
荧光蛋白真核表达载体的构建及其瞬时表达
孙婷婷a,高晓莲a,b
a浙江理工大学生物工程所,浙江杭州(310018)
b休斯顿大学生物学与生物化学研究所,美国TX77004-5001
E-mail:suntingting1218@https://www.doczj.com/doc/815564321.html,
摘要:荧光蛋白易于和其他蛋白融合,且融合后的荧光蛋白仍能保持荧光激发活性的优点广泛地应用于细胞学,医学和分子学领域。然而,随着荧光蛋白研究方法的发展对荧光蛋白的种类也提出了更多的要求。而传统的获得荧光蛋白的方式主要局限于从生物体内直接提取或通过定点诱变和突变的技术获得,而对利用基因合成的方法合成新型荧光蛋白的研究则很少。本文基于微流体芯片合成获得新型荧光蛋白的基础上,构建真核质粒载体pcDNA3.1(+)-G2-hexaHis,通过转染进入HeLa细胞内进行表达,结果显示G2在真核细胞中可以大量表达,且荧光强度很强,亚细胞定位结果显示荧光蛋白G2绝大部分存在于细胞质中,极少部分分布在细胞核中,故荧光蛋白G2将成为真核细胞质中蛋白的定位,细胞骨架标记和细胞分裂,胞质蛋白表达强度等同时也可以应用于细胞核中特异蛋白的标记及核质间物质运输应用中很有潜力的工具。
关键词:真核载体构建;转染;亚细胞定位
荧光蛋白被证实是活体生物成像的良好工具,被广泛的应用于生物学的不同领域。例如细胞骨架和细胞分裂,细胞器动力学和囊泡运输,细胞核和细胞质之间的物质运输,蛋白质之间的互作,蛋白质定位,基因转录强度等的测定[1-4]。近几十年来,绿色荧光蛋白通过和其他蛋白融合标记不同蛋白的在亚细胞中的分布及动力学或标记细胞中的不同分区和器官[5,6]。而且随着荧光蛋白变体类型和数量的扩充,其荧光蛋白涉及的新的应用技术也不断展现,如荧光共振能量转移(FRET)、荧光双分子和多分子标记技术、双分子荧光互补技术、荧光双杂交技术(F2H)等[7-10]。
传统获得荧光蛋白变体的方法局限于通过从生物体直接提取或利用定点诱变和突变的方法获得,此方法不仅费时耗资金,且不能高通量获得目的荧光蛋白变体。而生物合成方法解决了这一难题,利用生物合成方法可以合成一些自然界不存在或常规方法难以获得的生物大分子,而微流体芯片合成技术在生物合成法的基础上得到了增进,其是一种简便、快捷、准确率高的生物合成技术[11,12]。本实验基于此技术合成的新型荧光蛋白(此结果将在英文期刊进行发表),构建真核载体,在Hela细胞中进行表达,结果显示新型荧光蛋白在HeLa 细胞可以得到大量表达,故其将成为生物学领域重要的工具。
1 材料和方法
1.1 质粒、感受态、细胞系
载体pcDNA3.1(+)-G2-Hexa His由本实验室构建;E.coli BL21感受态本实验室利用0.1 M CaCl2制备;HeLa细胞由本校新元医药研究所保存;G2本实验组微流体芯片从头合成荧光蛋白。
1.2 主要试剂
Deep VentR? DNA聚合酶(2 unit/μl,NEB),dNTP mixture(2.5 mM,TaKaRa),dATP (100 mM,Songon),T4 DNA连接酶(40 U/μl,NEB), Bam HⅠ(10 U/μl,Fermentas),Xho I (10 U/μl,Fermentas),Xba I(10 U/μl,Fermentas);pcDNA3.1(+)载体系统(Promega),AxyPrep TM Gel Extraction Kit(Axygen),Microcon? YM-3(Millipore),DEME(GIBICO),
Fetal bovine serum(GIBICO),HE染色液(Beyotime),Hoechst染液(Beyotime),甘油及其他试剂均是国产分析纯。
1.3 仪器设备
PCR仪,水浴锅(Bioer),离心机(Eppendorf),电泳仪(Bio-RAD),凝胶成像系统(SynGENE),台式恒温振荡器(Vortex Genie2),恒温培养箱(Salvis Lab),超净台(Thermo electron corporation),二氧化碳孵箱(Thorma),荧光显微镜(Olympus),激光共聚焦显微镜(Nikon)。
1.4 真核载体的构建
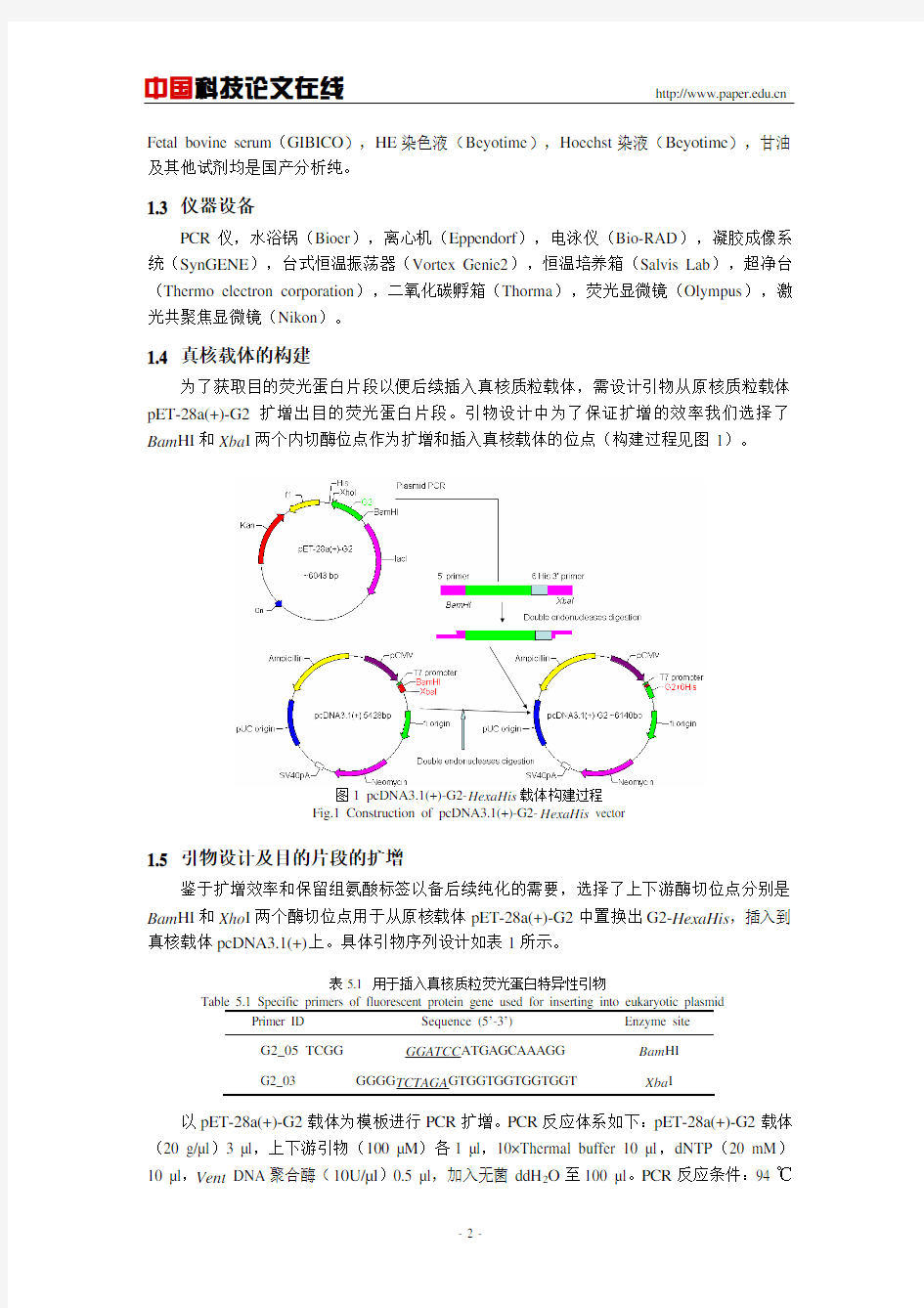
为了获取目的荧光蛋白片段以便后续插入真核质粒载体,需设计引物从原核质粒载体pET-28a(+)-G2扩增出目的荧光蛋白片段。引物设计中为了保证扩增的效率我们选择了Bam HI和Xba I两个内切酶位点作为扩增和插入真核载体的位点(构建过程见图1)。
图1 pcDNA3.1(+)-G2-HexaHis载体构建过程
Fig.1 Construction of pcDNA3.1(+)-G2-HexaHis vector
1.5 引物设计及目的片段的扩增
鉴于扩增效率和保留组氨酸标签以备后续纯化的需要,选择了上下游酶切位点分别是Bam HI和Xho I两个酶切位点用于从原核载体pET-28a(+)-G2中置换出G2-HexaHis,插入到真核载体pcDNA3.1(+)上。具体引物序列设计如表1所示。
表5.1 用于插入真核质粒荧光蛋白特异性引物
Table 5.1 Specific primers of fluorescent protein gene used for inserting into eukaryotic plasmid Primer ID Sequence (5’-3’) Enzyme site
G2_05 TCGG GGATCC ATGAGCAAAGG Bam HI
G2_03 GGGG TCTAGA GTGGTGGTGGTGGT Xba I
以pET-28a(+)-G2载体为模板进行PCR扩增。PCR反应体系如下:pET-28a(+)-G2载体(20 g/μl)3 μl,上下游引物(100 μM)各1 μl,10×Thermal buffer 10 μl,dNTP(20 mM)10 μl,Vent DNA聚合酶(10U/μl)0.5 μl,加入无菌ddH2O至100 μl。PCR反应条件:94 ℃
预变性3 min;94 ℃变性30 s,59 ℃退火 40 s,72 ℃延伸1 min,30个循环;最后72 ℃延伸10 min。PCR产物用1%琼脂糖凝胶电泳进行鉴定,然后用DNA纯化试剂盒(AxyPrep TM Cleanup Kit,Axygen)进行纯化去除引物及dNTP等物质。
1.6 目的片段与pcDNA3.1(+)载体的双酶切、连接与鉴定
纯化后的PCR产物和载体pcDNA3.1(+)分别用Bam HI和Xba I进行双酶切,反应体系:纯化PCR产物96 μl,Bam HI和Xba I各1 μl(10 U/μl),10×buffer 11 μl,无菌ddH2O定容至110 μl;pcDNA3.1(+)(234.6 ng/μl)27.5 μl,Bam HI和Xba I各2.5 μl(10 U/μl),10×buffer 27 μl,无菌ddH2O定容至270 μl。将上述反应体系分别混匀,于37 ℃恒温箱中孵育5 h,最后,将酶切后的片段和表达载体按AxyPrep TM Cleanup Kit(Axygen)进行快速纯化回收,并用1%琼脂糖凝胶电泳进行鉴定。
分别取纯化后的酶切目的片段和pcDNA3.1(+)20 μl和10 μl,加入10×ligation buffer 4 μl,T4 DNA连接酶1 μl,加入无菌ddH2O至总体积40 μl,16 ℃连接12 h。将连接产物与E.coli BL21感受态细胞充分混合,冰浴30 min后42 ℃热激90 s,再冰浴4 min。然后加入700 μl LB培养基并置于37 ℃恒温振荡器150 r/min孵育45 min,5000 rpm离心5 min,去除上层培养基后用150 μl LB培养基重新悬浮,混匀后涂于含50 μg/ml氨苄青霉素的LB平板上。同时设置阴性对照即除不加入目的片段,其余操作过程同上。37 ℃培养16 h后挑取单菌落接种于3 ml含50 μg/ml氨苄青霉素的液体LB培养基中培养过夜。第二天用去除内毒素的质粒试剂盒提取质粒,提取的质粒用Bam HI和Xba I进行酶切,然后进行琼脂糖凝胶电泳检测有无目的片段的插入。为了进一步鉴定插入片段的正确性,将提取质粒样品寄送上海英骏科技有限公司进行测序。
1.7 HeLa细胞培养及瞬时转染
转染前一天将细胞进行铺板,铺板密度控制在40~80%,使细胞处于最佳的生理状态。利用Effectene转染试剂进行转染,首先将100 μl EC缓冲液与400 ng左右的转化质粒混合,并加入3.2 μl的增强子,振荡1~5 s后室温孵育10 min,然后加入10 μl的Effectene试剂,振荡10 s后室温孵育20 min,最后加入600 μl培养基后用枪头上下吹打混合液,同时吸除培养皿中培养基,将转染混合液加入细胞培养皿中并加入含有5%胎牛血清的新鲜DMEM 培养基1.5 ml,混合均匀后于37 ℃含5%二氧化碳孵育箱中培养。转染6 h更换培养基,并于转染48 h后进行亚细胞定位以检测目的基因表达情况和细胞形态。
1.8 荧光蛋白G2在HeLa细胞中的亚细胞定位
先用荧光染料对转染后细胞进行染色,分别用细胞核荧光染料DAPI(终浓度1 μg/ml,染色20 min)和细胞膜荧光染料DiI(终浓度9.34 μg/ml,染色10 min)进行染色,然后利用激光共聚焦显微镜对荧光蛋白G2在细胞中的分布进行检测。
2 结果
2.1 目的片段PCR扩增
用G2荧光蛋白基因的特异性引物以pET28a(+)-G2为模板扩增的产物经过1%琼脂糖凝胶电泳分离后,仅在750 bp附近显示出一条特异性条带,大小与理论值763 bp相符合(图2),说明G2片段从原质粒载体pET-28a(+)-G2中扩增成功。
图2 G2荧光蛋白基因扩增结果(1% Agarose, TAE buffer)
Fig.2 PCR results of fluorescent protein of G2
M: Marker; S: G2 PCR product from pET-28a(+)-G2
2.2 重组pcDNA
3.1(+)-G2-HexaHis载体的鉴定
对经过T4 DNA ligase连接和转化获得的克隆斑进行培养,然后对抽提的质粒进行双酶切鉴定,结果是重组质粒双酶切后的片段和理论值相符,一条为5000 bp左右的载体片段(与空载体质粒酶切相符)和另一条为750 bp左右的目的荧光蛋白基因片段(与目的片段大小相符)(图3),同时重组子克隆测序结果和设计序列完全相同,说明真核载体构建成功(图4)。
图3重组质粒构建鉴定结果( 1% Agarose gel TAE buffer)
Fig.3 Verification of constructive combination vector
Lane 1: pcDNA3.1(+) plasmid; Lane 2: pcDNA3.1(+) plasmid digested by Bam HI and Xba I; Lane 3: pcDNA3.1(+)-G2 plasmid; Lane 4: pcDNA3.1(+)-G2 plasmid digested by Bam HI and Xba I; Lane 5: G2 PCR product digested by Bam HI and Xba I; Lane 6: pcDNA3.1(+) and G2 ligation product.
图4重组质粒pcDNA3.1(+)-G2- Hexa His测序结果
Fig.4 Sequencing result of the recombinant plasmid of pcDNA3.1(+)-G2 -Hexa His
2.3 荧光蛋白G2的亚细胞定位
利用荧光染料DAPI和DiI分别对细胞核和细胞膜染色后可以发现,荧光蛋白G2主要在细胞质中,细胞核内含量极低(图5)。对于DAPI染色结果中的B、H和L由于其中一个细胞处于分裂期所以可以看到两个DAPI细胞核染色区域。当细胞处于分裂期时,G2主要分布在细胞核周围,而当细胞处于G期和分裂期末期时,G2在细胞中的分布趋向均匀(图5A、E、K)。
图 5 G2在HeLa细胞中的定位
Fig.5 Location of G2 in HeLa cells. A.E.K.
G2-HexaHis expression in HeLa cells; B.H.L. nucleus dyed with DAPI; D.G.J. cell membrane dyed with DiI;
C.F.I.M merged images; Scar bars indicated 10 μm.
空载体pcDNA3.1(+)转染HeLa细胞后观测不出绿色荧光,且空载体转染后细胞形态和pcDNA3.1(+)-G2- Hexa His有较大差别,空载体转染后细胞形态更接近于正常状态而含有荧光蛋白G2转染,随着G2的不段表达,细胞变圆,有可能是因为荧光蛋白G2的过量表达对HeLa产生毒性作用,这需要设置eGFP荧光蛋白的转染作为对照,如果eGFP转染结果HeLa细胞形态也变圆则细胞形态变化不是由于荧光蛋白表达产生的,反之,荧光蛋白的表达对细胞产生毒性(图6)。
图 6 pcDNA3.1(+)-G2-HexaHis和pcDNA3.1(+)在HeLa细胞中的表达
Fig.6 The expression of pcDNA3.1(+)-G2-HexaHis and pcDNA3.1(+) in HeLa cells.
An-Dn expression of pcDNA3.1(+) in HeLa cells; A-E expression of pcDNA3.1(+)-G2-HexaHis in HeLa cells. An, A cells without any dye and fluorescent proteins; Bn, B. cell membrane dyed with DiI; Cn, D. nucleus dyed with DAPI; C G2-HexaHis expression in HeLa cells, Scar bars indicated 10 μm.
3 结论
荧光蛋白分子信标的功能范围与其在亚细胞中的定位密切相关。而真核细胞是荧光蛋白应用的重要领域,其在细胞内的不同部位就决定了其应用方式的不同。由于荧光蛋白几乎都分布在细胞质中,所以目前荧光蛋白可应用于细胞骨架的标记,细胞核与细胞质穿梭载体的标记[13,14]。
本论文主要就新荧光蛋白G2在HeLa细胞中的定位情况进行了研究。首先,通过双酶切,特异性引物PCR和重组子的测序证实了真核质粒pcDNA3.1(+)-G2- Hexa His的成功构建,后将载体在转染试剂的作用下瞬时转入HeLa细胞中,荧光显微镜下可以看到很强的绿色荧光,说明荧光蛋白G2可在HeLa细胞中成功表达。接着对细胞进行染色和亚细胞定位,发现荧光蛋白G2绝大部分存在于细胞质中,极少部分分布在细胞核中,且对不同细胞形态的细胞进行观测后,发现当细胞处于分裂(M)期时,荧光蛋白G2在细胞核周围的荧光强度比胞质中的荧光强度强,而当细胞处于G期或M末期时,荧光蛋白G2则较为均匀地分布在细胞质中。这是因为在细胞处于分裂期时,核外质粒也同时进行复制和表达,当细胞处于静止期时,核外质粒也随之静止,而其表达的荧光蛋白G2则在细胞质的流动性和细胞骨架的作用下在细胞中进行迁移,趋向于均质[15]。结果显示,荧光蛋白G2将成为真核细胞质中蛋白的定位,细胞骨架标记和细胞分裂,胞质蛋白表达强度,细胞核中特异蛋白的标记及核质间物质运输应用中很有潜力的工具。
参考文献
[1] Shannon, C., Salter, M., Fern, R. GFP imaging of live astrocytes: regional differences in the effects of ischaemia upon astrocytes [J].Journal of anatomy 2007 210(6):684-692
[2] Gurniak, C. B., Witke, W. HuGE, a novel GFP-actin-expressing mouse line for studying cytoskeletal dynamics [J].European journal of cell biology 2007 86(1):3-12
[3] Dillingham, M. S., Tibbles, K. L., Hunter, J. L., et al. Fluorescent single-stranded DNA binding protein as a probe for sensitive, real-time assays of helicase activity [J].Biophysical journal 2008 95(7):3330-3339
[4] Rolls, M. M., Stein, P. A., Taylor, S. S., et al. A visual screen of a GFP-fusion library identifies a new type of nuclear envelope membrane protein [J].The Journal of cell biology 1999 146(1):29-44
[5] Tsien, R. Y., Miyawaki, A. Seeing the machinery of live cells [J].Science (New York, N.Y 1998 280(5371):1954-1955
[6] Hoffman, R. M. The multiple uses of fluorescent proteins to visualize cancer in vivo [J].Nature reviews 2005 5(10):796-806
[7] Takanishi, C. L., Bykova, E. A., Cheng, W., et al. GFP-based FRET analysis in live cells [J].Brain research 2006 1091(1):132-139
[8] Citovsky, V., Gafni, Y., Tzfira, T. Localizing protein-protein interactions by bimolecular fluorescence complementation in planta [J].Methods (San Diego, Calif 2008 45(3):196-206
[9] Wan, H., He, J., Ju, B., et al. Generation of two-color transgenic zebrafish using the green and red fluorescent protein reporter genes gfp and rfp [J].Marine biotechnology (New York, N.Y 2002 4(2):146-154
[10] Zolghadr, K., Mortusewicz, O., Rothbauer, U., et al. A fluorescent two-hybrid (F2H) assay for direct visualization of protein interactions in living cells [J].Mol Cell Proteomics 2008
[11] Zhou, X., Cai, S., Hong, A., et al. Microfluidic PicoArray synthesis of oligodeoxynucleotides and simultaneous assembling of multiple DNA sequences [J].Nucleic acids research 2004 32(18):5409-5417
[12] Gao, X., Pellois, J. P., Na, Y., et al. High density peptide microarrays. In situ synthesis and applications [J].Molecular diversity 2004 8(3):177-187
[13] Fischer, M., Haase, I., Simmeth, E., et al. A brilliant monomeric red fluorescent protein to visualize cytoskeleton dynamics in Dictyostelium [J].FEBS letters 2004 577(1-2):227-232
[14] Ohashi, T., Kiehart, D. P., Erickson, H. P. Dual labeling of the fibronectin matrix and actin cytoskeleton with green fluorescent protein variants [J].Journal of cell science 2002 115(Pt 6):1221-1229
[15] Kim, K., Eaton, M. S., Schubert, W., et al. Optimized expression of green fluorescent protein in Toxoplasma gondii using thermostable green fluorescent protein mutants [J].Molecular and biochemical parasitology 2001 113(2):309-313
The construction and contemporary expression of
eukaryotic vector of fluorescent protein
Sun Tingting a, Gao Xiaolian a,b
a Institute of Bioengineering, Zhejiang Science and Technology University, Hangzhou (310018)
b Department of Biology and Biochemistry, University of Houston, TX 77004-5001, USA
Abstract
Fluorescent proteins are used intensively in cytology, medicine biology and molecular fields as they remain the property of fluorescence excitable activity after fused with other proteins fluently. However, with the advancement of fluorecent proteins study techonlogies, it demands more fluorescent variants’ occurrence. Neverthless, fluorecent proteins obtained traditionally restricted in extracting them directly from orgnism or site-directed mutagenesis and random mutation methods and little attentions payed on gene synthesis technology to get the novel fluorescent proteins.
Here, we constructed the eukaryotic plasmid of pcDNA3.1(+)-G2-HexaHis based on the novel fluorescent proteins which were synthesized by Microfluidic PicoArray method. The constructed eukaryotic plasmid was then transferred into HeLa cells and the result showed that it can express considerably in the HeLa cells with intensive fluorescence. Besides, the subcelullar location result presented G2 mainly located in plasma and scarcely in nucleolus, so the fluorescent protein of G2 can be used in localization of proteins in plasma, marking cytoskeleton, cell mitosis and plasma protein expression intensity, meantime, it can also be used in marking specific protein of nucleus and proteins transferring between plasma and nucleus.
Key words: eukaryotic plasmid construction;transfection;subcellular location
北京华越洋?生物?首页关于我们新闻中?心产品展?示在线留?言加?入我们 联系我们 pCambia1391Z pCambia1391Z 产品编号载体名称北京华越洋?生物VECT0010 pCambia1391Z pCambia 1391Z 载体基本信息 出品公司:Cambia 载体名称: pCambia1391Z, pCambia 1391Z 质粒类型: 植物表达载体?高拷贝/低拷贝:低拷贝启动?子:CAMV 35S 克隆?方法 :多克隆位点,限制性内切酶 载体?大?小: 11227 bp 5' 测序引物及序列:M13-F: TGTAAAACGACGGCCAGT 3' 测序引物及序列 : M13-R: CAGGAAACAGCTATGAC 载体标签: GusA 载体抗性: 卡那和潮霉素筛选标记: HPTII 详情产品分类 ?生化试剂 精细化学品 中间体/标准品 病理实验试剂 其他关键词: 热线电话:400-818-1148150 1148 1284
备注:-- 产品?目录号:1391Z 稳定性:稳定表达 组成型:?非组成型 病毒/?非病毒:?非病毒 pCambia 1391Z载体质粒图谱和多克隆位点信息 pCambia 1391Z载体序列 LOCUS pCAMBIA1391Z 11227 bp ds-DNA circular SYN DEFINITION Agrobacterium binary vector for plant transformation, with hygromycin- and kanamycin-resistance and LacZ-GUS genes plus the pUC9 MCS. ACCESSION AF234312 VERSION . KEYWORDS pCAMBIA1391Z SOURCE synthetic DNA construct ORGANISM synthetic DNA construct REFERENCE 1 (bases 1 to 11227) AUTHORS Cambia TITLE Direct Submission JOURNAL Exported from SnapGene Viewer COMMENT The GenBank record was corrected by inserting a G at position 4743. FEATURES Location/Qualifiers
真核细胞常见表达载体 真核细胞, 表达载体 1、pCMVp-NEO-BAN载体 特点:该真核细胞表达载体分子量为6600碱基对,主要由CMVp启动子、兔β-球蛋白基因内含子、聚腺嘌呤、氨青霉素抗性基因和抗neo基因以及pBR322骨架构成,在大多数真核细胞内都能高水平稳定地表达外源目的基因。更重要的是,由于该真核细胞表达载体中抗neo 基因存在,转染细胞后,用G418筛选,可建立稳定的、高表达目的基因的细胞株。 插入外源基因的克隆位点包括Sal1、BamH1和EcoR1位点。注意在此载体中有二个EcoR1位点存在。 2、pEGFP, 增强型绦色荧光蛋白表达载体(Enhanced Fluorecent Protein Vector) 特点: pEGFP表达载体中含有绿色荧光蛋白,在PCMV启动子驱动下,在真核细胞中高水平表达。载体骨架中的SV40origin使该载体在任何表达SV40 T抗原的真核细胞内进行复制。Neo抗性盒由SV40早期启动子、Tn5的neomycin/kanamycin抗性基因以及HSV-TK基因的聚腺嘌呤信号组成,能应用G418筛选稳定转染的真核细胞株。此外,载体中的pUC origin 能保证该载体在大肠杆菌中的复制,而位于此表达盒上游的细菌启动子能驱动kanamycin抗性基因在大肠杆菌中的表达。 用途: 该表达载体EGFP上游有Nde1、Eco47111和Age1克隆位点,将外源基因扦入这些位点,将合成外源基因和EGFP的融合基因。借此可确定外源基因在细胞内的表达和/或组织中的定位。 亦可用于检测克隆的启动子活性(取代CMV启动子,Acet1-Nhe1)。 3、pEGFT-Actin, 增强型绿色荧光蛋白/人肌动蛋白表达载体 特点:pEGFP-Actin表达载体中含有绿色荧光蛋白和人胞浆β-肌动蛋白基因,在PCMV启动子驱动下,在真核细胞中高水平表达。载体骨架中的SV40origin使该载体在任何表达SV40 T 抗原的真核细胞内进行复制。Neo抗性盒由SV40早期启动子、Tn5的neomycin/kanamycin 抗性基因以及HSV-TK基因的聚腺嘌呤信号组成,能应用G418筛选稳定转染的真核细胞株。此外,载体中的pUC origin 能保证该载体在大肠杆菌中的复制,而位于此表达盒上游的细菌启动子能驱动kanamycin抗性基因在大肠杆菌中的表达。 用途: pEGFP-Actin载体在真核细胞表达EGFP-Actin融合蛋白,该蛋白能整合到胞内正在生的肌动蛋白,因而在活细胞和固定细胞中观察到细胞内含肌动蛋白的亚细胞结构。 4、pSV2表达载体 特点:该表达质粒是以病责SV40启动子驱动在真核细胞目的基因进行表达的,克隆位点为Hind111。SV40启动子具有组织/细胞的选择特异性。此载体不含neo基因,故不能用来筛选、建立稳定的表达细胞株。 5、CMV4 表达载体 特点:该真核细胞表达载体由CMV启动子驱动,多克隆区域酶切位点选择性较多。含有氨苄青霉素抗性基因和生长基因片段以及SV40复制原点和fl单链复制原点。但值得注意的是,该表达载体不含有neo基因,转染細胞后不能用G418筛选稳定的表达细胞株。 其他常用克隆Vector: pBluscript II KS DNA 15 ug pUC18 DNA 25 ug pUC19 DNA 25 ug 说明: pBluescript II kS、pUC18 &Puc19载体适合于DNA片段的克隆、DNA测序和对外源基因进行表达等。这些载体由于在lacZ基因中含有多克隆位点,当外源DNA片段扦入,转化lacZ基因缺乏细胞,并在含有IPTG和X-gal的培养基上培养时,含有外源DNA载体的细胞将
原核蛋白表达常见问题解析 1、为什么目的蛋白总是以包涵体的形式出现? 在原核蛋白表达纯化中目的蛋白经常发生错误的折叠,并聚集成为 包涵体。经过诱导,目的蛋白通常可达细胞总蛋白的50%以上。虽然有一定比例的蛋白以可溶的单体形式存在,而多达95%(甚至更多)的蛋白则在包涵体中。实验过程中,可以采取降低诱导温度,例如25–30°C,或降低IPTG浓度(0.01–0.1mM)并延长诱导时间,还有采用特别的 培养基等方法获得更多的可溶蛋白。 2、跨膜蛋白为什么很难表达? 跨膜蛋白的表达成功率相对较低是一个实验结果,究其原理,目前 众说纷纭很多种理论。以我们浅薄的理解层面来看,主要有以下几个 原因: 跨膜蛋白一般都是强疏水性的氨基酸分子和亲水性的分子跳跃式 的连接,形成的亲水疏水的一个最简单的跨膜化学结构,这种结构与 信号肽结构相似,对于原核细胞来说,简单的细胞器很难像真核细胞 一样完成信号肽识别及切除、引导内质网、高尔基体重新包装及分泌 这一复杂过程,有些蛋白是多次跨膜,对于原核细胞来说几乎是不可 能完成的任务。 另外,对于疏水性的片段,在原核细胞中极易形成包涵体,疏水 性多肽会抑制翻译过程,甚至与原核膜结构融合形成毒性,出于生物 自我保护的本能,所有的细胞器都会停止合成蛋白的过程。 3、如何选择蛋白表达宿主菌?
4、我们有哪些原核蛋白纯化方式?如何选择不同的纯化方式? 答:我们公司的蛋白纯化方法大致分为亲和纯化、离子交换、切胶 回收三类。 1、常规情况下,一般携带融合标签(His标签,GST标签,sumo标签,Fc标签),我们可以通过Ni柱、GST柱、Protein A等进行亲和纯化 获得融合蛋白,用亲和纯化的方法一般可以获得85%以上纯度的蛋白, 亲和纯化的方便快捷。 2、如果需要目的蛋白不含有任何标签,怎么选择纯化方式?。 (1)可表达融合蛋白,用蛋白工具酶切割融合蛋白,再进行纯化除去 工具酶。此方法能快速得到蛋白。 (2)可表达不含标签的蛋白,进行离子、分子筛、疏水等纯化,通过AKATA纯化设备获得蛋白。 3、如果需要获得蛋白作为抗原,可以直接通过切胶回收的方式,此方 法获得蛋白纯度较高,进行免疫动物后得到的抗体进行WB反应,灵敏 度较高。 5、表达得到的蛋白是有活性的么? 答:需要让蛋白有活性的条件很复杂,合适的缓冲液体系、盐浓度、蛋白的折叠状态甚至检测活性的方法的细微差别都可能导致活性 的强弱有无,一般情况下,上清表达的蛋白要比包涵体经过变复性纯 化后得到的蛋白活性要好,我们尽量从上清中获得蛋白,期许蛋白形 成的折叠最接近活性状态,这也是我们擅长的。但是在实际实验条件下,我们无法承诺表达纯化的蛋白一定具有客户期望的生理活性。
表达载体的构建方法及步骤 令狐采学 一、载体的选择及如何阅读质粒图谱 目前,载体主要有病毒和非病毒两大类,其中质粒DNA 是一种新的非病毒转基因载体。 一个合格质粒的组成要素: (1)复制起始位点Ori 即控制复制起始的位点。原核生物DNA 分子中只有一个复制起始点。而 真核生物DNA 分子有多个复制起始位点。 (2)抗生素抗性基因可以便于加以检测,如Amp+ ,Kan+ (3)多克隆位点MCS 克隆携带外源基因片段 (4)P/E 启动子/增强子 (5)Terms 终止信号 (6)加poly(A)信号可以起到稳定mRNA 作用 选择载体主要依据构建的目的,同时要考虑载体中应有合适的限制酶切位点。如果构建的目 的是要表达一个特定的基因,则要选择合适的表达载体。 载体选择主要考虑下述3点: 【1】构建DNA 重组体的目的,克隆扩增/基因表达,选择合适的克隆载体/表达载体。 【2】.载体的类型:
(1)克隆载体的克隆能力-据克隆片段大小(大选大,小选小)。如<10kb 选质粒。 (2)表达载体据受体细胞类型-原核/真核/穿梭,E.coli/哺乳类细胞表达载体。 (3)对原核表达载体应该注意:选择合适的启动子及相应的受体菌,用于表达真核蛋白质时注意克服4个困难和阅读框错位;表达天然蛋白质或融合蛋白作为相应载体的参考。 【3】载体MCS 中的酶切位点数与组成方向因载体不同而异,适应目的基因与载体易于链接,不能产生阅读框架错位。 综上所述,选用质粒(最常用)做载体的5点要求: (1)选分子量小的质粒,即小载体(1-1.5kb)→不易损坏,在细菌里面拷贝数也多(也有大载 体); (2)一般使用松弛型质粒在细菌里扩增不受约束,一般10个以上的拷贝,而严谨型质粒<10个。 (3)必需具备一个以上的酶切位点,有选择的余地; (4)必需有易检测的标记,多是抗生素的抗性基因,不特指多位Ampr(试一试)。 (5)满足自己的实验需求,是否需要包装病毒,是否需要加入荧光标记,是否需要加入标签蛋白,是否需要真核抗性(如Puro、G418)等等。 无论选用哪种载体,首先都要获得载体分子,然后采用适当的限制酶将载体DNA 进行切割,获得线性载体分子,以便于与
真核细胞表达系统的类型与常用真核细胞表达载体 标签:真核细胞酵母表达系统细胞表达载体真核表达系统昆虫表达系统动物表达系统 摘要: 原核表达系统是常被用来研究基因功能的成熟系统,由于原核表达系统具有包涵体蛋白不易纯化、蛋白修饰不完整等缺陷,人们也开始利用真核细胞表达系统来研究基因。 原核表达系统是常被用来研究基因功能的成熟系统,由于原核表达系统具有包涵体蛋白不易纯化、蛋白修饰不完整等缺陷,人们也开始利用真核细胞表达系统来研究基因。 自上世纪70年代基因工程技术诞生以来,基因表达技术已渗透到生命科学研究的各个领域。并随着人类基因组计划实施的进行,在技术方法上得到了很大发展,时至今日已取得令人瞩目的成就。随着人类基因组计划的完成,越来越多的基因被发现,其中多数基因功能不明。利用表达系统在哺乳动物细胞内表达目的基因是研究基因功能及其相互作用的重要手段。 在各种表达系统中,最早被采用进行研究的是原核表达系统,这也是目前掌握最为成熟的表达系统。该项技术的主要方法是将已克隆入目的基因DNA段的载体(一般为质粒)转化细菌(通常选用的是大肠杆菌),通过iptg诱导并最终纯化获得所需的目的蛋白。其优点在于能够在较短时间内获得基因表达产物,而且所需的成本相对比较低廉。但与此同时原核表达系统还存在许多难以克服的缺点:如通常使用的表达系统无法对表达时间及表达水平进行调控,有些基因的持续表达可能会对宿主细胞产生毒害作用,过量表达可能导致非生理反应,目的蛋白常以包涵体形式表达,导致产物纯化困难;而且原核表达系统翻译后加工修饰体系不完善,表达产物的生物活性较低。 为克服上述不足,许多学者将原核基因调控系统引入真核基因调控领域,其优点是: ①根据原核生物蛋白与靶DNA间作用的高度特异性设计,而靶DNA与真核基因调控序列基本无同源性,故不存在基因的非特异性激活或抑制; ②能诱导基因高效表达,可达105倍,为其他系统所不及; ③能严格调控基因表达,即不仅可控制基因表达的“开关”,还可人为地调控基因表达量。 因此,利用真核表达系统来表达目的蛋白越来越受到重视。目前,基因工程研究中常用的真核表达系统有酵母表达系统、昆虫细胞表达系统和哺乳动物细胞表达系统。 1.酵母表达系统 最早应用于基因工程的酵母是酿酒酵母,后来人们又相继开发了裂殖酵母、克鲁维酸酵母、甲醇酵母等,其中,甲醇酵母表达系统是目前应用最广泛的酵母表达系统。目前甲醇酵母主要有H Polymorpha,Candida Bodini,Pichia Pastris3种。以Pichia Pastoris应用最多。
载体质粒图谱和多克隆位点信息 载体简介 载体序列 LOCUS AF234298 10549 bp DNA circular SYN 24-APR-2000 DEFINITION Binary vector pCAMBIA-1302, complete sequence. ACCESSION AF234298 VERSION AF234298.1 GI:7638073
KEYWORDS . SOURCE Binary vector pCAMBIA-1302 ORGANISM Binary vector pCAMBIA-1302 other sequences; artificial sequences; vectors. REFERENCE 1 (sites) AUTHORS Hajdukiewicz,P., Svab,Z. and Maliga,P. TITLE The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation JOURNAL Plant Mol. Biol. 25 (6), 989-994 (1994) PUBMED 7919218 REFERENCE 2 (bases 1 to 10549) AUTHORS Roberts,C., Rajagopal,S., Smith,L.M., Nguyen,T.A., Yang,W., Nugrohu,S., Ravi,K.S., Vijayachandra,K., Harcourt,R.L., Dransfield,L., Desamero,N., Slamet,I., Hadjukiewicz,P., Svab,Z., Maliga,P., Mayer,J.E., Keese,P.K., Kilian,A. and Jefferson,R.A. TITLE A comprehensive set of modular vectors for advanced manipulations and efficient transformation of plants JOURNAL Unpublished REMARK Full description of constructs REFERENCE 3 (bases 1 to 10549) AUTHORS Roberts,C., Rajagopal,S., Smith,L.M., Nguyen,T.A., Yang,W., Nugrohu,S., Ravi,K.S., Vijayachandra,K., Harcourt,R.L., Dransfield,L., Desamero,N., Slamet,I., Hadjukiewicz,P., Svab,Z., Maliga,P., Mayer,J.E., Keese,P.K., Kilian,A. and Jefferson,R.A. TITLE Direct Submission
过表达慢病毒载体构建和包装手册 Version1.0 吉凯基因 二零一一年五月
目录 简介 (3) 第一部分过表达慢病毒载体的制备 实验流程 (4) 实验材料 (5) 过表达克隆制备 (6) 第二部分慢病毒包装与滴度检测 实验流程 (17) 实验材料 (18) L e n t i v i r u s病毒包装 (21) 病毒的收获及浓缩 (22) L e n t i v i r u s滴度测定 (24) 参考文献 (33)
简介 慢病毒(Lentivirus)载体是以人类免疫缺陷型病毒(HIV)为基础发展起来的基因治疗载体,它对分裂细胞和非分裂细胞均具有感染能力,并可以在体内较长期的表达且安全性高。吉凯基因提供的慢病毒为“自杀”性病毒,即病毒感染目的细胞后不会再感染其他细胞,也不会利用宿主细胞产生新的病毒颗粒。慢病毒中的毒性基因已经被剔除并被外源性目的基因所取代,属于假型病毒。但该病毒仍然具有可能的潜在的生物学危险,吉凯基因建议不要使用编码已知或可能会致癌的基因的假型病毒,除非已经完全公认某个基因肯定没有致癌性,否则均不建议采用假型病毒进行生物学实验。 吉凯基因慢病毒载体系统由GV慢病毒载体系列、pHelper 1.0载体和pHelper 2.0载体三质粒组成。GV慢载体中含有HIV的基本元件5’LTR和3’LTR以及其他辅助元件,例如WRE (woodchuck hepatitis virus posttranscriptional regulatory element)。通常根据不同的实验目的针对GV载体改造以进行基因功能研究。pHelper 1.0载体中含有HIV病毒的gag基因,编码病毒主要的结构蛋白;pol基因,编码病毒特异性的酶;rev基因,编码调节gag和pol基因表达的调节因子。pHelper 2.0载体中含有单纯疱疹病毒来源的VSV-G基因,提供病毒包装所需要的包膜蛋白。 吉凯基因过表达慢病毒产品可通过对GV慢病毒载体的改造和病毒包装,获得带有特定基因序列的慢病毒颗粒,以满足不同的实验需求。 本手册为吉凯基因RNAi慢病毒载体的构建和病毒包装的通用操作流程,目的是为了方便大家交流使用,部分细节内容未能做到一一详述,敬请谅解。同时希望大家能够针对手册中的错误和问题,提出宝贵的意见。
真核生物基因表达的调控 一、生物基因表达的调控的共性 首先,我们来看看在生物基因表达调控这一过程中体现的共性和一些基本模式。 1、作用范围。生物体内的基因分为管家基因和奢侈基因。管家基因始终表达,奢侈基因只在需要的时候表达,但二者的表达都受到调控。可见,调控是普遍存在的现象。 2、调控方式。基因表达有两种调控方式,即正调控与负调控,原核生物和真核生物都离不开这两种模式。 3、调控水平。一种基因表达的调控可以在多种层面上展开,包括DNA水平、转录水平、转录后加工水平、翻译后加工水平等。然为节省能量起见,转录的起始阶段往往作为最佳调控位点。 二、真核生物基因表达调控的特点 真核生物与原核细胞在结构上就有着诸多不同,这决定了二者在运行方面的迥异途径。真核生物比原核生物复杂,转录与翻译不同时也不同地,基因组与染色体结构复杂,因而有着更为复杂的调控机制。 1、 2、 3、 4、多层次。真核生物的基因表达可发生在染色质水平、转录起始水平、无操纵子和衰减子。 大多数原核生物以负调控为主,而真核生物启动子以正调控为主。 个体发育复杂,而受环境影响较小。真核生物多为多细胞生物,在转录后水平、翻译水平以及翻译后水平。
生长发育过程中,不仅要随细胞内外环境的变化调节基因表达,还要随发育的不同阶段表达不同基因。前者为短期调控,后者属长期调控。 从整体上看,不可逆的长期调控影响更深远。 三、真核生物基因表达调控的机制 介于真核生物表达以多层次性为最主要特点,我们可以分别从它的几个水平着眼,剖析它的调控机制。 1、染色质水平。真核生物基因组DNA以致密的染色质形式存在,发生在染色质水平的调控也称作转录前水平的调控,产生永久性DNA序列和染色质结构的变化,往往伴随细胞分化。染色质水平的调控包括染色质丢失、基因扩增、基因重排、染色体DNA的修饰,等等。a.基因丢失:丢失一段DNA或整条染色体的现象。在细胞分化过程中,可以通过丢失掉某些基因而去除这些基因的活性。某些原生动物、线虫、昆虫和甲壳类动物在个体发育中,许多体细胞常常丢失掉整条或部分的染色体,只有将来分化产生生殖细胞的那些细胞一直保留着整套的染色体。如马蛔虫2n=2,但染色体上有多个着丝粒。第一次卵裂是横裂,产生上下2个子细胞。第二次卵裂时,一个子细胞仍进行横裂,保持完整的基因组,而另一个子细胞却进行纵向分裂,丢失部分染色体。目前,在高等真核生物(包括动物、植物)中尚未发现类似的基因丢失现象。 b.基因扩增:基因扩增是指某些基因的拷贝数专一性增大的现象,它使得细胞在短期内产生大量的基因产物以满足生长发育的需要,是基因活性调控的一种方式。如非洲爪蟾卵母细胞中rDNA的基因扩增是因发育需要而出现的基因扩增现象;基因组拷贝数增加,即多倍性,在植物中是非常普遍的现象。基因组拷贝数增加使可供遗传重组的物质增多,这可能构成了加速基因进化、基因组重组和最终物种形成的一种方式。 c.基因重排:将一个基因从远离启动子的地方移到距它很近的位点从而启动转录,这种方式被称为基因重排。通过基因重排调节基因活性的典型例子是免疫球蛋白结构基因的表达。在人类基因组中,所有抗体的重链和轻链都不是由固定的完整基因编码的,而是由不同基因片段经重排后形成的完整基因编码的。
红色荧光蛋白(RFP )的原核表达 生物学实验教学中心 报告题目 红色荧光蛋白(RFP )的原核表达 作者姓名 饶慧 班级学号 0802班/2008114010214 指导教师 王友如 完成时间 2011年02月
目录 引言 (1) 1实验材料及实验仪器 (4) 1.1实验材料 (4) 1.2实验仪器 (5) 2实验方法 (7) 2.1 重组质粒的构建 (7) 2.2工程菌株的活化 (7) 2.3诱导表达 (8) 2.4 SDS-PAGE检测表达蛋白 (8) 3 结果与分析 (9) 总结 (10) 参考文献 (11) 致谢 (13)
红色荧光蛋白的原核表达 饶慧 (指导老师:王友如) (湖北师范学院生命科学学院生物科学0802班湖北黄石435002) 摘要实验目的:研究红色荧光蛋白(Red Fluorescent Protein,RFP)基因在大肠杆菌中原核表达。实验方法:通过分别将DH-5ɑ(pDsRed-N1)和DH-5ɑ(pET-28ɑ)提取质粒、酶切并连接形成重组质粒pET-28a-RFP,将重组质粒通过转化的方法把含红色荧光蛋白(RFP)外源基因转入大肠杆菌体内进行表达,再用IPTG诱导RFP基因表达,可以看到显现红色,最后根据SDS-PAGE电泳结果,判断RFP基因在大肠杆菌中是否成功表达。实验结果:结果显示构建的重组质粒pET-28ɑ-RFP在E.coli中成功表达。 关键词红色荧光蛋白;质粒重组;原核表达;诱导表达
分子生物学综合实验 Prokaryote Expression of Red Fluorescent Protein (RFP) Rao Hui (Class 0802, College of Biology Science ,Hubei Normal University, Huangshi, Hubei,435002 ) Abstract Objective: To study the expression of the RFP gene in the E.coli. Methods: Extract the plasmid of the DH-5ɑ (pDsRed-N1) and DH-5ɑ (pET-28ɑ). Then the two plasmids are cut by enzyme and are connected to form pET-28a-RFP recombined plasmid. Guiding the recombined plasmid, which contains exogenous genes of RFP, into E.coli for expression, through transformative method. The expression of RFP gene can be induced by the IPTG and then we can see red. Finally, judging whether the RFP gene has expressed successfully in E.coli according to the results of SDS-PAGE electrophoresis. Results: The results suggest that pET-28ɑ-RFP recombined plasmid has successfully expressed in E.coli. Keywords Red Fluorescent Protein; Recombined Plasmid; Prokaryote Expression; Induced Expression
真核细胞常见表达载体 1、pCMVp-NEO-BAN载体 特点: 该真核细胞表达载体分子量为6600碱基对,主要由CMVp启动子、兔β-球蛋白基因内含子、聚腺嘌呤、氨青霉素抗性基因和抗neo基因以及pBR322骨架构成,在大多数真核细胞内都能高水平稳定地表达外源目的基因。更重要的是,由于该真核细胞表达载体中抗neo基因存在,转染细胞后,用G418筛选,可建立稳定的、高表达目的基因的细胞株。 插入外源基因的克隆位点包括Sal1、BamH1和EcoR1位点。注意在此载体中有二个EcoR1位点存在。 2、pEGFP, 增强型绦色荧光蛋白表达载体(Enhanced Fluorecent Protein Vector) 特点: pEGFP表达载体中含有绿色荧光蛋白,在PCMV启动子驱动下,在真核细胞中高水平表达。载体骨架中的SV40 origin使该载体在任何表达SV40 T 抗原的真核细胞内进行复制。Neo抗性盒由SV40早期启动子、Tn5的neomycin/kanamycin抗性基因以及HSV-TK基因的聚腺嘌呤信号组成,能应用G418筛选稳定转染的真核细胞株。此外,载体中的pUC origin 能保证该载体在大肠杆菌中的复制,而位于此表达盒上游的细菌启动子能驱动kanamycin抗性基因在大肠杆菌中的表达。 用途: 该表达载体EGFP上游有Nde1、Eco47111和Age1克隆位点,将外源基因扦入这些位点,将合成外源基因和EGFP的融合基因。借此可确定外源基因在细胞内的表达和/或组织中的定位。 亦可用于检测克隆的启动子活性(取代CMV启动子,Acet1-Nhe1)。 3、pEGFT-Actin, 增强型绿色荧光蛋白/人肌动蛋白表达载体 特点: pEGFP-Actin表达载体中含有绿色荧光蛋白和人胞浆β-肌动蛋白基因,在PCMV启动子驱动下,在真核细胞中高水平表达。载体骨架中的SV40 origin使该载体在任何表达SV40 T 抗原的真核细胞内进行复制。Neo抗性盒由SV40早期启动子、Tn5的neomycin/kanamycin抗性基因以及HSV-TK基因的聚腺嘌呤信号组成,能应用G418筛选稳定转染的真核细胞株。此外,载体中的pUC origin 能保证该载体在大肠杆菌中的复制,而位于此表达盒上游的细菌启动子能驱动kanamycin抗性基因在大肠杆菌中的表达。 用途: pEGFP-Actin载体在真核细胞表达EGFP-Actin融合蛋白,该蛋白能整合到胞内正在生的肌动蛋白,因而在活细胞和固定细胞中观察到细胞内含肌动蛋白的亚细胞结构。 4、pSV2表达载体 特点:该表达质粒是以病责SV40启动子驱动在真核细胞目的基因进行表达的,克隆位点为Hind111。SV40启动子具有组织/细胞的选择特异性。此载体不含neo基因,故不能用来筛选、建立稳定的表达细胞株。5、CMV4 表达载体 特点:该真核细胞表达载体由CMV启动子驱动,多克隆区域酶切位点选择性较多。含有氨苄青霉素抗性基因和生长基因片段以及SV40复制原点和fl单链复制原点。但值得注意的是,该表达载体不含有neo基因,转染細胞后不能用G418筛选稳定的表达细胞株。 其他常用克隆Vector: pBluscript II KS DNA 15 ug pUC18 DNA 25 ug pUC19 DNA 25 ug
几种新型植物基因表达载体的构建方法 摘要:利用基因工程技术手段研究基因功能过程中,构建基因表达载体处于转基因植物的主导地位,采用合适的构建方法会使实验效果事半功倍。植物基因表达载体的构建方法除了传统构建法、Gateway 技术、三段T-DNA 法、一步克隆法等,还有近年来出现的几种新型的载体构建方法:基于竞争性连接原理快速构建小片段基因表达载体;Micro RNA 前体PCR 置换法适用于构建小分子RNA 表达载体;重组融合PCR 法特别适用于插入片段中含有较多限制性酶切位点的载体构建;利用In-Fusion 试剂盒可以将任何目的片段插入一个线性化载体的某个区域;构建多片段复杂载体可采用不依赖序列和连接的克隆方法(Sequence and ligation-independent cloning, SLIC) 法;Gibson 等温拼接法。本文将在总结分析前人工作的基础上,分析这6种新方法的特点,期望通过这几种新的方法给植物基因工程表达载体的构建提供新的思路。 关键词: Micro RNA 前体PCR 置换法,In-Fusion 试剂盒法,重组融合PCR 法,Gibson 等温拼接法,Golden Gate 拼接法 基因克隆、载体构建是植物功能基因组研究中的常规步骤[ 1 ]。而载体构建是基因工程和分子生物学研究中常用的基础技术。随着植物基因工程技术的发展,适合于不同研究目的各种载体系统应运而生,其中在转基因植物中最常用的是质粒载体。传统的载体构建方法在进行构建多片段拼接的复杂载体时,需要精心选择酶切位点[ 2 ],有时还需要构建多个中间载体,操作比较麻烦,费时费力,因此寻找简单、高效、快捷的载体构建方法具有重要的现实意义。从1969 年Arber 等发现了限制性内切酶,载体的构建方法逐步发展,从传统构建方法到
过表达质粒的构建及转染 质粒及菌液说明过表达质粒为高纯度的DNA 粉末制品,经过真空冷冻干燥的质粒是呈薄膜状或粉末状附在离心管中,请适当离心(10000r/min 10~15s)后小心开启,以免飞扬丢失。请根据实验需要的质粒浓度和质粒的总量加入适量的ddH2O (通常建议储存液浓度500ng~1μg/μL),合上管盖充分震荡使其溶解后可用于细胞转染及其他分子生物学实验。粉末制品的DNA 可长期保存(建议最好不要超过6个月),质粒溶解后建议在-20℃的环境中存贮,避免多次冻融处理。 2、甘油菌液为含有重组质粒的菌液的过夜培养物与甘油的混合物,甘油的终浓度为20% 。客户收到甘油菌液建议即刻活化(例如取50~100μL到5mL 含有相应抗性(根据载体种类可以选择25~50μg/mL 卡那霉素或50~100μg/mL 氨苄霉素)的LB 培养基过夜活化(12~16h),活化后的菌液与适量甘油混匀后于-80℃保存。) 3、重组质粒测序峰值图文件结果请参见附件报告中的.abl 文件。测序序列文件参见与峰值图文件同名的.seq 文件。 基因过表达实验设计在使用载体法针对某一基因进行过表达研究过程中,通常会遇到如下几个问题:实验对照组的确立、细胞转染条件的确定、基因表达效率的检测。 1. 实验对照组的确立在一个完善的基因过表达实验设计中,必须考虑设立正确合理的实验对照组。通常,这些对照组包括阴性对照、转染试剂对照。阴性对照通常是用基因过表达选择的载体对应的空载体来作为对照。 2. 细胞转染条件的确定使用DNA 载体转染细胞时,为了选择合适的转染方法和确定转染效率,通
常采用报告基因来检测DNA 的导入情况。最常用的报告基因是绿色荧光蛋白。 3. 基因表达效率的检测通常用两大类方法来检测过表达质粒中目的基因表达的效率,一类方法是直接检测目的基因在不同水平如mRNA 和蛋白水平的变化,具体的方法如qPCR 和Western blot 等;另一类方法是通过检测目的基因的生物学效应和细胞效应来间接的反映目的基因的表达变化,这一类方法很多,不同的基因有不同的检测方法。通常多选用qPCR 和Western blot 等方法直接检测目的基因的变化情况。由于细胞种类和蛋白表达翻译及抗体因素,吉玛构建表达载体通常可保证qRCR 检测转染工程细胞293T,目的基因mRNA 表达水平有2倍上调。基因过表达实验操作由于在基因过表达实验中有多种条件选择,如试剂和细胞株等,在本实验操作中仅以pEX-1 空载体为阴性对照,Lipofectamin2000 为转染试剂,293T 细胞为实验细胞株,按照上述条件分步阐述过表达质粒转染实验的操作过程。如果用户使用不同的细胞株和转染试剂,可根据不同的目的基因和实验条件进行相应的调整。 1、293T 细胞在6cm dish 中培养至80-90%融合时,倾去培养液,用3mL PBS 洗涤细胞两次。 2、加1mL Trypsin-EDTA solution, 混匀后,小心吸去胰酶溶液,37℃ 放置3 分钟。 3、再加入2 mL 含10% FBS 的DMEM 培养液,吹打使细胞形成单细胞悬 液。 4、血球计数板计数,将细胞稀释至3×105 细胞/mL。 5、按5×103细胞/孔的浓度接种96 孔板,混匀后于37℃ 5% CO2培养24h。 6、转染试剂Lipofectamin2000 用于转染过表达质粒,剂量如下,每个剂量
MCB课程 真核细胞的基因表达和调控 一,生物体内遗传物质的基本结构和功能单位是基因 上个世纪70年代在细胞生物学,细胞遗传学和生物化学的基础上,经过一系列重大发现而奠定基础,逐步发展形成了分子生物学(molecular biology)这一现代生命学科。分子 生物学认为生物体内存在着决定生物体性状的遗传物质,其基本的结构和功能单位是基因(gene)。基因的本质是一段携带着能合成功能蛋白质所需的全部信息的DNA,其中包括着蛋白质的编码序列,也包括非编码的调控序列。基因主要具有两大功能。一是指导合成蛋白质,通过蛋白质发挥的功能将遗传信息转换成具体的细胞性状和功能;二是通过细胞有丝分裂过程中的DNA复制(replication),将遗传信息传递给子代细胞,从而保持子代细胞与母代细胞性状的一致性。基因在双螺旋结构的DNA长链组成的染色体 上呈线性排列。在哺乳动物的真核细胞中线性排列的基因以核小体 (nucleosome) 的形式被紧密包绕存在于细胞核中,组成核染质(chromatin)。核小体的核心是由H2a,H2b,H3和 H4四组组蛋白形成的八聚体,核心外包绕着1又3/4圈的DNA长链。因此在电镜下核 染质呈“串珠样“结构。由于基因的本质是呈双螺旋结构的方向相反的两条脱氧核糖核酸(DNA)分子,因此基因的排列具有方向性,其DNA分子的5’端为基因的上游,3’端为基因的下游。构成基因DNA分子序列的有腺嘌呤(A)胸腺嘧啶(T)胞嘧啶(C)和鸟嘌呤(G)4种碱基。在双链DNA分子中一条DNA分子上的A总是以两条氢键与另一条DNA分子上的T相结合,而C总是以三条氢键与G相结合。A与T,C与G 之间称为互补关系(complementary)。双链DNA分子中A,T,C,G的不同组合排列形成了三联密码,每一个三联密码都代表着一种相应的氨基酸。然而,基因中的编码序列往往并不连续,其中间隔着非编码的序列。这些编码的序列称为基因结构中的外显子,而非编码序列称为内含子。在基因的上游端具有启动基因表达作用的特殊序列称为启动子,它们的序列中富含A,T,C, 在基因的上游,下游较远处,乃至基因内部还有某些序列对基因的表达有明显的促进作用,称为增强子。基因的下游端往往还有基因表达的终止信号。上述基因本身的主要结构统称为基因的顺式元件,而参与基因表达过程的基因外的蛋白质因子称为基因的反式元件(见下节)。上述排列着基因的DNA成为基因组DNA,真 核细胞中除了基因组DNA携带遗传信息外,线粒体中能独立复制的DNA也携带着遗传 信息。
浅谈原核表达的技巧 摘要:原核表达是表达外源基因常用的方法,具有操作简单、快捷,需时较短,表达产量高,适合工业化等优点。本文作者根据自己的实践经验,总结了原核表达的一些技巧。 关键词:原核表达表达载体限制性内切酶 将植物、动物、微生物等的目的基因插入合适载体后导入大肠杆菌用于表达大量蛋白质的方法一般称为原核表达。这种方法在蛋白纯化、定位及功能分析等方面都有应用。大肠杆菌用于表达重组蛋白有以下优点:易于生长和控制;易于培养,实验耗费少;可选择多种大肠杆菌菌株及与之匹配的具各种特性的质粒。原核表达是近年来表达外源蛋白常用的方法,本文根据自己的实践经验,着重谈谈对原核表达中的技巧问题。 一、原核表达一般程序 表达前准备-获得目的基因-构建含目的片段的表达载体(测序验证)-转化表达宿主菌-诱导靶蛋白的表达-表达蛋白的分析。 二、原核表达中各操作步骤的关键因素及技巧 1.表达前的准备要素:原核表达注重表达前对目的片段、表达载体及表达菌株的分析、选择。正所谓“磨刀不误砍柴功”,经过细致、周全的分析、准备、设计可带来较为顺当的实验,可免去许多不必要的麻烦。 (1)对表达载体的分析 载体的选择:同样的载体,同样的系统,很可能表达这个蛋白表达量起高,但另外一个就是做不出来,所以表达载体的选择非常重要,没有万能的载体。选择载体通常我们关心质粒上的几个功能组件及所带来的问题:是否为诱导表达型载体,启动子的强弱、多克隆位点、限制性内切酶的位置、终止密码子的有无及位置,融合Tag的有无,筛选报告基因的位置等。所选载体一定要保持原来的遗传背景(有些载体经过多次交换已变异)。选择表达载体时,要根据所表达蛋白的最终应用考虑,如果为了方便纯化,可选择融合表达;如果为了获得天然蛋白,可选择非融合表达。融合表达时在选择外源DNA同载体分子连接反应时,对转录和转译过程中密码结构的阅读不能发生干扰。 翻译的起始位点:要表达目的蛋白,在该基因的5’端必须有一起始位点,现在大部分的表达载体都提供起始位点,起始密码子与核糖体结合位点的距离都已被优化,一般情况下不需要自己再加,实际操作时要留意载体图谱上是否注明有起始密码子和终止密码子,如无,还得根据自己的实际情况加上。 在起始密码子附近的mRNA二级结构:外源基因其始转录后,保持mRNA的有效延伸、终止及稳定存在是外源基因有效表达的关键,尤其是在起始密码子附近的mRNA二级结构可能会抑制翻译的起始或者造成翻译暂停从而产生不完全的蛋白。如果利用Primer Premier软件分析DNA或RNA结构上有柄(stem)结构,并且结合长度超过8个碱基,这种结构会因为位点专一突变等因素而变得不稳定,影响正常的翻译。 (2)对目的片段的分析 基因(或蛋白)的大小:原核表达的成功与否与所要表达的蛋白(或基因)大小有关,一般说来小于5kD或者大于100kD的蛋白都是难以表达的。蛋白越小,越容易被内源蛋白水解酶所降解。在这种情况下可以采取串联表达,在每个表达单位(即单体蛋白)间设计蛋白水解或者是化学断裂位点。如果蛋白较小,那么加入融合标签GST、Trx、MBP或者其它较大的促进融合的蛋白标签就较有可能使蛋白正确折叠,并以融合形式表达。如果蛋白较大,大于60kD的蛋白建议使用较小的标签(如6×组氨酸标签)。对于结构研究较清楚的蛋白可以采取截取表达。当然表达时要根据目的进行截取,如果是要进行抗体制备而截取,那么一定要保证截取的部位抗原性较强。对于抗原性也可
原核、真核表达载体构建 真核表达载体和原核表达载体的区别:主要是因为原核和真核表达系统所需的表达元件不同。 比如说启动子,终止子在两种表达系统中是不一样的。 带有真核表达元件的是真核载体,能在真核生物内表达; 带有原核表达元件的是原核载体,能在原核生物内表达。两者都具有的为穿梭载体。 ㈠原核表达载体指:能携带插入的外源核酸序列进入原核细胞中进行复制的载体。 原核表达载体调控原件 1.启动子 启动子是DNA链上一段能与RNA聚合酶结合并起始RNA合成的序列,它是基因表达不可缺少的重要调控序列。没有启动子,基因就不能转录。由于细菌RNA聚合酶不能识别真核基因的启动子,因此原核表达载体所用的启动子必须是原核启动子。原核启动子是由两段彼此分开且又高度保守的核苷酸序列组成,对mRNA的合成极为重要。在转录起始点上游5~10 bp处,有一段由6~8个碱基组成,富含A和T的区域,称为Pribnow 盒,又名TATA 盒或-10区。来源不同的启动子,Pribnow 盒的碱基顺序稍有变化。在距转录起始位点上游35 bp 处,有一段由10 bp组成的区域,称为-35区。转录时大肠杆菌RNA聚合酶识别并结合启动子。-35区与RNA聚合酶s亚基结合,-10区与RNA聚合酶的核心酶结合,在转录起始位点附近DNA被解旋形成单链,RNA聚合酶使第一和第二核苷酸形成磷酸二酯键,以后在RNA聚合酶作用下向前推进,形成新生的RNA 链。原核表达系统中通常使用的可调控的启动子有Lac(乳糖启动子)、Trp(色氨酸启动子)、Tac(乳糖和色氨酸的杂合启动子) 、lPL (l噬菌体的左向启动子)、T7噬菌体启动子等。 2. SD序列 1974年Shine和Dalgarno首先发现,在mRNA上有核糖体的结合位点,它们是起始密码子AUG和一段位于AUG上游3~10 bp处的由3~9 bp组成的序列。这段序列富含嘌呤核苷酸,刚好与16S rRNA 3¢末端的富含嘧啶的序列互补,是核糖体RNA的识别与结合位点。以后将此序列命名为Shine-Dalgarno序列,简称SD序列。它与起始密码子AUG之间的距离是影响mRNA转录、翻译成蛋白的重要因素之一,某些蛋白质与SD序列结合也会影响mRNA与核糖体的结合,从而影响蛋白质的翻译。另外,真核基因的第二个密码子必须紧接在ATG 之后,才能产生一个完整的蛋白质。 3.终止子 在一个基因的3¢末端或是一个操纵子的3'末端往往有特定的核苷酸序列,且具有终止转录功能,这一序列称之为转录终止子,简称终止子(terminator)。转录终止过程包括:RNA聚合酶停在DNA模板上不再前进,RNA的延伸也停止在终止信号上,完成转录的RNA从RNA聚合酶上释放出来。对RNA聚合酶起