
9101分析方法验证指导原则
分析方法验证(analytical method validation)的目的是证明建立的方法适合于相应检测要求。在建立药品质量标准、变更药品生产工艺和制剂组分、修订原分析方法时,需对分析方法进行验证。生物制品质量控制中采用的方法包括理化分析方法和生物学测定方法,其中理化分析方法的验证原则与化学药品基本相同,所以可参照本指导原则进行,但在进行具体验证时还需要结合生物制品的特点考虑;相对于理化分析方法而言,生物学测定方法存在更多的影响因素,因此本指导原则不涉及生物学测定方法验证的内容。
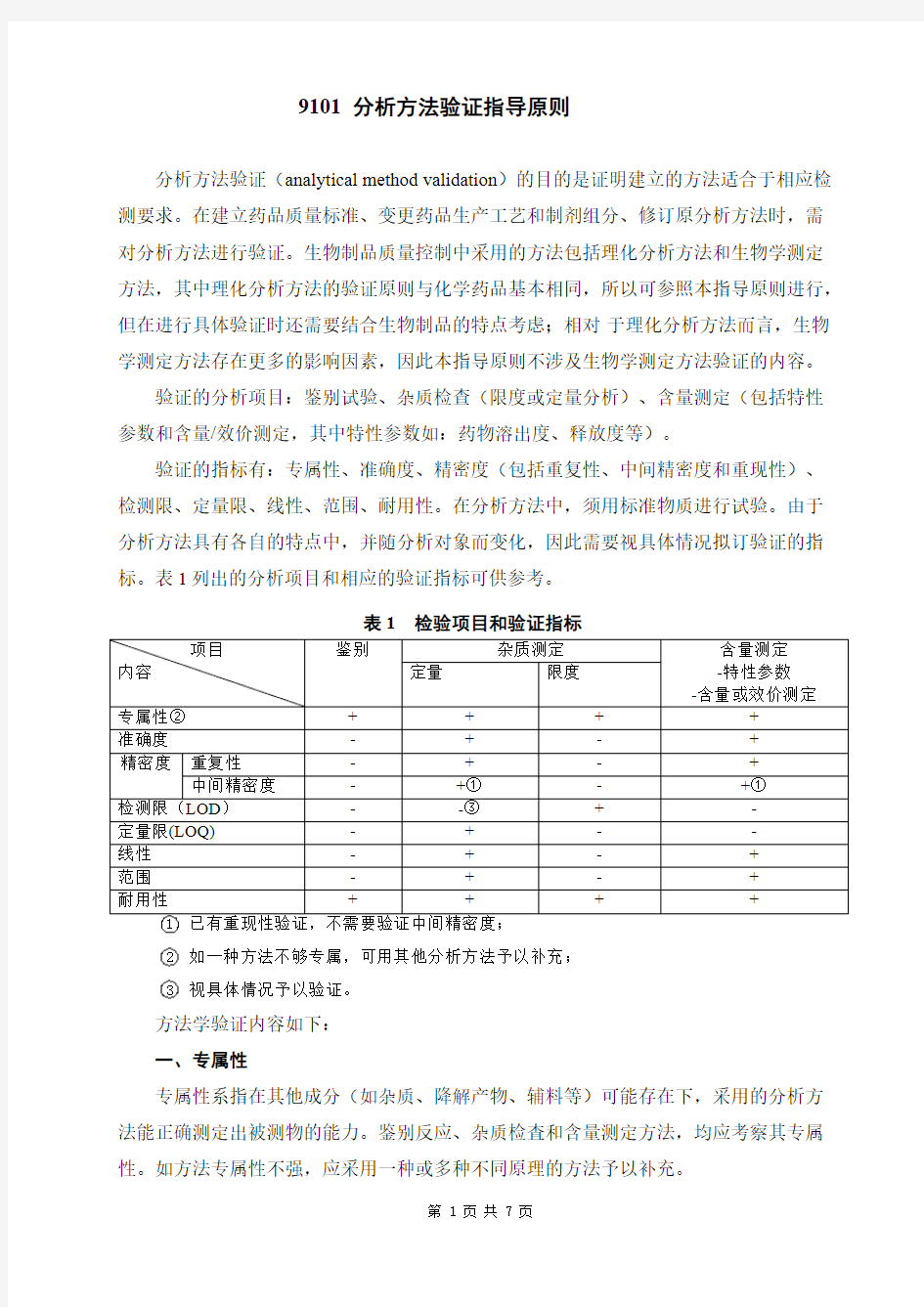
验证的分析项目:鉴别试验、杂质检查(限度或定量分析)、含量测定(包括特性参数和含量/效价测定,其中特性参数如:药物溶出度、释放度等)。
验证的指标有:专属性、准确度、精密度(包括重复性、中间精密度和重现性)、检测限、定量限、线性、范围、耐用性。在分析方法中,须用标准物质进行试验。由于分析方法具有各自的特点中,并随分析对象而变化,因此需要视具体情况拟订验证的指标。表1列出的分析项目和相应的验证指标可供参考。
表1检验项目和验证指标
项目内容鉴别杂质测定含量测定
-特性参数
-含量或效价测定定量限度
专属性②++++
准确度-+-+
精密度重复性-+-+中间精密度-+①-+①
检测限(LOD)--③+-
定量限(LOQ)-+--
线性-+-+
范围-+-+
耐用性++++ 1已有重现性验证,不需要验证中间精密度;
2如一种方法不够专属,可用其他分析方法予以补充;
3视具体情况予以验证。
方法学验证内容如下:
一、专属性
专属性系指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的分析方法能正确测定出被测物的能力。鉴别反应、杂质检査和含量测定方法,均应考察其专属性。如方法专属性不强,应采用一种或多种不同原理的方法予以补充。
1.鉴别反应
应能区分可能共存的物质或结构相似的化合物。不含被测成分的供试品,以及结构相似或组分中的有关化合物,应均呈阴性反应。
2.含量测定和杂质测定
采用的色谱法和其他分离方法,应附代表性图谱,以说明方法的专属性,并应标明诸成分在图中的位置,色谱法中的分离度应符合要求。
在杂质对照品可获得的情况下,对于含量测定,试样中可加入杂质或辅料,考察测定结果是否受干扰,并可与未加杂质或辅料的试样比较测定结果。对于杂质检查,也可向试样中加入一定量的杂质,考察杂质之间能否得到分离。
在杂质或降解产物不能获得的情况下,可将含有杂质或降解产物的试样进行测定,与另一个经验证的方法或药典方法比较结果。也可用强光照射、高温、高湿、酸(碱)水解或氧化的方法进行强制破坏,以研究可能的降解产物和降解途径对含量测定和杂质测定的影响。含量测定方法应比对两种方法的结果,杂质检査应比对检出的杂质个数,必要时可采用光电二极管阵列检测和质谱检测,进行峰纯度检查。
二、准确度
准确度系指用所建立方法测定的结果与真实值或参比值接近的程度,一般用回收率(%)表示。准确度应在规定的线性范围内试验。准确度也可由所测定的精密度、专属性推算出来。
在规定范围内,取同一浓度(相当于100%浓度水平)的供试品,用至少6份样品的测定结果进行评价;或设计至少3种不同浓度,每种浓度分别制备至少3份供试品溶液进行测定,用至少9份样品的测定结果进行评价,且浓度的设定应考虑样品的浓度范围。两种方法的选定应考虑分析的目的和样品的浓度范围。
1.化学药含量测定方法的准确度
原料药可用已知纯度的对照品或供试品进行测定,或用所测定结果与已知准确度的另一个方法测定的结果进行比较。制剂可在处方量空白辅料中,加入已知量被测物对照品进行测定。如不能得到制剂辅料的全部组分,可向待测制剂中加入已知量的被测物进行测定,或用所建立方法的测定结果与已知准确度的另一个方法测定结果进行比较。
2.化学药杂质定量测定的准确度
可向原料药或制剂中加入已知量杂质对照品进行测定。如不能得到杂质对照品,可用所建立的方法与另一成熟的方法(如药典标准方法或经过验证的方法)的测定结果进行比较。
3.中药化学成分测定方法的准确度
可用已知纯度的对照品进行加样回收率测定,即向已知被测成分含量的供试品中再精密加入一定量的已知纯度的被测成分对照品,依法测定。用实测值与供试品中含有量之差,除以加入对照品量计算回收率。在加样回收试验中须注意对照品的加入量与供试品中被测成分含有量之和必须在标准曲线线性范围之内;加入的对照品的量要适当,过小则引起较大的相对误差,过大则干扰成分相对减少,真实性差。
4.数据要求
对于化学药应报告已知加入量的回收率(%),或测定结果平均值与真实值之差及其相对标准偏差或置信区间(置信度一般为95%);对于中药应报告供试品取样量、供试品中含有量、对照品加入量、测定结果和回收率(%)计算值,以及回收率(%)的相对标准偏差(RSD%)或置信区间。样品中待测定成分含量和回收率限度关系可参考表2。在基质复杂、组分含量低于0.01%及多成分等分析中,回收率限度可适当放宽。
表2样品中待测定成分含量和回收率限度
待测定成分含量待测定成分质量分数回收率限
度(%)(%)(ppm或ppb)(mg/g或μg/g)(g/g)
100–1000mg/g 1.098~101
10100000ppm100mg/g0.195~102
110000ppm10mg/g0.0192~105
0.11000ppm1mg/g0.00190~108
0.01100ppm100μg/g0.000185~110
0.00110ppm10μg/g0.0000180~115
0.00011ppm1μg/g0.00000175~120
10ppb0.01μg/g0.0000000170~125 *此表源自AOAC《Guidelines for Single Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals》。
三、精密度
精密度系指在规定的测定条件下,同一份均匀供试品,经多次取样测定所得结果之间的接近程度。精密度一般用偏差、标准偏差或相对标准偏差表示。
在相同条件下,由同一个分析人员测定所得结果的精密度称为重复性;在同一个实验室内的条件改变、不同分析人员、不同设备测等测定结果之间的精密度,称为中间精密度;在不同实验室测定结果之间的精密度,称为重现性。
含量测定和杂质的定量测定应考察方法的精密度。
1.重复性
在规定范围内,取同一浓度(分析方法拟定的样品测定浓度,相当于100%浓度水平)的供试品,用至少6份的测定结果进行评价;或设计至少3种不同浓度,每种浓度分别制
备至少3份供试品溶液进行测定,用至少9份样品的测定结果进行评价。采用至少9份测定
结果进行评价时,浓度的设定应考虑样品的浓度范围。
2.中间精密度
考察随机变动因素如不同日期、不同分析人员、不同仪器对精密度的影响,应进行
中间精密度试验。
3.重现性
国家药品质量标准采用的分析方法,应进行重现性试验,如通过不同实验室协同检
验获得重现性结果。协同检验的目的、过程和重现性结果均应记载在起草说明中。应注
意重现性试验所用样品质量的一致性及贮存运输中的环境对该一致性的影响,以免影响
重现性
试验结果。
4.数据要求
均应报告标准偏差、相对标准偏差或置信区间。样品中待测定成分含量和精密度
RSD可接受范围参考表3(可接受范围可在给出数值0.5~2倍区间,计算公式,重复性:RSD r=C-0.15;重现性:RSD R=2C-0.15,其中C为待测定成分含量)。在基质复杂、组分含
量低于0.01%及多成分等分析中,精密度限度可适当放宽。
表3样品中待测定成分的含量与精密度可接受范围关系
待测定成分含量待测定成分
质量分数重复性
(RSD r%)
重现性
(RSD R%)
(%)(ppm或
ppb)
(mg/g或
μg/g)
(g/g)
100–1000mg/g 1.012 10100000ppm100mg/g0.1 1.53 110000ppm10mg/g0.0124 0.11000ppm1mg/g0.00136 0.01100ppm100μg/g0.000148 0.00110ppm10μg/g0.00001611
0.00011ppm1μg/g0.000001816
10ppb0.01μg/g0.000000011532 *此表源自AOAC《Guidelines for Single Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals》。
四、检测限
检测限系指试样中被测物能被检测出的最低量。检测限仅作为限度试验指标和定性鉴别的依据,没有定量意义。常用的方法如下。
1.直观法
用已知浓度的被测物,试验出能被可靠地检测出的最低浓度或量。
2.信噪比法
用于能显示基线噪声的分析方法,即把已知低浓度试样测出的信号与空白样品测出的信号进行比较,计算出能被可靠地检测出的被测物质最低浓度或量。一般以信噪比为3:1
或时相应浓度或注入仪器的量确定检测限。
3.基于响应值标准偏差和标准曲线斜率法
按照LOD=3.3δ/S公式计算。式中LOD:检测限;δ:响应值的偏差;S:标准曲线的斜率。
δ可以通过下列方法测得:(1)测定空白值的标准偏差;(2)标准曲线的剩余标准偏差或是截距的标准偏差。
4.数据要求
上述计算方法获得的检测限数据须用含量相近的样品进行验证。应附测定图谱,说明试验过程和检测限结果。
五、定量限
定量限系指试样中被测物能被定量测定的最低量,其测定结果应符合准确度和精密度要求。对微量或痕量药物分析、定量测定药物杂质和降解产物时,应确定方法的定量限。常用的方法如下。
1.直观法
用已知浓度的被测物,试验出能被可靠地定量测定的最低浓度或量。
2.信噪比法
用于能显示基线噪声的分析方法,即已知低浓度试样测出的信号与空白样品测出的
信号进行比较,计算出能被可靠地定量的被测物质的最低浓度或量。一般以信噪比为10:1时相应浓度或注入仪器的量确定定量限。
3.基于响应值标准偏差和标准曲线斜率法
按照LOQ=10δ/S公式计算。式中LOQ:定量限;δ:响应值的偏差;S:标准曲线的斜率。
δ可以通过下列方法测得:(1)测定空白值的标准偏差;(2)采用标准曲线的剩余标准偏差或是截距的标准偏差。
4.数据要求
上述计算方法获得的定量限数据须用含量相近的样品进行验证。应附测试图谱,说明测试过程和定量限结果,包括准确度和精密度验证数据。
六、线性
线性系指在设计的范围内,线性试验结果与试样中被测物浓度直接呈比例关系的能力。
应在设计的范围内测定线性关系。可用同一对照品贮备液经精密稀释,或分别精密称取对照品,制备一系列对照品溶液的方法进行测定,至少制备5种不同浓度水平。以测得的响应信号作为被测物浓度的函数作图,观察是否呈线性,再用最小二乘法进行线性回归。必要时,响应信号可经数学转换,再进行线性回归计算。或者可采用描述浓度-响应关系的非线性模型。
数据要求:应列出回归方程、相关系数、残差平方和、线性图(或其它数学模型)。
七、范围
范围系指分析方法能达到精密度、准确度和线性要求时的高低限浓度或量的区间。
范围应根据分析方法的具体应用及其线性、准确度、精密度结果和要求确定。原料药
和制剂含量测定,范围一般为测定浓度的80%~120%;制剂含量均匀度检査,范围一般为测定浓度的70%~130%,特殊剂型,如气雾剂和喷雾剂,范围可适当放宽;溶出度或释放度中的溶出量测定,范围一般为限度的±30%,如规定了限度范围,则应为下限的-20%至上限的+20%;杂质测定,范围应根据初步实际测定数据,拟订为规定限度的
±20%。如果一个试验同时进行含量测定与纯度检查,且仅使用100%的对照品,线性范围应覆盖杂质的报告水平至规定含量的120%。
在中药分析中,范围应根据分析方法的具体应用和线性、准确度、精密度结果及要求确定。对于有毒的、具特殊功效或药理作用的成分,其验证范围应大于被限定含量的
区间。溶出度或释放度中的溶出量测定,范围一般为限度的±30%。
八、耐用性
耐用性系指在测定条件有小的变动时,测定结果不受影响的承受程度,为所建立的方法用于常规检验提供依据。开始研究分析方法时,就应考虑其耐用性。如果测试条件要求苛刻,则应在方法中写明,并注明可以接受变动的范围,可以先采用均匀设计确定主要影响因素,再通过单因素分析等确定变动范围。典型的变动因素有被测溶液的稳定性、样品的提取次数、时间等。液相色谱法中典型的变动因素有流动相的组成和pH值、不同品牌或不同批号的同类型色谱柱、柱温、流速等。气相色谱法变动因素有不同品牌或批号的色谱柱、不同类型的担体、载气流速、柱温、进样口和检测器温度等。
经试验,测定条件小的变动应能满足系统适用性试验要求,以确保方法的可靠性。
指导原则编号: 【H】G P H 5-1 化学药物质量控制分析方法验证 技术指导原则 二○○五年三月
目 录 一、概述 (1) 二、方法验证的一般原则 (2) 三、方法验证涉及到的三个主要方面 (2) (一)需要验证的检测项目 (2) (二)分析方法 (3) (三)验证内容 (3) 四、方法验证的具体内容 (3) (一)专属性 (3) 1、鉴别反应 (4) 2、杂质检查 (4) 3、含量测定 (4) (二)线性 (5) (三)范围 (5) 1、含量测定 (6) 2、制剂含量均匀度 (6) 3、溶出度或释放度 (6) 4、杂质 (6) (四)准确度 (6) 1、含量测定 (7) 2、杂质定量试验 (7) (五)精密度 (7) 1、重复性 (8) 2、中间精密度 (8) 3、重现性 (8)
(六)检测限 (8) 1、直观法 (8) 2、信噪比法 (9) (七)定量限 (9) 1、直观法 (9) 2、信噪比法 (9) (八)耐用性 (10) (九)系统适用性试验 (10) 五、方法再验证 (11) 六、方法验证的评价 (12) (一)有关方法验证评价的一般考虑 (12) (二)方法验证的整体性和系统性 (12) 七、参考文献 (13) 八、著者 (13)
化学药物质量控制分析方法验证技术指导原则 一、概述 保证药品安全、有效、质量可控是药品研发和评价应遵循的基本原则,其中,对药品进行质量控制是保证药品安全有效的基础和前提。为达到控制质量的目的,需要多角度、多层面来控制药品质量,也就是说要对药物进行多个项目测试,来全面考察药品质量。一般地,每一测试项目可选用不同的分析方法,为使测试结果准确、可靠,必须对所采用的分析方法的科学性、准确性和可行性进行验证,以充分表明分析方法符合测试项目的目的和要求,这就是通常所说的对方法进行验证。 方法验证的目的是判断采用的分析方法是否科学、合理,是否能有效控制药品的内在质量。从本质上讲,方法验证就是根据检测项目的要求,预先设置一定的验证内容,并通过设计合理的试验来验证所采用的分析方法能否符合检测项目的要求。 方法验证在分析方法建立过程中具有重要的作用,并成为质量研究和质量控制的组成部分。只有经过验证的分析方法才能用于控制药品质量,因此方法验证是制订质量标准的基础。方法验证是药物研究过程中的重要内容。 本指导原则重点探讨方法验证的本质,将分析方法验证的要求与所要达到的目的结合起来进行系统和规律性的阐述,重点阐述如何科学合理地进行论证方案的设计。 本指导原则主要包括方法验证的一般原则、方法验证涉及的三个主要方
1、《中国药典》2015年修订情况介绍。 答:“中国药典”是国家为保证药品质量可控和人民群众用药安全有效而制定的药品法典。是药品开发、生产、经营、使用、管理的法律依据,是国家药品标准体系的核心。2015年版《中国药典》是新中国成立以来的第10版。2010年3月,第十届药典委员会成立,历时5年完成新版药典编制工作。编写期间,将修订后的药典内容全部在网上公示并征求意见,共收到网上反馈意见4000余条,远远超过前几版药典收到的反馈意见数量,体现了社会和公众对新版药典编写的关注度和参与度不断提高。 针对各种反馈意见,药典委员会各专业委员会逐一研究讨论,组织召开标准评审会700余次,向社会反馈意见。可以说,2015年版《中国药典》不仅凝聚了第十届药典委员会全体委员、广大专家学者、药品检验机构、科研院所、高校和药品生产企业的心血,更蕴含着社会公众的共同智慧。 2、2015年版药典实施细则。 答:新版《中国药典》于2015年12月1日起施行,新版《药典》每5年发布一次。自实施之日起,上市药品质量标准应符合2015年版《中国药典》品种质量标准。该品种已列入2015年版药典但未收录的质量标准,也应符合《中国药典总则》的相关要求。对于那些已提交注册、未获批准的品种,在批准时也要符合2015年版药典标准
的相关要求。 3、2015年版《中国药典》主要有哪些变化? 答:首先,收到的品种数量增加了27.4%。2015年版药典计划收录5800个品种,比2010年版药典增加1200多个品种,修订品种751个。 二是通过对《药典》总则、总则、总则的全面增补和修订,整体上进一步提高了对药品质量控制的要求,完善了《药典》标准的技术规定,使《药典》标准更加系统化、规范化。 三是完善了药品标准体系。 特别是药用辅料品种增加到260种,增加了相关指导原则;在归纳、验证、规范的基础上,实现了《中国药典》不同部分常用检测方法的协调统一。 四是2015年版药典附录(总则)和辅料独立卷成册,构成《中国药典》四个部分的主要内容。 五是药用辅料品种明显增加。计划新增128家,共计260家,增速高达97%。 六是安全治理工程大幅提升。 中医药:制定中草药、饮片二氧化硫残留限量标准,建立健全重金属和有害元素、黄曲霉毒素、农药残留等物质检测限量标准;加强中草药重金属和有毒有害物质管控。化学药品:有关物质加强杂质定性定量检测方法研究,实现已知杂质和未知杂质的差异化控制,优化抗生
2015版《中国药典》考卷 一、填空题(20分) 1、《中华人民共和国药典》(以下简称《中国药典》)2015年版已由国家食品药品监督管理总局2015年第67号公告(2015年07月15日)发布,自起实施。 2、2015版药典将分为四部出版,每部的主要内容分别是一部;二部;三部;四部、。 3、山药等10种传统习用硫磺熏蒸的中药材及其饮片,二氧化硫残留量不得过,其他中药材及其饮片的二氧化硫残留量不得过。 4、“”项下明确列出的有机溶剂或未在正文中列有此项检查的品种,如生产过程中引入或产品中残留有机溶剂,均应按附录“”检查并应符合相应溶剂的限度要求。 5、微生物计数方法:1:;2:;3:最可能数法。 6、常用的鉴别方法包括和。 7、含量测定中常用的方法有和。 8、药品的灰分测定主要是指和。 9、重金属测定主要的测试方法有和。 10、SO2的测定方法有、和离子色谱法。 二、选择题(20分) 1、在《中国药典》检定通则中规定,以下哪种中药材的SO2残留量不得超过400 mg/kg。() A、山药 B、山药片 C、天冬 D、白芍 2、2015版《中国药典》四部通则2331 二氧化硫残留量测定法中规定三种方法,以下哪种不属于规定的方法。() A、酸碱滴定法 B、离子色谱法 C、液相色谱法 D、气相色谱法 3、以下哪种元素不属于重金属元素。() A、铅 B、钙 C、砷 D、磷 4、《中国药典》中通则0832水分测定法中明确了5种方法,除烘干法、减压干燥法外,以下哪种方法不是水分测定的方法。() A、费休氏法 B、甲苯法 C、气相色谱法 D、液相色谱法 5、在《中国药典》中规定除矿物、动物、海洋类以外的中药材中,铜的限值是。() A、10 mg/kg B、5 mg/kg C、1 mg/kg D、20 mg/kg 6、以下哪种测定方法不是《中国药典》规定的方法。() A、水溶浸出物测定法 B、醇溶性浸出物测定法 C、挥发性醚浸出物测定法 D、酯溶性浸出物测定法 7、下面哪种化学物质不是农药。() A、六六六 B、艾氏剂 C、氯丹 D、DNT 8、下面哪种农药不是有机氯类农药。() A、艾氏剂 B、狄氏剂 C、七氯 D、乐果 9、茯苓的SO2限值要求是。() A、150 mg/kg B、400 mg/kg C、10 mg/kg D、100 mg/kg 10、以下哪个选项不是气相色谱仪中的组件。() A、色谱柱 B、流动相 C、氦气 D、进样器 三、判断题(20分) 1、人参对农药残留量只有六六六、滴滴涕、五氯硝基苯有限定要求。()
药品微生物检验替代方法验证指导原则 本指导原则是为所采用的试验方法能否替代药典规定的方法用于药品微生物的检验提供指导。 随着微生物学的迅速发展,制药领域不断引入了一些新的微生物检验技术,大体可分为三类:(1)基于微生物生长信息的检验技术,如生物发光技术、电化学技术、比浊法等;(2)直接测定被测介质中活微生物的检验技术,如固相细胞技术法、流式细胞计数法等;(3)基于微生物细胞所含有特定组成成分的分析技术,如脂肪酸测定技术、核酸扩增技术、基因指纹分析技术等。这些方法与传统检查方法比较,或简便快速,或具有实时或近实时监控的潜力,使生产早期采取纠正措施及监控和指导优良生产成为可能,同时新技术的使用也促进了生产成本降低及检验水平的提高。 在控制药品微生物质量中,微生物实验室出于各种原因如成本、生产量、快速简便及提高药品质量等需要而采用非药典规定的检验方法(即替代方法)时,应进行替代方法的验证,确认其应用效果优于或等同于药典的方法。 微生物检验的类型及验证参数 药品微生物检验方法主要分两种类型:定性试验和定量试验。定性试验就是测定样品中是否存在活的微生物,如无菌检查及控制菌检査。定量试验就是测定样品中存在的微生物数量,如菌落计数试验。 由于生物试验的特殊性,如微生物检验方法中的抽样误差、稀释误差、操作误差、培养误差和计数误差都会对检验结果造成影响,因此,药品质量标准分析方法验证指导原则(附录XIX A)不完全适宜于微生物替代方法的验证。药品微生物检验替代方法的验证参数见表1。 表1 不同微生物检验类型验证参数 注: 尽管替代方法的验证参数与药品质量标准分析方法验证参数有相似之处,但是其具体的内容是依据微生物检验特点而设立的。替代方法验证的实验结果需进行统计分析,当替代方法属于定性检验时,一般采用非参数的统计技术;当替代方法属于定量检验时,需要采用参数统计技术。 进行微生物替代方法的验证时,若替代方法只是针对药典方法中的某一环节进行技术修改,此时,需要验证的对象仅是该项替代技术而不是整个检验方法。如无菌试验若改为使用含培养基的过滤器,然后通过适宜的技术确认活的微生物存在,那么,验证时仅需验证所用的微生物回收系统而不是整个无菌试验方法。 替代方法验证的一般要求 在开展替代方法对样品检验的适用性验证前,有必要对替代方法有一个全面的了解。首先,所选用的替代方法应具备必要的方法适用性证据,表明在不含样品的情况下,替代方法
该版药典中现代分析技术得到进一步扩大应用,除在附录中扩大收载成熟的新技术方法外,品种正文中进一步扩大了对新技术的应用;药品的安全性保障得到进一步加强,除在凡例和附录中加强安全性检查总体要求外,在品种正文标准中增加或完善安全性检查项目;对药品质量可控性、有效性的技术保障得到进一步提升,除在附录中新增和修订相关的检查方法和指导原则外,在品种正文标准中增加或完善有效性检查项目;为适应药品监督管理的需要,制剂通则中新增了药用辅料总体要求;积极引人了国际协调组织在药品杂质控制、无菌检查法等方面的要求和限度。此外,该版药典也体现了对野生资源保护与中药可持续发展的理念,不再收载濒危野生药材。 第九届药典委员会还完成了《中国药典》2005年版增补本、《药品红外光谱集》(第四卷)、《临床用药须知》(中药材和饮片第一版、中成药第二版、化学药第五版)、《中药材显微鉴别彩色图鉴》及《中药材薄 层色谱彩色图集》(第一册、第二册)的编制工作。 2015年版(第十版)2010年12月国家食品药品监督管理局(2013年3月22日更名为国家食品药品监督管理总局)组建第十届药典委员会。本届药典委员遴选工作按照新修订的《新增委员遴选办法》和《第十届药典委员会委员遴选工作方案》,向全社会公开征集新增委员候选人,并采取差额选举、无记名投票的方式选举新增委员。本届委员会共有委员351名,其中续聘委员248名,新增委员103名。时任第十一届全国人大常委会副委员长桑国卫任名誉主任委员,时任卫生部部长陈竺任主任委员,时任卫生部副部长、国家药品监督管理局局长邵明立任常务副主任委员。本届委员会下设执行委员会和23个专业委员会。执行委员会委员共计67名,其中院士委员28名、资深专家3名、各专业委员会主任20名、相关部委专家4名、总局相关技术单位负责人7名。根据药典标准工作需要,本届委员会以第九届药典委员会专业委员会设置为基础,对专业委员会的设立进行了适当调整;为加强化学药标准的制定工作,增设了化学药品第三专业委员会,扩大化学药委员的人数;同时,根据实际工作需要,取消政策与发展委员会、标准信息工作委员会和注射剂工作委员会。 2010年12月第十届药典委员会成立暨全体委员大会召开。会议审议通过了“《中国药典》2015年版编制大纲”,编制大纲明确了《中国药典》2015年版编制工作的指导思想、基本原则、发展目标和主要任务。 按照《国家药品安全“十二五”规划》的要求,国家药典委员会以实施“国家药品标准提高行动计划”为基础,组织各专业委员会和相关机构开展药典编制工作。药典委员会常设机构首次将I S O 9001质量管理体系引入药典编制的全过程管理,按照规范的“中国药典编制工作程序”开展品种遴选、课题立项、试验研究、标准起草、复核和审定等各项工作,稳步推进本版药典编制工作。2015年2月4日《中国药典》2015年版经第十届药典委员会执行委员会全体会议审议通过,于2015年6月5日经国家食品药品监督管理总局批准颁布,自2015年12月1日起实施。 本版药典进一步扩大药品品种的收载和修订,共收载品种5608种。一部收载品种2598种,其中新增品种440种、修订品种517种、不收载品种7种。二部收载品种2603种,其中新增品种492种、修订品种415种、不收载品种28种。三部收载品种137种,其中新增品种13种、修订品种105种、新增生物制品通则1个、新增生物制品总论3个、不收载品种6种。本版药典首次将上版药典附录整合为通则,并与药用辅料单独成卷作为《中国药典》四部。四部收载通则总数317个,其中制剂通则38个、检测方法240个(新增27个)、指导原则30个(新增15个)、标准品、标准物质及试液试药相关通则9个。药用辅料收载270种,其中新增137种、修订97种、不收载2种。 本版药典完善了药典标准体系的建设,整体提升质量控制的要求,进一步扩大了先进、成熟检测技术的应用,药用辅料的收载品种大幅增加,质量要求和安全性控制更加严格,使《中国药典》的引领作用和技术导向作用进一步体现。 在编制本版药典的过程中,还完成了《中国药典》2010年版第一、二、三增补本,《红外光谱集》(第五卷),《中国药品通用名称》,《国家药品标准工作手册》(第四版),《中国药典注释》的编制和修订工作,组织开展了《中国药典》2015年版英文版、《临床用药须知》2015年版的编制工作。
I. INTRODUCTION This guidance provides recommendations to applicants on submitting analytical procedures, validation data, and samples to support the documentation of the identity, strength, quality, purity, and potency of drug substances and drug products. 1. 绪论 本指南旨在为申请者提供建议,以帮助其提交分析方法,方法验证资料和样品用于支持原料药和制剂的认定,剂量,质量,纯度和效力方面的文件。 This guidance is intended to assist applicants in assembling information, submitting samples, and presenting data to support analytical methodologies. The recommendations apply to drug substances and drug products covered in new drug applications (NDAs), abbreviated new drug applications (ANDAs), biologics license applications (BLAs), product license applications (PLAs), and supplements to these applications. 本指南旨在帮助申请者收集资料,递交样品并资料以支持分析方法。这些建议适用于NDA,ANDA,BLA,PLA及其它们的补充中所涉及的原料药和制剂。 The principles also apply to drug substances and drug products covered in Type II drug master files (DMFs). If a different approach is chosen, the applicant is encouraged to discuss the matter in advance with the center with product jurisdiction to prevent the expenditure of resources on preparing a submission that may later be determined to be unacceptable. 这些原则同样适用于二类DMF所涉及的原料药和制剂。如果使用了其它方法,鼓励申请者事先和FDA药品评审中心的官员进行讨论,以免出现这种情况,那就是花了人力物力所准备起来的递交资料后来发现是不可用的。 The principles of methods validation described in this guidance apply to all types of analytical procedures. However, the specific recommendations in this guidance may not be applicable to certain unique analytical procedures for products such as biological, biotechnological, botanical, or radiopharmaceutical drugs. 本指南中所述的分析方法验证的原则适用于各种类型的分析方法。但是,本指南中特定的建议可能不适用于有些产品所用的特殊分析方法,如生物药,生物技术药,植物药或放射性药物等。 For example, many bioassays are based on animal challenge models, 39 immunogenicity assessments, or other immunoassays that have unique features that should be considered when submitting analytical procedure and methods validation information. 比如说,许多生物分析是建立在动物挑战模式,免疫原性评估或其它有着独特特性的免疫分析基础上的,在递交分析方法和分析方法验证资料时需考虑这些独特的性质。Furthermore, specific recommendations for biological and immunochemical tests that may be necessary for characterization and quality control of many drug substances and drug products are beyond the scope of this guidance document. 而且,许多原料药和制剂的界定和质量控制所需的生物和免疫化学检测并不在本指南的范围之内。 Although this guidance does not specifically address the submission of analytical procedures and validation data for raw materials, intermediates, excipients, container closure components, and other materials used in the production of drug
浅谈2015年版中国药典的变更 1.基本情况: 1950年1月卫生部成立第一届国家药典委员会,组成8个专家的小组团队,展开中国药典的编制,亦是我国最早的标准化机构。第一部<中国药典>1953年版由卫生部编印发行。至今已组建十屇药典委员会,并经已编制共九版中国药典(英文名称为Pharmacopoeia of The People’s Republic of China; 英文简称为Chinese Pharmacopoeia; 英文缩写为Ch.P.)。中国药典是为保证药品产量、保障人民群众用药安全、有效、稳定、质量可控的技术法典,亦是药品研究、生产、经营、使用和监管的法定依据。 作为国家药品标准体系的核心及对外的竞争 力,药典收载范围遂步扩大,由1953年(第 一版)共531品种增加至现有的2010版(第九 版)共4567种(包括有中药: 2165种(一部),化 学药: 2271种(二部)及生物药制品: 131种(三 部)),当中涵盖了中药材、中药饮片、中药 饮片、中成药、生物制品、药用辅料、凡例、 通则及附录等等。 国家药品标准 国家药品标准是由凡例与正文及其引用的附录共同构成。并且对药典以外的其他国家标准具同等效力。由此可见,药典是国家对药品监控及为企业建立质量体系的重要手段。 药典的法律地位: 依照《药品管理法》规定: 药品必须符合国家药品标准。。” “药品必须符合国家药品标准 管理部门颁布的药典和药品标准为国家药品标准。。” “国务院药品监 国务院药品监督督管理部门颁布的药典和药品标准为国家药品标准 2.基本结构: 凡例: 为正确使用<中国药典>进行药品质控的基本原则,是对正文、附录及与质量检定有关的共性问题的统一规定。 正文: 各品种项下收载的内容统称正文,是根据药物自身的理化与生物学特性,按照批准的来源、处方、制法、和运输、贮藏等条件所制定的、用以检测药品是否达到用药要求,并衡量其质量是否稳定均一的技术规定。
201507 FDA行业指南:分析方法验证(中英文)(下) VII. STATISTICAL ANALYSIS AND MODELS 统计学分析和模型 A. Statistics 统计学 Statistical analysis of validation data can be used to evaluate validation characteristics against predetermined acceptance criteria. All statistical procedures and parameters used in the analysis of the data should be based on sound principles and appropriate for the intended evaluation. Several statistical methods are useful for assessing validation characteristics, for example, an analysis of variance (ANOVA) to assess regression analysis R (correlation coefficient) and R squared (coefficient of determination) or linear regression to measure linearity. Many statistical methods used for assessing validation characteristics rely on population normality, and it is important to determine whether or not to reject this assumption. There are many techniques, such as histograms, normality tests, and probability plots that can be used to evaluate the observed distribution. It may be appropriate to transform the data to better fit the normal distribution or apply distribution-free (nonparametric) approaches when the observed data are
药品及生物制品的分析方法和方法验证指导原则 目录 1.介绍...................... (1) 2.背景..................... .. (2) 3.分析方法开发. ..................... . (3) 4.分析程序内容.............................................. ......... ..................................... .. 3 A.原则/范围 (4) B.仪器/设备............................................. . (4) C.操作参数.............................................. .. (4) D.试剂/标准............................................. . (4) E.样品制备.............................................. .. (4) F.标准对照品溶液的制备............................................ .. (5) G.步骤......... ....................................... (5) H.系统适应性..... (5) I.计算 (5) J.数据报告 (5) 5.参考标准和教材............................................ (6) 6分析方法验证用于新药,仿制药,生物制品和DMF (6) A.非药典分析方法............................................. (6) B.验证特征 (7) C.药典分析方法............................................. .. (8) 7.统计分析和模型 (8) A.统计 (8) B.模型 (8) 8.生命周期管理分析程序 (9) A.重新验证 (9) B.分析方法的可比性研究............................................ . (10) 1.另一种分析方法............................................... .. (10) 2.分析方法转移的研究 (11) C.报告上市后变更已批准的新药,仿制药,或生物制品 (11) 9.美国FDA方法验证............................................... . (12) 10.参考文献
一、准确度 准确度系指采用该方法测定的结果与真实值或参考值接近的程度,一般用回收率(%)表示。准确度应在规定的范围内测定。 1.化学药含量测定方法的准确度 原料药采用对照品进行测定,或用本法所得结果与已知准确度的另一个方法测定的结果进行比较。制剂可在处方量空白辅料中,加入已知量被测物对照品进行测定。如不能得到制剂辅料的全部组分,可向待测制剂中加人已知量的被测物对照品进行测定,或用所建立方法的测定结果与已知准确度的另一种方法测定结果进行比较。准确度也可由所测定的精密度、线性和专属性推算出来。 2.化学药杂质定量测定的准确度 可向原料药或制剂处方量空白辅料中加人已知量杂质进行测定。如不能得到杂质或降解产物对照品,可用所建立方法测定的结果与另一成熟的方法进行比较,如药典标准方法或经过验证的方法。在不能测得杂质或降解产物的校正因子或不能测得对主成分的相对校正因子的情况下,可用不加校正因子的主成分自身对照法计算杂质含量。应明确表明单个杂质和杂质总量相当于主成分的重量比(%) 或面积比(% )。 3.中药化学成分测定方法的准确度 可用对照品进行加样回收率测定,即向已知被测成分含量的供试品中再精密加人一定量的被测成分对照品,依法测定。用实测值与供试品中含有量之差,除以加入对照品量计算回收率。在加样回收试验中须注意对照品的加人量与供试品中被测成分含有量之和必须在标准曲线线性范围之内;加入对照品的量要适当,过小则引起较大的相对误差,过大则干扰成分相对减少,真实性差。 回收率:%= (C - A ) /S X 100% 式中:A为供试品所含被测成分量;B 为加入对照品量; C 为实测值。 4.校正因子的准确度 对色谱方法而言,绝对(或定量)校正因子是指单位面积的色谱峰代表的待测物质的量。待测定物质与所选定的参照物质的绝对校正因子之比,即为相对校正因子。相对校正因子计算法常应用于化学药有关物质的测定、中药材及其复方制剂中多指标成分的测定。校正因子的表示方法很多,本指导原则中的校正因
中华人民共和国药典: 《中国药典》分为四部出版:一部收载药材和饮片、植物油脂和提取物、成方制剂和单味制剂等;二部收载化学药品、抗生素、生化药品以及放射性药品等;三部收载生物制品。 《中华人民共和国药典》2015年版,药典包括凡例、正文及通则,是药品研制、生产、经营、使用和监督管理等均应遵循的法定依据。所有国家药品标准应当符合中国药典凡例及附录的相关要求。 新版药典进一步扩大药品品种的收载和修订,共收载品种5608种。一部收载品种2598种,其中新增品种440种。二部收载品种2603种,其中新增品种492种。三部收载品种137种,其中新增品种13种、修订品种105种。首次将上版药典附录整合为通则,并与药用辅料单独成卷作为新版药典四部。四部收载通则总数317个,其中制剂通则38个、检测方法240个、指导原则30个、标准物质和对照品相关通则9个;药用辅料收载270种,其中新增137种、修订97种。 1949年10月1日中华人民共和国成立后,党和政府十分关怀人民的医药卫生保健工作,当年11月卫生部召集在京有关医药专家研讨编纂药典问题。1950年1月卫生部从上海调药学专家孟目的教授负责组建中国药典编纂委员会和处理日常工作的干事会,筹划编制新中国药典。 1950年4月在上海召开药典工作座谈会,讨论药典的收载品种原则和建议收载的品种,并根据卫生部指示,提出新中国药典要结合
国情,编出一部具有民族化、科学化、大众化的药典。随后,卫生部聘请药典委员49人,分设名词、化学药、制剂、植物药、生物制品、动物药、药理、剂量8个小组,另聘请通讯委员35人,成立了第一届中国药典编纂委员会。卫生部部长李德全任主任委员。 1951年4月24日至28日在北京召开第一届中国药典编纂委员会第一次全体会议,会议对药典的名称、收载品种、专用名词、度量衡问题以及格式排列等作出决定。干事会根据全会讨论的意见,对药典草案进行修订,草案于1952年底报卫生部核转政务院文教委员会批准后,第一部《中国药典》1953年版由卫生部编印发行。 1953年,版药典共收载药品531种,其中化学药215种,植物药与油脂类65种,动物药13种,抗生素2种,生物制品25种,各类制剂211种。药典出版后,于1957年出版《中国药典》1953年版第一增补本。 1955年,卫生部成立第二届药典委员会,聘请委员49人,通讯委员68人,但这届委员会因故未能进行工作。1957年成立第三届药典委员会,聘请委员80人,药学专家汤腾汉教授为这届委员会主任委员(不设通讯委员),同年7月28日至8月5日在北京召开第一次全体委员会议,卫生部李德全部长作了药典工作报告,特别指出第一版中国药典没有收载广大人民习用的中药,是个很大的缺陷。会议在总结工作的基础上,通过了制订药典的原则,讨论了药典的性质和作用,并修改了委员会章程,会议一致认为应把合乎条件的中药收载到药典中。8月27日卫生部批准委员会分设药理与医学、化学
美国FDA分析方法验证指南中英文对照 美国FDA分析方法验证指南中英文对 照 I. INTRODUCTION This guidance provides recommendations to applicants on submitting analytical procedures, validation data, and samples to support the documentation of the identity, strength, quality, purity, and potency of drug substances and drug products. 1. 绪论 本指南旨在为申请者提供建议,以帮助其提交分析方法,方法验证资料和样品用 于支持原料药和制剂的认定,剂量,质量,纯度和效力方面的文件。 This guidance is intended to assist applicants in assembling information, submitting samples, and presenting data to support analytical methodologies. The recommendations apply to drug substances and drug products covered in new drug applications (NDAs), abbreviated new drug applications (ANDAs), biologics license applications (BLAs), product license applications (PLAs), and supplements to these applications. 本指南旨在帮助申请者收集资料,递交样品并资料以支持分析方法。这些建议适 用于NDA,ANDA,BLA,PLA及其它们的补充中所涉及的原料药和制剂。 The principles also apply to drug substances and drug products covered in Type II drug master files (DMFs). If a different approach is chosen, the applicant is encouraged to discuss the matter in advance with
化学药物质量控制分析方法验证技术指导原则【】HGPH 5-1指导原则编号: 化学药物质量控制分析方法验证 技术指导原则 二??四年十一月 目录 一、概 述 ..................................................................... ............................................ 1 二、方法验证的一般原 则 ..................................................................... ................ 2 三、方法验证涉及到的三个主要方 面 (2) ,一,需要验证的检测项 目 ..................................................................... . (2) ,二,分析方 法 ..................................................................... .. (3) ,三,验证内 容 ..................................................................... .......................... 3 四、方法验证的具体内 容 ..................................................................... . (3)
,一,专属 性 ..................................................................... (3) 1、鉴别反 应 ..................................................................... (3) 2、杂质检 查 ..................................................................... (4) 3、含量测 定 ..................................................................... (4) ,二,线 性 ..................................................................... . (5) ,三,范 围 ..................................................................... . (5) 1、含量测 定 ..................................................................... (5) 2、制剂含量均匀度...................................................................... (5)
《中国药典》2015年版实施公告 有关问题的解读(一) 1. 问:国家食品药品监督管理总局关于实施《中华人民共和国 药典》2015年版(以下简称“2015年版药典”)有关事宜的公告(以下简称“公告”)(2015年第105号)中规定,为符合2015年版药典而需进行补充申请的,应在2015年 12月1日前进行申报,2015年12月1日后是否仍可提交相应补充申请? 答:对2015年版药典发布前已上市药品,生产企业应在2015年12月1日前完成原标准与新版药典相关要求的研究和比对,并应按公告要求进行相应的备案或补充申报。2015年12月1日以后仍可以提交相应补充申请。 2. 问:企业的注册标准已经对2010年版药典相关品种进行评估 的,且2010年版与2015年版药典品种质量标准和检测方法无变化的,是否需要重新对产品进行评估? 答:虽然品种正文内容与2015年版药典品种规定无变化,但由于2015年版药典通用性要求,包括凡例、通则、制剂通则以及通用性检验方法等进行了全面的增修订,因此,生产企业仍需针对2015年版药典通用性要求方面对本产品进行相应的评估。 3. 问:关于药品执行标准的表述方式的问题
答:对于注册标准不低于《中国药典》项目的制品,执行注册标准,其执行标准表示方式为:“执行药品注册标准且符合《中国药典》2015年版要求”。 4. 问:对于进口药品生产企业,能否使用注册代理公司出具的 说明信来代替国外的声明信,进行备案或补充申请的申报?答:原则上注册代理公司应出具持证商的声明信。如使用说明信代替国外的声明信,应同时提供进口药品生产企业出具的委托注册代理公司办理该事项的委托书。 5. 问:国家食药总局2015年第67号公告中规定,2015年版药 典自2015年12月1日起实施”。如何界定产品的执行日期?答:按是历版药典执行惯例要求,自2015年12月1日起生产或进口的药品应符合2015年版药典的相关规定。 6. 问:按照实施公告要求提出备案或补充申请的品种,审评审 批期间是否仍可执行原标准,期间若有进口再注册申请的是否可按原注册标准核发新证。 答:申请人应按105号实施公告第五款规定执行。出现补充申请与再注册申请交叉情形者,建议补充申请与进口再注册合并审评,如2015年12月1日起前已提交补充申请,可在补充申请期间执行原标准的要求。 7. 问:制剂中间体是否也需要按照制剂的药典标准进行提高?
9012 生物样品定量分析方法验证指导原则
1. 范围
准确测定生物基质(如全血、血清、血浆、尿)中的药物浓度,对于药物和 制剂研发非常重要。这些数据可被用于支持药品的安全性和有效性,或根据毒动 学、药动学和生物等效性试验的结果做出关键性决定。因此,必须完整地验证和 记录应用的生物分析方法,以获得可靠的结果。
本指导原则提供生物分析方法验证的要求,也涉及非临床或临床试验样品实 际分析的基本要求,以及何时可以使用部分验证或交叉验证,来替代完整验证。
生物样品定量分析方法验证和试验样品分析应符合本指导原则的技术要求。 应该在相应的生物样品分析中遵守 GLP 原则或 GCP 原则。
2. 生物分析方法验证
2.1 分析方法的完整验证
分析方法验证的主要目的是,证明特定方法对于测定在某种生物基质中分析 物浓度的可靠性。此外,方法验证应采用与试验样品相同的抗凝剂。一般应对每 个物种和每种基质进行完整验证。当难于获得相同的基质时,可以采用适当基质 替代,但要说明理由。
一个生物分析方法的主要特征包括:选择性、定量下限、响应函数和校正范 围(标准曲线性能)、准确度、精密度、基质效应、分析物在生物基质以及溶液 中储存和处理全过程中的稳定性。
有时可能需要测定多个分析物。这可能涉及两种不同的药物,也可能涉及一 个母体药物及其代谢物,或一个药物的对映体或异构体。在这些情况下,验证和 分析的原则适用于所有涉及的分析物。
对照标准物质 在方法验证中,含有分析物对照标准物质的溶液将被加入到空白生物基质 中。此外,色谱方法通常使用适当的内标。 应该从可追溯的来源获得对照标准物质。应该科学论证对照标准物质的适用 性。分析证书应该确认对照标准物质的纯度,并提供储存条件、失效日期和批号。 对于内标,只要能证明其适用性即可,例如显示该物质本身或其相关的任何杂质 不产生干扰。 当在生物分析方法中使用质谱检测时,推荐尽可能使用稳定同位素标记的内 标。它们必须具有足够高的同位素纯度,并且不发生同位素交换反应,以避免结 果的偏差。
1