxillin,ILK,PIP3,S6,p70S6K,RTK,Gab1,Gab2,IRS-1,PI3K,PTEN,AKT,PDK1,Cytokine Receptor,Jak1,CD19,BCR,Ag,BCAP,Syk,Lyn,GPCR,TSC1,TSC2,Gβγ,GαGTP,PP2A,PHLPP,CTMP,PDCD4,4E-BP1,ATG13,mTORC1,TSC1,TSC2,PRAS40,XIAP,FoxO1,Bim,Bcl-2,Bax,MDM2,p53,Bax,Bad,14-3-3,Wee1,Myt1,p27Kip1,p21Waf1/Cip1,CyclinD1,GSK-3,GS,Bcl-2,mTORC2,LaminA,Tpl2,IKKα,eNOS,GABAAR,Huntingtin,Ataxin-1,PFKFB2,PIP5K,AS160等。
信号通路9—MAPK Signaling APExBIO 图▲ MAPK信号通路图 丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK, MAP kinase)是一种对丝氨酸,苏氨酸和酪氨酸特异的蛋白激酶(即丝氨酸/苏氨酸特异性蛋白激酶)。由于MAPK是培养细胞在受到生长因子等丝裂原刺激时被激活而被鉴定的,因而得名。MAPKs参与引导细胞反应至各类刺激物,如有丝分裂原,渗透压,热休克和促炎细胞因子。MAPKs调节多种细胞功能,包括增殖,基因表达,分化,有丝分裂,细胞存活和凋亡。 MAPKs仅在真核生物中发现。MAPKs属于CMGC(CDK / MAPK / GSK3 / CLK)激酶组。CDK相关程度最大。
MAPK链由3类蛋白激酶组成:上游激活蛋白→MAPK激酶激酶(MAPKKK)→MAPK激酶(MAPKK)→MAPK,通过依次磷酸化将上游信号传递至下游应答分子。 经典的MAPK通路激活开始于细胞膜,在这里,小GTP酶和各种蛋白激酶磷酸化并激活MAPKKK(MAP kinase kinase kinase,MAP3K或MKKK,MAPK激酶激酶)。随后,MAPKKK直接磷酸化MAPKK(MAP kinase kinase,MAP2K 或MKK,MAPK激酶),MAPKK一旦被激活就会磷酸化并激活MAPK。MAPK 的激活导致特异性MAPK激活蛋白激酶(MAPKAPK,MAPK-activated protein kinase)的磷酸化和活化,例如RSK,MSK或MNK家族成员和MK2/3/5。 MKKK的4个亚族已得到鉴定: A. Raf亚族。研究的最为透彻,包括B-Raf、A-Raf、Raf1。 B. MEKK亚族。由4种MEKK构成:MEKK1~MEKK4。
干货细胞信号通路图解之MAPK通路【值得珍藏】 科研小助手原创,转载请注明来源。公众号内回复“Cell Signaling Pathway”获取全套信号通路图本文由百度贴吧nosce吧吧主黄杰投稿一、MAPK信号通路: (1)有丝分裂原激活的蛋白激酶(MAPK)是一族在真核生物中非常保守的丝/苏氨酸蛋白激酶,在许多细胞活动中起作用,如生长增殖,细胞分化,细胞运动或死亡。MAPK级联信号传导由3 个不同层次的分子所组成。MAPK被MAPK的激 酶( MAPKK)磷酸化后激活,MAPKK被MAPKK的激酶(MAPKKK )磷酸化而激活。而MAPKKK通过与小GTPase 和/或其他蛋白酶相互作用而被激活,从而将MAPK和细胞 表面的受体以及胞外的信号联系在一起。 (2)许多参与生长和分化的受体都能够激活MAPK/ERK信号通路,比如说受体酪氨酸激酶(RTK),整合素,和离子通道。响应特定信号所涉及到的具体分子会相差很大,但通路的结构是一致的,那就是接头分子(adaptor,如Shc, GRB2, Crk等)将鸟苷酸交换因子(SOS, C3G 等)和受体连接在一起,然后把信号向小GTP 结合蛋白(Ras, Rap1)传递,后者又激活核心的级联反应,这是由一个MAPKKK( Raf) ,一个MAPKK( MEK1/2)和MAPK( Erk)所构成的。活化的ERK 二聚体能调节胞浆中的目标分子,也可以转移到细胞核中,然
后对一系列转录因子进行磷酸化以调节基因表达。SciRes(3)很多外部的刺激都能够激活G蛋白偶联受体(GPCR)。在受体活化以后,G 蛋白将GDP 转换成GTP ,然后结合了GTP的α和β/γ亚基从受体脱离开,启动信号向胞内的传导。与不同亚型的异质三聚体G 蛋白结合的受体可以采取不同 的手段激活小G 蛋白/MAPK级联反应,至少有三个不同家族的酪氨酸激酶参与其中。Src家族激酶响应活化的PI3Kγ,而后者被β/γ亚基激活。它们还能够响应受体的内化,受体酪氨酸激酶的交叉活化,以及有Pyk2 和/或FAK参与的整 合素途径信号。GPCRs同样可以通过PLCβ去激活PKC 和CaMKII ,对下游的MAPK通路可以有激活或抑制的影响。SciRes(4)压力激活的蛋白激酶(Stress-activated protein kinase, SAPK)或称Jun氨基端激酶(Jun amino-terminal kinase, JNK) 是MAPK的家族成员,能被一系列的环境压力,炎症细胞因子,生长因子和GPCR激动剂所激活。压力信号通过Rho家族的小GTP 酶(small GTPase)向这条级联通路传导,这些小GTP酶包括(Rac, Rho, cdc42) 。和其他的MAPK情况一样,靠近膜的激酶是一个MAPKKK,一般 是MEKK1-4 ,或者是一个混合激酶去磷酸化并激活 MKK4(SEK)或MKK7,它们是SAPK/JNK的激酶。另外,MKK4/7也可以被生发中心激酶(germinal center kinase, GCK)以一种GTPase 依赖的方式激活。活化后的
1 PPAR信号通路:过氧化物酶体增殖物激活受体( PPARs) 是与维甲酸、类固醇 和甲状腺激素受体相关的配体激活转录因子超家族核激素受体成员。它们作为脂 肪传感器调节脂肪代谢酶的转录。PPARs由PPARα、PPARβ和PPARγ 3种亚型组成。PPARα主要在脂肪酸代谢水平高的组织,如:肝、棕色脂肪、心、肾和骨骼肌表达。他通过调控靶基因的表达而调节机体许多生理功能包括能量代谢、生 长发育等。另外,他还通过调节脂质代谢的生物感受器而调节细胞生长、分化与 凋亡。PPARa同时也是一种磷酸化蛋白,他受多种磷酸化酶的调节包括丝裂原激活蛋白激酶( ERK-和p38.M APK) ,蛋白激酶A和C( PKA,PKC) ,AM PK和糖原合成酶一3( G SK3) 等调控。调控PPARa生长信号的酶报道有M APK、PKA和G SK3。PPARβ广泛表达于各种组织,而PPAR γ主要局限表达在血和棕色脂肪,其他组织如骨骼肌和心肌有少量表达。PPAR-γ在诸如炎症、动脉粥样硬化、胰岛素抵抗和糖代谢调节,以及肿瘤和肥胖等方面均有着举足轻重的作用, 而其众多生物学效应则是通过启动或参与的复杂信号通路予以实现。鉴于目前人 们对PPAR—γ信号通路尚不甚清,PPARs通常是通过与9-cis维甲酸受体( RXR)结合实现其转录活性的。 2 MAPK信号通路:mapk简介:丝裂原激活蛋白激酶(mitogen—activated protein kinase,MAPK)是广泛存在于动植物细胞中的一类丝氨酸/苏氨酸蛋白激酶。作用主要是将细胞外刺激信号转导至细胞及其核内,并引起细胞的生物化学反应(增殖、分化、凋亡、应激等)。 MAPKs家族的亚族 :ERKs(extracellular signal regulated kinase):包括ERK1、ERK2。生长因子、细胞因子或激素激活此通路,介导细胞增殖、分化。 JNKs(c-Jun N-terminal kinase)包括JNK1、JNK2、JNK3。此亚族成员能使 Jun转录因子N末端的两个氨基酸磷酸化而失活,因此称为Jun N末端激酶(JNKs)。物理、化学的因素引起的细胞外环境变化以及致炎细胞因子调节此通路。P38 MAPKs:丝氨酸/络氨酸激酶,包括p38 α、p38β、p38γ、p38δ。p38 MAP K参与多种细胞内信息传递过程 ,能对多种细胞外刺激发生反应,可磷酸化其它细胞质蛋白,并能从胞浆移位至细胞核而调节转录因子的活性来改变基因的表达水平 ,从而介导细胞生长、发育、分化及死亡的全过程。 ERK5:是一种非典型的MAPK通路,也叫大MAPK通路,只有一个成员。它可被各种刺激因素激活。不仅可以通过磷酸化作用使底物活化,并且通过C端的物理性结合作用激活底物。 3 ERBB信号途径:ErbB 蛋白属于跨膜酪氨酸激酶的 EGF 受体家族成员。ErbB 的命名来源于在禽红白血病 B( v-Erb-B) 发现的 EGF 受体的突变体,因而 EGF 受体 亦称为“ ErbB1”。人源 ErbB2 称为HER2, 特指人的 EGF 受体。ErbB 家族的
1JAK-STAT信号通路 1)JAK与STAT蛋白 JAK-STAT信号通路是近年来发现的一条由细胞因子刺激的信号转导通路,参与细胞的增殖、分化、凋亡以及免疫调节等许多重要的生物学过程。与其它信号通路相比,这条信号通路的传递过程相对简单,它主要由三个成分组成,即酪氨酸激酶相关受体、酪氨酸激酶JAK和转录因子STAT。(1)酪氨酸激酶相关受体(tyrosinekinaseassociatedreceptor) 许多细胞因子和生长因子通过JAK-STAT信号通路来传导信号,这包括白介素2?7(IL-2?7)、GM-CSF(粒细胞/巨噬细胞集落刺激因子)、GH(生 长激素)、EGF(表皮生长因子)、PDGF(血小板衍生因子)以及IFN(干扰素)等等。这些细胞因子和生长因子在细胞膜上有相应的受体。这些受体的共同特点是受体本身不具有激酶活性,但胞内段具有酪氨酸激酶JAK 的结合位点。受体与配体结合后,通过与之相结合的JAK的活化,来磷酸化各种靶蛋白的酪氨酸残基以实现信号从胞外到胞内的转递。 (2)酪氨酸激酶JAK(Januskinase) 很多酪氨酸激酶都是细胞膜受体,它们统称为酪氨酸激酶受体(receptor tyrosinekinase,RTK),而JAK却是一类非跨膜型的酪氨酸激酶。JAK是英文Januskinase的缩写,Janus在罗马神话中是掌管开始和终结的两面神。之所以称为两面神激酶,是因为JAK既能磷酸化与其相结合的细胞因子受体,又能磷酸、JAK1个成员:4蛋白家族共包括JAK结构域的信号分子。SH2化多个含特定
JAK2、JAK3以及Tyk2,它们在结构上有7个JAK同源结构域(JAKhomologydomain,JH),其中JH1结构域为激酶区、JH2结构域是“假”激酶区、JH6和JH7是受体结合区域。 (3)转录因子STAT(signaltransducerandactivatoroftranscription)STAT被称为“信号转导子和转录激活子”。顾名思义,STAT在信号转导和转录激活上发挥了关键性的作用。目前已发现STAT家族的六个成员,即STAT1-STAT6。STAT蛋白在结构上可分为以下几个功能区段:N-端保守序列、DNA结合区、SH3结构域、SH2结构域及C-端的转录激活区。其中,序列上最保守和功能上最重要的区段是SH2结构域,它具有与酪氨酸激酶Src的SH2结构域完全相同的核心序列“GTFLLRFSS”。 2)JAK-STAT信号通路 与其它信号通路相比,JAK-STAT信号通路的传递过程相对简单。信号传 递过程如下:细胞因子与相应的受体结合后引起受体分子的二聚化,这使得与受体偶联的JAK激酶相互接近并通过交互的酪氨酸磷酸化作用而活化。JAK激活后催化受体上的酪氨酸残基发生磷酸化修饰,继而这些磷酸化的酪氨酸位点与周围的氨基酸序列形成“停泊位点”(dockingsite),同时含有SH2结构域的STAT蛋白被招募到这个“停泊位点”。最后,激酶JAK 催化结合在受体上的STAT蛋白发生磷酸化修饰,活化的STAT蛋白以二 聚体的形式进入细胞核内与靶基因结合,调控基因的转录。值得一提的是,一种JAK激酶可以参与多种细胞因子的信号转导过程,一种细胞因子的信号通路也可以激活多个JAK激酶,但细胞因子对激活的STAT分子却具有一定的选择性。例如IL-4激活STAT6,而IL-12 。STAT4却特异性激活
MAPK 细胞最基本的生命活动是细胞的生长、分化与分裂。 细胞分裂周期可分为DNA 及蛋白质合成作准备的G1 期、DNA 合成的S 期、为有丝分裂作准备的G2 期与有丝分裂的M 期以及细胞呈相对稳定状态的G0 期。 生物信息通过一系列复杂的信号传递过程来诱导相关基因的表达、调控细胞分裂,决定细胞的转归。衰老细胞的细胞周期常阻滞于G1/ S 期或G2/M期,尤其是G1 末期的限制性调控点“R”点的阻滞。 促分裂素原活化蛋白激酶(mitogen-activated protein kinases,MAP激酶,MAPK)链是真核生物信号传递网络中的重要途径之一,在基因表达调控和细胞质功能活动中发挥关键作用。MAPK 链由3类蛋白激酶MAP3K-MAP2K-MAPK组成,通过依次磷酸化将上游信号传递至下游应答分子. MAPK信号通路包括:MAP激酶(MAPK)、MAPK激酶(MEK、MKK或MAPK 激酶)和MEK 激酶(MEKK、MKKK或MAPK激酶激酶)。在哺乳动物机体中,已经发现五种不同的MAPK 信号转导通路。其中ERK1/2信号转导通路调控细胞生长和分化,JNK和p38 MAPK信号转导通路在炎症与细胞凋亡等应激反应中发挥重要作用。使用这一芯片试剂盒检测RNA实验标本,操作者通过杂交反应技术,即可研究实验系统中与MAPK信号通路相关基因表达水平改变。 MAPK属于一种Ser/Thr蛋白激酶,可在多种不同的信号转导途径中充当一种共同的信号转导成份,且在细胞周期调控中发挥重要的作用。目前MAPK家族中至少有4个成员已被纯化和深入研究。如p42mapk,p44erk1,p54MAPK及p44mpk。 MAPK可促进血管内皮细胞增殖和新血管生成。新血管生成后可为肿瘤提供更多的营养,加速肿瘤的生长,促进癌细胞的扩散。 MAPK有4个主要亚族:ERK、JNK、p38MAPK和ERK5。
目录 actin肌丝 (5) Wnt/LRP6 信号 (7) WNT信号转导 (7) West Nile 西尼罗河病毒 (8) Vitamin C 维生素C在大脑中的作用 (10) 视觉信号转导 (11) VEGF,低氧 (13) TSP-1诱导细胞凋亡 (15) Trka信号转导 (16) dbpb调节mRNA (17) CARM1甲基化 (19) CREB转录因子 (20) TPO信号通路 (21) Toll-Like 受体 (22) TNFR2 信号通路 (24) TNFR1信号通路 (25) IGF-1受体 (26) TNF/Stress相关信号 (27) 共刺激信号 (29) Th1/Th2 细胞分化 (30) TGF beta 信号转导 (32) 端粒、端粒酶与衰老 (33) TACI和BCMA调节B细胞免疫 (35) T辅助细胞的表面受体 (36) T细胞受体信号通路 (37) T细胞受体和CD3复合物 (38) Cardiolipin的合成 (40) Synaptic突触连接中的蛋白 (42) HSP在应激中的调节的作用 (43) Stat3 信号通路 (45) SREBP控制脂质合成 (46) 酪氨酸激酶的调节 (48) Sonic Hedgehog (SHH)受体ptc1调节细胞周期 (51) Sonic Hedgehog (Shh) 信号 (53) SODD/TNFR1信号 (56) AKT/mTOR在骨骼肌肥大中的作用 (58) G蛋白信号转导 (59) IL1受体信号转导 (60) acetyl从线粒体到胞浆过程 (62) 趋化因子chemokine在T细胞极化中的选择性表达 (63) SARS冠状病毒蛋白酶 (65) SARS冠状病毒蛋白酶 (67) Parkin在泛素-蛋白酶体中的作用 (69)
THE P38 SIGNALING PATHWAY p38 MAPK is phosphorylated and activated by either MKK3 or MKK6. Similar to the MAPKKs in the JNK andERK pathways, MKK3 and MKK6 phosphorylate the MAPK component, in this case p38, on both a tyrosine and threonine residue. MKK3 and MKK6 are directly downstream of a kinase known as MLK3 in this pathway. MLK3 is activated by the small G-proteins Rac1 and cdc42 (162). Both growth factor receptors and members of the TNF family of receptors are known to activate this pathway. The TNF family of receptors activate the p38 pathway via the activation of cdc42 (95), whereas growth factor receptors have been proposed to active this pathway via the sequential activation of RAS and Rac1 (63, 151). Thus, many of the initial proteins and activation events in the JNK pathway are also involved in the activation of the p38 pathway. ASK1 is also able to induce the activation of the p38 pathway. This activation is thought to occur via ASK1 phosphorylation of MKK3 and 6 (75). In some cases growth factor removal can result in the activation of the p38 pathway (9). Targets of p38 kinase activity include multiple transcription factors such as MEF2 (184), ATF-2 (106), Elk-1 (188), and indirectly CREB (138, 154). The p38 pathway is the only MAPK pathway that does not induce an antioxidant response via the phosphorylation of Nrf2. In fact, signaling via the p38 pathway may actually inhibit Nrf2 phosphorylation by other MAPK pathways (126, 190). This finding may explain the ability of this pathway to strongly promote apoptosis (182). The ability of RAS to activate Rho, and subsequently the p38 signaling pathway, may be the reason that transfection with RAS can lead to or augment apoptosis in some cases (54, 168, 173). Removal of IL-3 from cultures of the cytokine-dependent TF-1 hematopoietic cell line results in the induction of apoptosis, and activation of the JNK and p38 pathways (9). The p38 pathway under these conditions appeared to be important for the induction of apoptosis because inhibitors of p38 prevented IL-3-deprived TF-1 cells from undergoing apoptosis. To determine if the balance between the ERK and p38 signaling pathways determines the fate of the cell, Birkenkamp et al. incubated cells with IL-1 (9). IL-1 will induce the activation of the ERK, JNK, and p38 signaling pathways, whereas IL-3 removal only induced JNK and p38 expression. They found that IL-1, unlike cytokine withdrawal, did not induce apoptosis in these cells. These investigators then demonstrated that inhibition of the ERK signaling pathway with PD98059 allowed IL-1 to induce apoptosis in these cells. These data suggest that although the activation of the p38 pathway may be required for growth factor withdrawal-induced apoptosis, in the presence of high enough levels of ERK activation, p38 activation may not be sufficient in itself for apoptosis to occur. These data also demonstrate that the effects of the ERK signaling pathway can overcome the pro-apoptotic effects of the p38 signaling pathways, at least in certain experimental conditions (Fig. 3) ACTIVATION OF THE P38 PATHWAY BY OXIDATIVE STRESS Singlet oxygen (25, 91, 195), hydrogen peroxide (65), nitric oxide (98, 99), and peroxynitrite (143) all activate the p38MAPK pathway. The p38 MAPK pathway is known to be activated in a number of different cell types in response to reactive oxygen intermediates. These cell types include: Jurkat, 3T3, HeLa, fibroblasts, and endothelial cells (90). The mechanism by which this occurs is likely very similar to the mechanisms by which JNK activation occurs, as many of the same signals activate both pathways concurrently and in many of the same cell types. RAS activation and subsequent signaling via Rho can also activate this pathway as does ligation of the TNF receptor (75, 121, 162). Thus, the ability of oxygen radicals to induce receptor signaling by the TNF receptor in the absence of any receptor ligand binding could also have a potential role in activating the p38 pathway. The ability of nitric oxide to increase RAS activity indicates a potential mechanism by which reactive nitrogen intermediates can induce signaling via the p38 pathway (98). Similar to the JNK pathway, ASK1 has a role in oxidant-induced activation of the p38 pathway (112, 114) and is yet another mechanism by which oxygen radicals may induce p38 activation. Deletion of ASK1 protects against hydrogen peroxide-induced apoptosis in fibroblasts and also prevents prolonged p38 activation, suggesting an apoptotic role for p38 in response to oxidative stress (164). These data also suggest that the kinetics of p38 activation may also be important in determining the fate of the cell.
mTOR信号通路图 mTOR可对细胞外包括生长因子、胰岛素、营养素、氨基酸、葡萄糖等多种刺激产生应答。它主要通过PI3K/Akt/mTOR途径来实现对细胞生长、细胞周期等多种生理功能的调控作用。正常情况下,结节性脑硬化复合物-1(TSC-1)和TSC-2形成二聚体复合物,是小GTP 酶Rheb(Ras-homolog enriched in brain)的抑制剂,而Rheb是mTOR活化所必需的刺激蛋白,因此TSC-1/TSC-2在正常情况下抑制mTOR的功能。当Akt活化后,它可磷酸化TSC-2的Ser939和Thr1462,抑制了TSC-1/TSC-2复合物的形成,从而解除了对Rheb 的抑制作用,使得mTOR被激活。活化的mTOR通过磷酸化蛋白翻译过程中的某些因子来参与多项细胞功能,其中最主要的是4EBP1和P70S6K。
在整个PI3K/Akt/mTOR信号通路中,有一条十分重要的负反馈调节剂就是10号染色体上缺失与张力蛋白同源的磷酸酶基因(phosphatase and tensin homology deleted on chromosome 10, PTEN)。PTEN是一个肿瘤抑制基因,位于人染色体10q23。它有一个蛋白酪氨酸磷酸酶结构域,在这条通路中可以将PI-3,4-P2与PI-3,4,5-P3去磷酸化,从而负调节PI3K下游AKt/mTOR信号通路的活性。 本信号转导涉及的信号分子主要包括 IRS-1,PI3K,PIP2,PIP3,PDK1,PTEN,Akt,TSC1,TSC2,Rheb,mTOR,Raptor,DEPTOR,GβL,p70S6K,ATG13,4E-BP1,HIF-1,PGC-1α,PPARγ,Sin1,PRR5,Rictor,PKCα,SGK1,PRAS40,FKBP12,Wnt,LRP,Frizzled,Gαq/o,Dvl,Erk,RSK,GSK-3,REDD1,REDD2,AMPK,LKB1,RagA/B,RagC/D等。
MAPK信号通路 2008-06-04 21:50 MAPK,丝裂原活化蛋白激酶(mitogen-activated protein kinases,MAPKs)是细胞内的一类丝氨酸/苏氨酸蛋白激酶。研究证实,MAPKs信号转导通路存在于大多数细胞内,在将细胞外刺激信号转导至细胞及其核内,并引起细胞生物学反应(如细胞增殖、分化、转化及凋亡等)的过程中具有至关重要的作用。研究表明,MAPKs信号转导通路在细胞内具有生物进化的高度保守性,在低等原核细胞和高等哺乳类细胞内,目前均已发现存在着多条并行的MAPKs信号通路,不同的细胞外刺激可使用不同的MAPKs信号通路,通过其相互调控而介导不同的细胞生物学反应。 1并行MAPKs信号通路的组成及其活化特点 在哺乳类细胞目前已发现存在着下述三条并行的MAPKs信号通路[1]。 1.1ERK(extracellular signal-regulated kinase)信号通路1986年由Sturgill等人首先报告的MAPK。最初其名称十分混乱,曾根据底物蛋白称之为MAP2K、ERK、MBPK、RSKK、ERTK等。此后,由于发现其具有共同的结构和生化特征,而被命名为MAPK。近年来,随着不同MAPK家族成员的发现,又重新改称为ERK。 在哺乳类动物细胞中,与ERK相关的细胞内信号转导途径被认为是经典MAPK信号转导途径,目前对其激活过程及生物学意义已有了较深入的认识。研究证实,受体酪氨酸激酶、G蛋白偶联的受体和部分细胞因子受体均可激活ERK信号转导途径。如:生长因子与细胞膜上的特异受体结合,可使受体形成二聚体,二聚化的受体使其自身酪氨酸激酶被激活;受体上磷酸化的酪氨酸又与位于胞膜上的生长因子受体结合蛋白2(Grb2)的SH2结构域相结合,而Grb2的SH3结构域则同时与鸟苷酸交换因子SOS(Son of Sevenless)结合,后者使小分子鸟苷酸结合蛋白Ras的GDP解离而结合GTP,从而激活Ras;激活的Ras进一步与丝/苏氨酸蛋白激酶Raf-1的氨基端结合,通过未知机制激活Raf-1;Raf-1可磷酸化MEK1/MEK2(MAP kinase/ERK kinase)上的二个调节性丝氨酸,从而激活MEKs;MEKs为双特异性激酶,可以使丝/苏氨酸和酪氨酸发生磷酸化,最终高度选择性地激活ERK1和ERK2(即p44MAPK和p42MAPK)。
KEGG上的信号通路图怎么看? 提示:请点击标题下方蓝色“实验万事屋”,添加关注后,发“嗯”可以查看我们之前的文章。未经允许,其他公众号不得转载哦! 想要把自己研究的分子扯上明星分子或者明星通路?那是不难,难的是具体到底要怎么去扯,芯片结果啊,生信结果啊,都会给你提示,但真的要具体扯上去,还得看懂那些七七八八的信号通路图。 KEGG Pathway上有着大量的信号通路图,画得一个复杂啊!巨坑爹有没有?曾经有师弟说我之前曾经把Wnt通路描述错了,他师兄告诉他,应该是GSK-3β磷酸化抑制β-Catenin降解,并促进它入核的。在这里,我们只能默默地祝福这位师兄了…… 那我们就用Wnt通路来做例子吧。先上KEGG下载一个Wnt的信号通路图,如下: 绝壁是很高大上的不是么?这要咋看呢?其实这张图上把三个Wnt通路都画上去了,也就是Wnt/β-Catenin(经典Wnt通路),Wnt/PCP(平面的细胞极性途径)和Wnt/Ca2+(Wnt/钙离子)三条信号通路组成,我们就删减一下,就光看经典的Wnt通路,就变成了下面这个模样:
感觉还是很高大上有木有?那就再删减一下,把它变成经典Wnt信号通路的骨架会是什么样呢?就是这样: 简洁明快了吧,但要怎么来看懂这样的图呢?我们来看一下KEGG Pathway的具体图例:
把这些图例用来解释经典Wnt信号通路骨架图,就变成了: 看懂了么?那给你从左到右解释一下: 1)Wnt激活膜上受体,将信号传递到第二信使Dvl,活化的Dvl抑制由Axin、APC 和GSK-3β组成的复合物的活性,使β-catenin不能被GSK-3β磷酸化。 2)磷酸化的β-catenin才可通过泛素化(ubiquitination)而被胞浆内的蛋白酶体所降解,由于非磷酸化的β-catenin不能被蛋白酶体降解,从而导致β-catenin在胞浆内积聚,并移向核内。
MAPK 信号通路2008-06-04 21:50 MAPK, 丝裂原活化蛋白激酶( mitogen-activated protein kinases,MAPKs )是细胞内的一类丝氨酸/苏氨酸蛋白激酶。研究证实,MAPKs 信号转导通路存在于大多数细胞内,在将细胞外刺激信号转导至细胞及其核内,并引起细胞生物学反应(如细胞增殖、分化、转化及凋亡等)的过 程中具有至关重要的作用。研究表明,MAPKs 信号转导通路在细胞内具有生物进化的高度保守性,在低等原核细胞和高等哺乳类细胞内,目前均已发现存在着多条并行的MAPKs 信号通路,不同的细胞外刺激可使用不同的MAPKs 信号通路,通过其相互调控而介导不同的细胞生物学反应。 1 并行MAPKs 信号通路的组成及其活化特点在哺乳类细胞目前已发现存 在着下述三条并行的MAPKs 信号通路 [1]。1.1 ERK (extracellular signal-regulated kinase)信号通路1986 年由Sturgill 等人首先报告的MAPK 。最初其名称十分混乱,曾根据底物蛋白称之为MAP2K 、ERK、MBPK 、RSKK 、ERTK 等。此后,由于发现其具有共同的结构和生化特征,而被命名为MAPK 。近年来,随着不同MAPK 家族成员的发现,又重新改称为ERK 。 在哺乳类动物细胞中,与ERK 相关的细胞内信号转导途径被认为是经典MAPK 信号转导途径,目前对其激活过程及生物学意义已有了较深入的认识。研究证实,受体酪氨酸激酶、G 蛋白偶联的受体和部分细胞因子受体均可激活ERK 信号转导途径。如:生长因子与细胞膜上的特异受体结合,可使受体形成二聚体,二聚化的受体使其自身酪氨酸激酶被激活;受体上磷酸化的酪氨酸又与位 于胞膜上的生长因子受体结合蛋白2( Grb2)的SH2 结构域相结合,而Grb2 的SH3 结构域则同时与鸟苷酸交换因子SOS( Son of Sevenless)结合,后者使小分子鸟苷酸结合蛋白Ras的GDP 解离而结合GTP,从而激活Ras;激活的Ras 进一步与丝/苏氨酸蛋白激酶Raf-1 的氨基端结合,通过未知机制激活Raf-1;Raf-1 可磷酸化MEK1 /MEK2 (MAP kinase/ERK kinase)上的二个调节性丝氨酸,从而激活MEKs ;MEKs 为双特异性激酶,可以使丝/苏氨酸和酪氨酸发生 磷酸化,最终高度选择性地激活ERK1和ERK2(即p44MAPK 和p42MAPK )。ERKs 为脯氨酸导向的丝/苏氨酸激酶,可以磷酸化与脯氨酸相邻的丝/苏氨酸 在丝裂原刺激后,ERKs接受上游的级联反应信号,可以转位进入细胞核。因此,ERKs 不仅可以磷酸化胞浆蛋白,而且可以磷酸化一些核内的转录因子如c-
Toll样受体信号通路图 TLR家族成员(TLR3除外)诱导的炎症反应都经过一条经典的信号通路(图1),该通路起始于TLRs的一段胞内保守序列—Toll/IL-1受体同源区(Toll/IL-1receptorhomologousregion,TIR).TIR可激活胞内的信号介质—白介素1受体相关蛋白激酶(IL-1Rassociatedkinase,IRAK)IRAK-1和IRAK-4、肿瘤坏死因子受体相关因子6(TNFR-associatedfactor6,TRAF-6)、促分裂原活化蛋白激酶(mitogenactivatedproteinkinase,MAPK)和IκB激酶(IκBkinase,IκK),进而激活核因子κB(nuclearfactorκB,NF-κB),诱导炎症因子的表达。 Toll-liker Receptor Signaling 本信号转导涉及的信号分子主要包括: CD14,MD-2,TRAM,TRIF,TIRAP,MyD88,TLR1,TLR2,TLR3,TLR4,TLR5,TLR6,TLR7,TLR8,TLR9,IRAK-1,IRAK-2,IRAK-4,IRAK-M,TRAF6,TRIAD3A,ST2L,SOCS1,RIG-I,FADD,TOLLIP,RIP1,A20,UEV1A,Ubc13,ECSIT,MEKK-1,TAK1,
TBK1,MKK3/6,p38,TAB1/2,MKK4/7,JNK,IKKα,IKKβ,IKKγ,IKKε,NEMO,IκBα,NF-κB,p65/RelA,Casp-8,IRF-3,IRF-7,MA VS等
OPEN ORIGINAL ARTICLE Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation P Daniel 1,G Filiz 1,DV Brown 1,F Hollande 1,M Gonzales 1,2,G D’Abaco 3,N Papalexis 4,WA Phillips 5,6,J Malaterre 6,7,RG Ramsay 6,7and T Mantamadiotis 1,4 The cyclic-AMP response element binding (CREB)protein has been shown to have a pivotal role in cell survival and cell proliferation.Transgenic rodent models have revealed a role for CREB in higher-order brain functions,such as memory and drug addiction behaviors.CREB overexpression in transgenic animals imparts oncogenic properties on cells in various tissues,and aberrant CREB expression is associated with tumours.It is the central position of CREB,downstream from key developmental and growth signalling pathways,which gives CREB this ability to in?uence a spectrum of cellular activities,such as cell survival,growth and differentiation,in both normal and cancer cells.We show that CREB is highly expressed and constitutively activated in patient glioma tissue and that this activation closely correlates with tumour grade.The mechanism by which CREB regulates glioblastoma (GBM)tumour cell proliferation involves activities downstream from both the mitogen-activated protein kinase and phosphoinositide 3-kinase (PI3K)pathways that then modulate the expression of three key cell cycle factors,cyclin B,D and proliferating cell nuclear antigen (PCNA).Cyclin D1is highly CREB-dependent,whereas cyclin B1and PCNA are co-regulated by both CREB-dependent and -independent mechanisms.The precise regulatory network involved appears to differ depending on the tumour-suppressor phosphatase and tensin homolog status of the GBM cells,which in turn allows CREB to regulate the activity of the PI3K itself.Given that CREB sits at the hub of key cancer cell signalling pathways,understanding the role of glioma-speci?c CREB function may lead to improved novel combinatorial anti-tumour therapies,which can complement existing PI3K-speci?c drugs undergoing early phase clinical trials. Oncogenesis (2014)3,e108;doi:10.1038/oncsis.2014.21;published online 30June 2014 INTRODUCTION Patients diagnosed with malignant glioblastoma (GBM)show a median survival of 14months;a statistic largely unchanged over the past decades.For gliomas,the only drug used as part of the standard therapy is the DNA alkylating/methylating agent temozolomide,which has led to an improvement in median overall survival,ranging between 0and 7months,depending on the methylation status of the patient’s DNA repair gene,MGMT.1Attempts at targeting speci?c factors in GBM have so far been unsuccessful,with such attempts exempli?ed by clinical trials conducted recently:the use of the promising angiogenesis inhibitor bevacizumab (Avastin)2or a combination of bevacizumab and a phosphoinositide 3-kinase (PI3K)pathway inhibitor provided no bene?t to patients.3Therefore there is a need to develop better approaches for treating gliomas to improve patient survival.To progress the discovery and testing of novel drugs and combinations of drugs,the understanding of the molecular genetic mechanisms and factors driving GBM development,growth and drug resistance must be clari?ed.Among the factors and pathways implicated in glioma development and growth,the kinases PI3K and mitogen-activated protein kinase (MAPK)are among the most studied.Highlighting the critical role of these kinases in cancer,480%of GBM patients harbour alterations such as epidermal growth factor receptor (EGFR)ampli?cation,EGFRvIII-activating mutation and/or downstream PIK3CA-activating mutations or phosphatase and tensin homolog (PTEN)deletions,4contributing to the hyperactivation of the downstream effectors such as extracellular signal–regulated kinase and AKT,key drivers of pathogenesis in GBM.5Although aspects of the immediate upstream and downstream components of these pathways are relatively well understood,the feedback loops and nuclear target networks controlling these pathways in GBM biology are not as well de?ned. As many cancer signalling pathways converge on nuclear transcription factors,which then orchestrate the expression of a tumour-promoting transcriptome,targeting these transcription factors in combination with upstream-activating factors may be an attractive approach.Indeed,this has come to the fore in terms of emerging anti-tumour strategies 6–9In cancer cells,one of the transcription factors that sit at the hub of tumour cell signalling pathways is the cyclic-AMP response element binding (CREB)protein,a serine/threonine kinase-regulated transcription factor in which phosphorylation of CREB in the N-terminal kinase-inducible domain recruits transcriptional co-activators such as CREB-binding protein and transducers of regulated CREB activity to activate CREB target gene transcription.10,11CREB has been implicated in the growth and progression of multiple cancers,including 1 Department of Pathology,The University of Melbourne,Parkville,Victoria,Australia;2Department of Anatomical Pathology,The Royal Melbourne Hospital,Parkville,Victoria,Australia;3NICTA Victorian Research Laboratories,Centre for Neural Engineering,The University of Melbourne,Carlton,Australia;4Laboratory of Physiology,Faculty of Medicine,University of Patras,Patras,Greece;5Surgical Oncology Research Laboratory,Peter MacCullum Cancer Centre,Melbourne,Victoria,Australia;6Sir Peter MacCallum Department of Oncology,The University of Melbourne,Parkville,Victoria,Australia and 7Differentiation and Transcription Laboratory,Peter MacCallum Cancer Centre,Melbourne,Victoria,Australia.Correspondence:Dr T Mantamadiotis,Department of Pathology,Faculty of Medicine,Dentistry and Health Sciences,The University of Melbourne,Parkville,Victoria 3010,Australia. E-mail:theom@https://www.doczj.com/doc/cd2335331.html,.au Received 3February 2014;revised 29April 2014;accepted 15May 2014 Citation:Oncogenesis (2014)3,e108;doi:10.1038/oncsis.2014.21 &2014Macmillan Publishers Limited All rights reserved 2157-9024/https://www.doczj.com/doc/cd2335331.html,/oncsis