
取代基效应
张巧燕
赤峰学院化学化工学院,赤峰024000
摘要:取代基不同而对分子性质产生的影响。取代基效应可以分为两大类。一类是电子效应,包括诱导效应、共轭效应和场效应。电子效应是通过键的极性传递所表现的分子中原子或基团间的相互影响,取代基通过影响分子中电子云的分布而起作用。另一类是空间效应,是由于取代基的大小和形状引起分子中特殊的张力或阻力的一种效应,空间效应也对分子的反应性产生一定影响。本文主要讨论电子效应。
关键字:诱导效应共轭效应场效应空间效应
取代基效
应
电子效应
空间效应(位阻效应,物理的相互作用
)
诱导效应(σ,π)
共轭效应(π-π, p-π)超共轭效应(σ- π,σ- p )
场效应空间传递
一、电子效应
电子效应是通过键的极性传递所表现的分子中原子或基团间的相互影响,取代基通过影响分子中电子云的分布而起作用。
有机化学中的电子效应有诱导效应(inductive effect)、共轭效应(conjugation)、超共轭效应(hyperconjugation)和场效应(field effect)。
1、诱导效应(inductive effect)
1.1共价键的极性与静态诱导效应
(1)共价键的极性(polarity):
成键原子间因电负性不同而使电子云不对称分布,电子云偏向电负性较高的原子一边,从而使共价键有了极性,称为极性共价键或极性键。
(2)诱导效应(inductive effect):
由于原子或基团电负性的影响沿着分子中的键传导,引起分子中电子云按一定方向转移或键的极性通过键链依次诱导传递的效应称为诱导效应或I效应。
(3)静态诱导效应(Is):
存在于未发生反应的分子中的诱导效应(电负性决定)
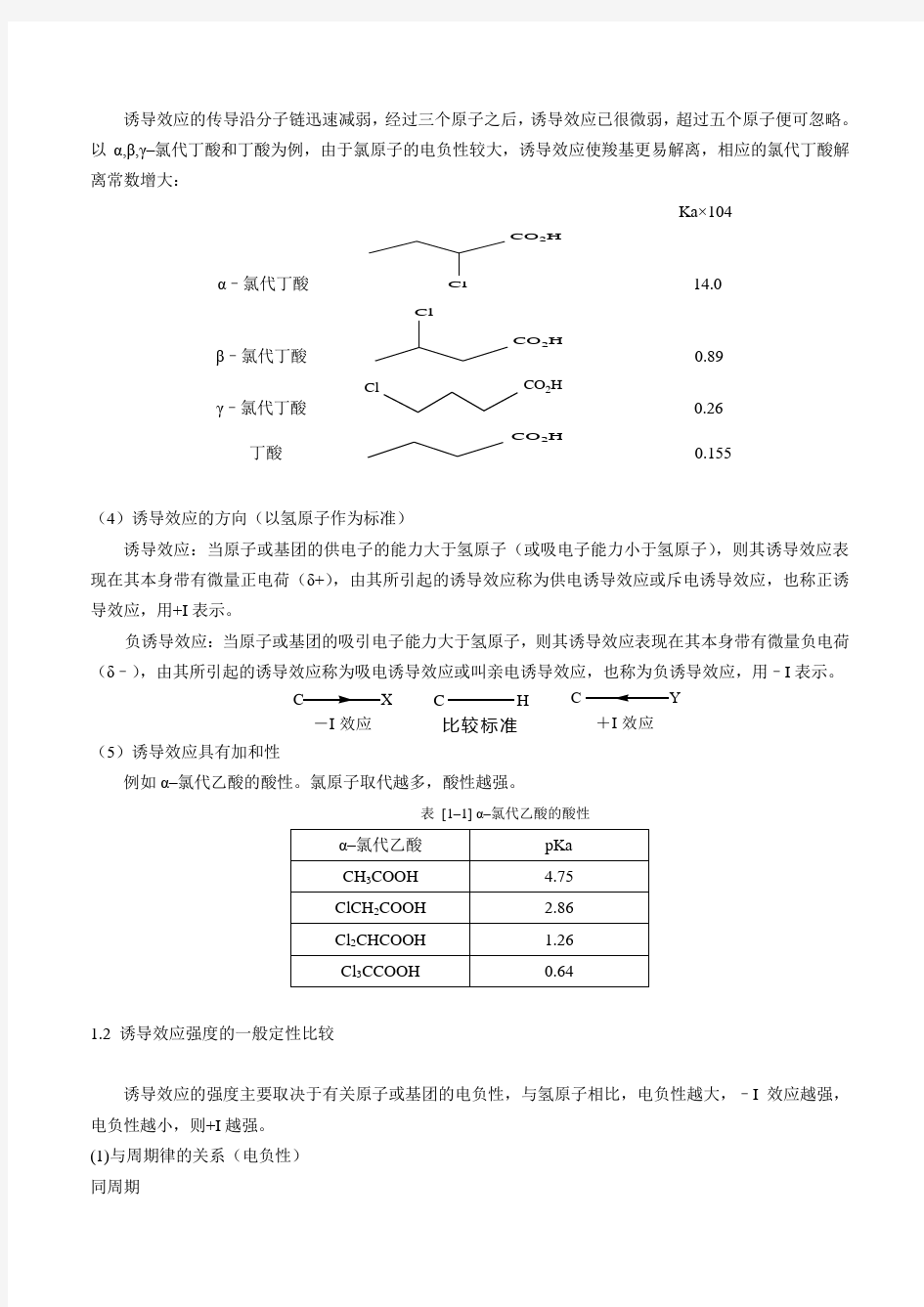
诱导效应的传导沿分子链迅速减弱,经过三个原子之后,诱导效应已很微弱,超过五个原子便可忽略。以α,β,γ–氯代丁酸和丁酸为例,由于氯原子的电负性较大,诱导效应使羧基更易解离,相应的氯代丁酸解离常数增大:
Ka×104
α–氯代丁酸
CO2H
Cl14.0
β–氯代丁酸
CO2H
Cl
0.89
γ–氯代丁酸
CO2H
Cl
0.26
丁酸
CO2H
0.155
(4)诱导效应的方向(以氢原子作为标准)
诱导效应:当原子或基团的供电子的能力大于氢原子(或吸电子能力小于氢原子),则其诱导效应表现在其本身带有微量正电荷(δ+),由其所引起的诱导效应称为供电诱导效应或斥电诱导效应,也称正诱导效应,用+I表示。
负诱导效应:当原子或基团的吸引电子能力大于氢原子,则其诱导效应表现在其本身带有微量负电荷(δ–),由其所引起的诱导效应称为吸电诱导效应或叫亲电诱导效应,也称为负诱导效应,用–I表示。
C X
-I 效应C H
比较标准
C
+I 效应
(5)诱导效应具有加和性
例如α–氯代乙酸的酸性。氯原子取代越多,酸性越强。
表[1–1] α–氯代乙酸的酸性
1.2 诱导效应强度的一般定性比较
诱导效应的强度主要取决于有关原子或基团的电负性,与氢原子相比,电负性越大,–I效应越强,电负性越小,则+I越强。
(1)与周期律的关系(电负性)
同周期
– I 效应: — F > — OH > — NH 2 > — CH 3 ;
﹥—OR 2—NR 3
+I 效应: ﹥—O —NR
同主族
–I 效应: —F > —C l > —Br > —I ;—OR > —SR > SeR
+ I 效应: —Oˉ> —Sˉ> —Seˉ> —Teˉ
在有机化合物中,非金属元素的+I 效应情形较少,同族非金属元素一般不会比较其+I 效应。 (2) 带电状况不同的情形比较
中心原子带有正电荷的比不带正电荷的同类基团的吸电诱导效应强,而中心原子带有负电荷的比同类不带负电荷的基团供电诱导效应要强。
–I 效应:中心原子
带正电荷的原子(团)>不带电荷的同类原子团
+ I 效应:中心原子
带有负电荷的原子(团)>同类不带电荷的基团 —Oˉ> —OR (3) 中心原子相同而不饱和程度不同 不饱和程度增大,–I 效应增强:
=O > –OR ; ≡N > =NR > –NR 2
通常通过测定取代酸碱的解离常数、偶极矩及反应速率等来比较诱导效应的强度。
1.3通过理化参数比较诱导效应
(1)根据酸碱的强度比较
选取适当的酸或碱,以不同的原子或基团取代其中某一个氢原子,测定取代酸碱的离解常数,可以估算出这些原子或基团的诱导效应次序。
各种取代乙酸的离解常数,可以得出下列基团诱导效应的强度次序:
+ I 效应:—NO 2 > —N +(CH 3)3> —CN > —F > —Cl > —Br > —I > —OH > —OCH 3 > —C 6H 5 > —CH=CH 2> —H > —CH 3 > —CH 2CH 3 >—C(CH 3)3 (–I 效应的方向与此相反)
NR 3>
NR 2
+
+
O R 2SR 2SeR 2
+
+
>
>
N R 3PR 3AsR 3
+
+
+
>
>
(2)根据偶极矩比较
表 [1–2] 单取代甲烷衍生物的偶极矩(D,德拜)
–I 效应 :CN > NO 2 > Cl > F > Br > I >H
上述两种不同方法所得的两个系列顺序不完全一致,在酸碱比较中F>Cl ,而在偶极比较中Cl>F ,一般认为F>Cl 。
比较具有不同分支程度的烷基的氯代烷及溴代烷的偶极矩,发现烷基分支愈多,偶极矩愈大,即+I 效应越大。
烷基的+I 效应顺序:
(CH 3)3C — > CH 3CH 2CH(CH 3)— > (CH 3)2CHCH 2— ≈ CH 3CH 2CH 2CH 2— (CH 3)3C — ≈(CH 3)2CH — > CH 3CH 2— > CH 3— > H (3)由核磁共振化学位移粗略比较
表 [1–3] X —CH 3中甲基的δ值:
1.4动态诱导效应
静态诱导效应,是分子本身所固有的性质,是与键的极性及其基态时的永久极性有关的。当某个外来的极性核心接近分子时,能够改变共价键电子云的分布。由于外来因素的影响引起分子中电子云分布状态的暂时改变,称为动态诱导效应,用Id 表示。
A
B
A
B[X]+
[X]+的作用
去[X]+的作用
●
●●
●
正常状态(静态) 试剂作用下的状态
1.5 动态诱导效应与静态诱导效应的不同。
(1)引起的原因不同。静态诱导效应是由于键的永久极性引起的,是一种永久的不随时间变化的效应,而动态诱导效应是由于键的可极化性而引起的,是一种暂时的随时间变化的效应。
(2)动态诱导效应是由于外界极化电场引起的,电子转移的方向符合反应的要求,即电子向有利于反应进行的方向转移,所以动态诱导效应总是对反应起促进或致活作用,而不会起阻碍作用。而静态诱导效应是分子的内在性质,并不一定向有利于反应的方向转移,其结果对化学反应也不一定有促进作用。
1.6动态诱导效应的比较
动态诱导效应是一种暂时的效应,不一定反映在分子的物理性质上,不能由偶极矩等物理性质的测定来比较强弱次序。比较科学、可靠的方法是根据元素在周期表中所在的位置来进行比较。
(1)在同一族元素,由上到下原子序数增加,电负性减小,电子受核的约束减小,电子的活动性、可极化性增加,动态诱导效应增强。如:
I d:—I > —Br > —Cl > —F
—TeR > —SeR > —SR > —OR
(2) 如果同一元素的原子或基团带有电荷,带正电荷的原子或基团比相应的中性原子或基团对电子的约束性大,而带负电荷的原子或基团则相反,所以I d效应随着负电荷的递增而增强。如:
I d :—O–> —OR > +OR2;
—NR2> —+NR3 ;
—NH2> —+NH3
(3)在同一周期中,随着原子序数的增加,元素的电负性增大,对电子的约束性增大,因此极化性变小,故动态诱导效应随原子序数的增加而降低。
I d:—CR3>—NR2>—OR>—F
1.7诱导效应对反应的影响
(1) 对反应方向的影响
[例如]
(a)丙烯与卤化氢加成,遵守马氏规则,而3,3,3–三氯丙烯加卤化氢则按反马氏规则的方向加成。
Cl3C←CH=CH2 + HCl Cl3—CH2—CH2Cl
(b)在苯环的定位效应中,+N(CH3)3具有强烈的–I效应,所以是很强的间位定位基,在苯环亲电取代反应中主要得到间位产物,而且使亲电取代比苯难于进行。
(2) 对反应机理的影响
在一些反应中,由于诱导效应等因素可以改变其反应机理。如溴代烷的水解反应,伯溴代烷如CH3—Br 主要按S N2历程进行,而叔溴代烷如(CH3)3C—Br则主要遵从S N1历程进行。
(3) 对反应速率的影响
[例] 羰基的亲核加成反应,羰基碳原子的电子云密度越低,就越容易和亲核试剂发生加成反应,在这种情况下,分子所需要的活化能就比较小,容易进入活化状态,因而反应速率较大。故取代基的–I效应愈强,愈有利于亲核加成;取代基的+I效应愈强,对亲核加成愈不利。
如下列化合物发生亲核加成的活性顺序为:
Cl3C—CHO > Cl2CHCHO > ClCH2CHO > CH3CHO
(4) 对化学平衡的影响
[例1] 酸碱的强弱是由其解离平衡常数的大小来衡量的,在酸碱的分子中引入适当的取代基后,由于取代基诱导效应的影响,使酸碱离解平衡常数增大或减小。如乙酸中的一个α–氢原子被氯原子取代后,由于氯的–I效应,使羧基离解程度加大,而且使生成的氯乙酸负离子比乙酸负离子稳定,所以K2>K1 :
[例2] 乙醛的水合反应是可逆的,形成的水化物很不稳定,只能存在于稀水溶液中。而三氯乙醛的水合反应则比较容易,能生成稳定的水合物并能离析和长期存在。主要是由于三氯甲基强烈的–I效应使羰基碳原子带部分正电荷,亲核反应容易进行,同时水合三氯乙醛因形成氢键也增加了稳定性。
2、共轭效应(conjugative effect)
分子轨道理论认为共轭效应是轨道或电子离域于整个共轭体系乃至整个分子所产生的一种效应。
2.1电子离域与共轭效应
[现象1]
在1,3–丁二烯CH2=CH-CH=CH2中的键长不是简单的单键和双键的键长,存在着平均化的趋势。
表[2–1] 1,3–丁二烯的键长
体系能量降低,化合物趋于稳定。
[现象2]
氯乙烯与氯乙烷比较,从诱导效应考虑,由于π键的电子云流动性较大,氯乙烯(μ=1.44D)的偶极矩应该加大,但实际上却比氯乙烷(μ=2.05D)的偶极矩小。
氯乙烯也同样存在着单双键平均化的趋势。
表[2–2] 氯乙烯的偶极矩
这些现象说明,在单双键交替排列的体系中,或具有未共用电子对的原子与双键相连的体系中,π轨道与π轨道或p轨道与π轨道之间存在着相互的作用和影响。电子云不再定域于成键原子之间,而是离域于整个分子形成了整体的分子轨道。每个成键电子不仅受到成键原子的原子核的作用,而且也受到分子中其他原子核的作用,因而分子整体能量降低,体系趋于稳定。
这种现象称为电子的离域(delocalization),这种键称为离域键,由此而产生的额外的稳定能称为离域能(也叫共轭能或共振能)。包含着这样一些离域键的体系通称为共轭体系,在共轭体系中原子之间的相互影响的电子效应叫共轭效应(conjugative effect)。
按照共轭效应的起源,可以将共轭效应分为静态共轭效应与动态共轭效应。
2.2 静态共轭效应
静态共轭效应是在没有外来因素的影响,分子本身就存在固有的一种永久效应。
共轭效应的主要表现:
(1)电子密度发生了平均化,引起了键长的平均化,
(2)共轭体系的能量降低。各能级之间能量差减小,分子中电子激发能低,以致使共轭体系分子的吸收光谱向长波方向移动。随着共轭链增长,吸收光谱的波长移向波长更长的区域,进入可见光区。
表[2–3] 某些化合物吸收峰波长与颜色
2.3共轭效应与诱导效应的区别
(1)共轭效应起因于电子的离域,而不仅是极性或极化的效应。
(2)共轭效应只存在于共轭体系中,不象诱导效应那样存在于一切键中。
(3)诱导效应是由于键的极性或极化性沿σ键传导,而共轭效应则是通过π电子的转移沿共轭链传递,
是靠电子离域传递;共轭效应的传导可以一直沿着共轭键传递而不会明显削弱,不象诱导效应削弱得那么快,取代基相对距离的影响不明显,而且共轭链愈长,通常电子离域愈充分,体系能量愈低愈稳定,键长平均化的趋势也愈大。
2.4共轭效应的相对强度
共轭效应分为供电共轭效应(即+C效应)和吸电共轭效应(即-C效应)。
通常将共轭体系中给出π电子的原子或原子团显示出的共轭效应称为+C效应,吸引π电子的原子或原子团的共轭效应称为-C效应。
H2
+C效应:
-C效应:
(1)+C效应
在同一周期中随原子序数的增大而减弱。
如:—NR2 > —OR > —F
在同一族中随原子序数的增加而减小。
如:—F > —Cl > —Br > —I ;
—OR > —SR > —SeR > TeR ;
—O–> —S–> —Se–> —Te –
带负电荷的元素将具有相对更强的+C效应:
—O–> —OR > —O+R2
主量子数相同元素的p 轨道大小相近,可以更充分地重迭,离域程度也较大。如果取代基中心原子的主量子数与碳的主量子数不同,则轨道重迭程度相对较小,离域程度相应减弱,而且主量子数差值越大影响越明显。因此,一般讲,+C效应在同族中随中心原子的原子序数增大而降低。
(2)-C效应
在同周期元素中,原子序数越大,电负性越强,–C效应越强。
如:=O > =NH > =CH2
对于同族元素,随着原子序数的增加,原子半径变大,能级升高,即与碳原子差别变大,使π键与π键的重迭程度变小,故–C效应变弱。
如:C=O > C=S
带正电荷将具有相对更强的–C效应。
如:=N+R2 > =NR
但共轭效应的影响因素是多方面、比较复杂的,不仅取决于中心原子的电负性和p轨道的相对大小,而且其强弱还受其他原子及整个分子结构的制约,同时共轭效应和诱导效应是并存的,是综合作用于分子的结果,通常是难以严格区分的。
2.5 动态共轭效应
动态共轭效应是共轭体系在发生化学反应时,由于进攻试剂或其他外界条件的影响使p 电子云重新分布,实际上往往是静态共轭效应的扩大,并使原来参加静态共轭的p 电子云向有利于反应的方向流动。 [例如]1,3–丁二烯在基态时由于存在共轭效应,表现体系能量降低,电子云分布发生变化,键长趋于平均化,这是静态共轭效应的体现。而在反应时,例如在卤化氢试剂进攻时,由于外电场的影响,电子云沿共轭链发生转移,出现正负交替分布的状况,这就是动态共轭效应。
CH 3
CH
3H
+CH 2
CH
CH
CH 3δδδ
动态共轭效应是在帮助化学反应进行时才会产生。静态共轭效应是一种永久效应,对化学反应有时可能会起阻碍作用。与诱导效应类似,动态因素在反应过程中,往往起主导作用。 [例如]氯苯,在静态下从偶极矩的方向可以测得 -I > + C 。
μ=1.86 D μ=1.70 D 动态时:氯苯发生亲电取代是邻对位为主 + C > -I
2.6 共轭体系
按参加共轭的化学键或电子类型,共轭效应包括π–π共轭体系、p –π共轭体系、σ–π共轭体系和σ–P 共轭体系等。
(1) π–π共轭体系
π–π共轭体系,是指由π轨道与π轨道电子离域的体系,一般由单键和不饱和键(双键和叁键)交替排列组成。这些体系中参与共轭的原子数与π电子数相等。如: CH 2=CH —CH=CH 2 CH 2=CH —CH=O CH 2=CH —CH=CH —CH=CH 2
(2) p –π共轭体系
具有处于p 轨道的未共用电子对的原子与π键直接相连的体系,称为p –π共轭体系。
[如]氯乙烯,当氯原子的p 轨道的对称轴与π键中的p 轨道对称轴平行时,电子发生离域。
CH C
H
Cl H
H
3、超共轭效应 (Hyperconjugation effect)
3.1 超共轭体系
(1) σ–π 超共轭体系
H
H
C -H 键上的σ电子发生离域,形成σ-π共轭,使体系能量下降。σ电子已经不再定域在原来的C 、H 两原子之间,而是离域在C 3-C 2之间,使H 原子容易作为质子离去。
这种共轭强度远远弱于π–π, p –
π 共轭。
烯烃的氢化热
△H /KJ mol –1
H 2
+-132
CH 2CH 2CH 3CH 3
H +-126
CH 2
CHCH 2CH 3CH 3
CH 3
H C
C
H CH 3
H 2
+
CH 3CH 2CH 2CH 3
-120
CH 3
H
C
C
H CH 3
H 2
+
CH 3CH 2CH 2CH 3
-115
CH 3
CH 3
C C
CH 3
CH 3
H 2
CH 3
CH 3
CH
CH
CH 3
CH 3
+-112
α-H 的活泼性
H
H
2
H
CH
H
H
H
CH
(2) σ–P 超共轭体系
H
C H H
C C H H H C H
H
H
H
C H H
C H C H
H
H >>>H
H
H C C H
H H
C H
H ++++
正碳离子稳定性:叔 > 仲 > 伯 > +CH 3
3.2 缺电子共轭体系与多电子共轭体系
缺电子共轭体系:
CH 2=CH —CH 2+ C 6 H 5CH 2+ 多电子共轭体系: CH 2=CH —CH 2 –
3.3 共轭效应与反应性
(1). 对化合物酸碱性的影响
羧酸的酸性是因为羧酸分子中具有p –π共轭,增大了O —H 键的极性,促使氢容易离解,且形成的羧基负离子共轭效应增强,更稳定。醇一般为中性,苯酚由于p –π共轭,有一定的酸性。
O 2N
NO 2
OH
NO 2
(2).对反应方向和反应产物的影响
在α,β–不饱和羰基化合物分子中,C=0与C=C 形成共轭体系,对反应方向和反应产物带来很大影响,使这些醛、酮具有一些特殊的化学性质。如丙烯醛与HCN 主要发生1,4加成。
CH 2
CH
CH
O +HCN
CH 2CH
CN
CH
O
CH 2CH
CN
CH
O O HCN CH 2CH
CH
CH 2CH 2
CH
CN
OH
CN
(3) 对反应速度的影响
共轭效应对化合物的反应速率影响很大。如在卤代苯的邻、对位上连有硝基,对碱性水解反应活性的影响很大。这是由于—NO 2具有很强的-C 效应,当它连在邻、对位时能得到很好的传递,而当它处在间位时,-C 效应不能传递,仅有-I 效应,对反应活性的影响不如在邻、对位时大。
4、场效应(Field effect)
分子中原子之间相互影响的电子效应,不是通过键链而是通过空间传递的,称为场效应。场效应和诱导效应通常是难以区分的,因为它们往往同时存在而且作用方向一致。但在共些场合场效应的作用还是很明显的。(同性斥,不稳定,异性吸,稳定)
如邻氯苯基丙炔酸的酸性比氯在间位或对位的弱
O
C
Cl
COOH
C
δ
比较下列化合物酸性强弱:(1)比(2)弱,用场效应解释。
场效应与距离的平方成反比,距离越远,作用越小。
氯代苯丙炔酸:
C C C
O
H
Cl
O
C C
C
O
H
O
Cl
pKa: 大小
场效应是依赖分子的几何构型的。
Cl
Cl
H
H
C
O
O
H
Cl
Cl
H
H
COOH
酸性较弱 酸性较强
Cl
C
O
H
O
C
HO
O
pKa: 6.25 pKa: 6.04
二、 空间效应 (Space effect)
分子内或分子间不同取代基相互接近时,由于取代基的体积大小、形状不同,相互接触而引起的物理的相互作用 -空间效应 (位阻效应)。
OH
CH 3
2
H 3C
H 3C
NO 2
CH 3
OH
空间效应的体现:
1. 化合物(构象)的稳定性
H H
CH 3
H
CH 3
2. 化合物的酸碱性
2
C
O CH 3
3
C
C
O
pike 邻 < pKa 对
当t –Butyl 在邻位时, 把羧基挤出了与苯环所在平面,羧基的 -C 效应消失。
3. 对反应活性的影响
伯卤代烷的乙醇解的相对速度是与中心碳原子连接的烷基大小相关的:
EtOH +
C Br
EtO
C R HBr
+
表 [3–1] RCH 2Br 乙醇解的相对速率
张力对反应活性的影响
非水溶液中,胺与质子酸作用,其碱性强度顺序为: R
3N > R 2NH > RNH 2 > NH 3
当胺与体积较大的Lewis 酸作用时,碱性强度顺序为: R 3
N < R 2NH < RNH 2 < NH 3
:
R'R'
3
3
R
X
X +°109
28
sp 3-四面体 sp 2-平面三角型
来自于离去基团背后的张力 —— B –张力 (Back Strain)
小环分子中,分子内部固有的张力——I –张力(Internal strain),也叫角张力 (Angle strain)
X R 60°
°
°
°1206010928'
三、总结
取代基电子效应是影响反应活性的重要因素,能使试剂的亲核性或亲电性强弱改变或受体的活性变化。芳环上取代基对酚、芳醛、芳酸反应活性的改变可折射出取代基的电子效应,取代基在芳环上的影响是共轭效应和诱导效应的综合结果。
由此可以看出,有机化合物是一个统一的整体,组成该化合物的原子是相互联系的,互相影响和制约的,它们对整个化合物都有贡献,分子所表现的性质,也正是各种因素共同作用的结果。因此,正确运用和掌握取代基效应,对于有机化学的学习,具有重要的作用。由于各个反应底物的化学结构不同,影响有机反应的因素很多,除了诱导效应、共轭效应外,还有立体效应和场效应等因素。因此,在研究化合物的性质时,应全面地看,从各方面的影响因素去观察,才能得出正确的结果。
参考文献:
[1] 傅相锴,《高等有机化学》,高等教育出版社,2003,1–19
[2] 赵六德,《诱导效应和共轭效应》,郑州轻工业学院学报,1987,(2):107-113,91 [3] 邢其毅 裴伟伟 徐瑞秋 裴 坚,《基础有机化学》,高等教育出版社,2005,243
–247
[4] 刘汉文 唐子龙 裴文丑
刘 狄,《论取代基立体效应对有机化合物性质的影响》,当代教育理论与
实践,2011(9):162-164
[5] 韩秋萍 王红霞,《浅析取代基对有机反应的影响》,运城学院学报,2007(2):29-30,55 [6] 万有志 王幸宜,《应用化学专业英语》,化学工业出版社,2008,
Substituent effect
Zhang Qiqo-Yan
College of chemistry and chemical engineering, Chifeng University, Chifeng 024000
Abstract: Different substituents for molecular properties influence. Replace the effect can be classified into two categories. One kind is electronic effects, including induced effect, conjugate effect and field effect. Electronic effect is through the key the polarity of the transfer of the performance in the atomic or group molecular interactions, substituents effects through molecular of the electron hull of distribution and work. Another kind is the space effect, is due to replace the size and shape of the cause of the special tension or molecules of resistance an effect, the space effect of reactive molecules also has certain influence. This paper mainly discusses the electronic effect.
Keywords: inductive effect; conjugation; hyperconjugation; field effect
在21世纪,物理有机化学家将会在更广阔的范围内,在相关的前沿交叉领域中寻找新的学科生长点,运用自己在理论、方法、概念和思维方式方面的特长和优势,研究新问题,发现新规律,为有机化学乃至整个科学事业的发展作出贡献。其中主要包括生命过程中的化学问题,分子聚集体化学中的结构/活性关系和反应规律,新分子和新材料的分子设计、合成和构效关系,计算化学和理论有机化学,自由基化学,有机光化学等领域。 内容提要 §1-1 诱导效应 一、共价键的极性与静态诱导效应 二、静态诱导效应的强度 三、静态诱导效应的强度比较 四、烷基的诱导效应 五、动态诱导效应 六、诱导效应对反应活性的影响 §1-2 共轭效应 一、电子离域与共轭效应 二、静态共轭效应 三、动态共轭效应 四、共轭体系 五、共轭效应与反应性 §1-3 超共轭效应 一、超共轭效应的特点和方向 二、超共轭效应的表现和作用 §1-4 场效应和空间效应 一、场效应 二、空间效应 第一章 取代基效应 (Substituent Effects) 反应的本质: 有机化合物的反应本质是旧键的断裂,新键的生成,这直接或间接与共价键的极性,即共价键上电子云的分布有关。例: C C C O 1 23 4 取代基效应 分子中的某个原子或原子团对整个分子或分子中其它部分产生的影 响(包括对共价键极性及整个分子物理性质和化学的影响)。 取代基效应的分类 取代基效应 电子效应: 诱导效应 共轭效应 超共轭效应 场效应: 空间传递 空间效应: 空助效应 位阻效应 §1-1 诱导效应(Inductive effect) 一、共价键的极性与静态诱导效应 C CH CH 2
定义含有取代基的苯衍生物,在进行芳香族亲电取代反应时,原有的取代基,对新进入的取代基主要进入位置,存有一定指向性的效应。这种效应称为取代基定位效应。 单取代的苯衍生物的定位效应①如苯环上的取代基为-NH2(-NHR、-NR2,R 为烷基)、-OH、-OCH3(-OC2H5等)、-NHCOCH3、-C6H5、-CH3(-C2H5等)等(按定位效应由强到弱次序排列)时,其亲电取代的反应性较苯高。在取代反应中,此类取代基导致得到大部分为邻位和对位取代的异构体。此类取代基称为有活化作用的邻、对位取代基。 取代基的定位效应是个反应速率问题。上邻、对位反应快而上间位慢,就显示邻、对位定位效应;上间位反应快而上邻、对位慢,就显示间位定位效应。 稳定的活性中间体的能量低,与之相应的过渡状态的能量也就低,活化能低,反应速率就快;过渡状态能量高,活化能高,反应速率就慢。因此,不同的反应速率实质上反映了活性中间体的稳定性,而活性中间体的稳定性,可以用共振论的方法加以分析。例如用甲苯进行亲电取代反应时,亲电试剂E+可以进攻邻、对位和间位。当亲电试剂进攻邻、对位时,有比较稳定的极限式(a,b)参与共振,CH3与带正电荷的碳相连,CH3有给电子效应,可以中和部分正电荷,使正碳离子稳定,杂化产生的活性中间体也比较稳定。亲电试剂进攻间位时,没有比较稳定的极限式,没有CH3与带正电荷的碳相连的极限式参与杂化。因此,甲基是邻、对位定位基。 ② 如苯环上的取代基为 -F、-Cl、-Br、-I、-CH2Cl、-CH匉CHNO2等时,则具有这些取代基的苯的亲电取代反应性较苯低,即这些基使苯环钝化。邻位和对位钝化程度较间位小,有利于形成邻位和对位的取代异构体。此类取代基称为有钝化作用的邻、对位取代基。 这类取代基的情况比较特殊。如在氯苯中,氯原子是强的吸引电子的取代基,在进行亲电取代反应时,它使苯环正碳离子的电荷更加集中,正碳离子不稳定,对苯环起钝化作用。 如果亲电试剂进攻邻、对位,有比较稳定的极限式(c、e),这是由于氯原子的非共享电子对向苯环转移,使(c、e)的每个原子均具有稳定的八隅体结构,由稳定极限式参与共振杂化所产生的活化中间体也较稳定。如亲电试剂进攻间位,极限式(d)有六电子的碳,不如极限式(c、e)稳定。因此,氯原子是邻、对位定位基。 ③ 如苯环上的取代基为-NO2、-+NH3、-+NR3、-CF3、-+PR3、-+SR2、-SO3H、-SO2R、-COOH、-COOR、-CONH2、-CHO、-COR、-CN等时,则具有这些取代基的苯的亲电取代反应性不如苯,即这些基团使苯环钝化。邻位和对位钝化程度较间位大,在取代反应中,新取代基大多进入间位,形成间位异构体。这类取代基称为有钝化作用的间位取代基。 这些取代基都有吸电子作用。例如当三氟甲基取代苯上的氢后,由于三氟甲
取代基效应对有机化合物性质的影响 应用化学09-2 29号王竹青 摘要:取代基效应是有机结构理论的重要组成部分. 取代基效应对有机化合物的物理性质、化学性质和反映活性都有重要影响。有机化学中的取代基效应涉及化合物的物理性质、酸碱性、反应活性以及反应的位置、类型、速度、平衡、产物等。在熟知官能团一般特性的基础上, 利用取代基效应可将各系列有机化合物千差万别的物理、化学性质有机地联系在一起, 易于学习和掌握。 关键词:取代基效应、有机化合物 取代基效应是分子中某些基团或原子对整个分子或分子中其它部分产生的影响。取代基效应对有机分子性质的影响是多方面的, 归纳起来, 可以分为三个方面: 电子效应、空间效应和场效应。 (1 电子效应,它是由于取代基的作用而导致的共用电子对沿共价键转移的结果,包括诱导效应, 共轭效应和超共轭效应。电子效应是通过影响分子中电子云的分布来影响分子的性质的。(2 空间效应, 是由于取代基的大小或形状引起分子中特殊的张力或阻力的一种效应。空间效应是通过几何结构来影响化合物分子的性质,当分子内的原子或基团处于范德华半径不许可的范围时产生的排斥作用,或两个分子相互接近时由于基团之间的非键作用所引起的化学效应,都是空间效应的具体表现,所以空间效应是通过几何结构来影响化合物分子的性质的。(3)场效应,当分子中原子或原子团间相互作用,通过空间传递的电子效应。 1 取代基效应对有机化合物物理性质的影响 1.1 取代基对化合物酸碱性质的影响 1.1.1 一般来讲,负诱导效应(原子或原子团吸引电子的能力大于氢原子,通常用-I 表示)将增强化合物的酸性,而正诱导效应(原子或原子团吸电子的能力小于氢原子,通常用+I 表示)将减小化合物的酸性。
取代基定位效应 开放分类:化学、效应、芳香烃 目录 ? 定义 ? 单取代的苯衍生物的定位效应 ? 苯环上有两个取代基的定位效应 ? 取代基定位效应解析 取代基定位效应 英文名称:Substituent??positioning??effects定义 ? ?? ?含有取代基的苯衍生物,在进行芳香族亲电取代反应时,原有的取代基,对新进入的取代基主要进入位置,存有一定指向性的效应。这种效应称为取代基定位效应。 单取代的苯衍生物的定位效应 ? ? ①如苯环上的取代基为-NH2(-NHR、-NR2,R为烷基)、-OH、
-OCH3(-OC2H5等)、-NHCOCH3、-C6H5、-CH3(-C2H5等)等(按定位效应由强到弱次序排列)时,其亲电取代的反应性较苯高。在取代反应中,此类取代基导致得到大部分为邻位和对位取代的异构体。此类取代基称为有活化作用的邻、对位取代基。 取代基的定位效应是个反应速率问题。上邻、对位反应快而上间位慢,就显示邻、对位定位效应;上间位反应快而上邻、对位慢,就显示间位定位效应。 ? ?? ? 稳定的活性中间体的能量低,与之相应的过渡状态的能量也就低,活化能低,反应速率就快;过渡状态能量高,活化能高,反应速率就慢。因此,不同的反应速率实质上反映了活性中间体的稳定性,而活性中间体的稳定性,可以用共振论的方法加以分析。例如用甲苯进行亲电取代反应时,亲电试剂E+可以进攻邻、对位和间位。当亲电试剂进攻邻、对位时,有比较稳定的极限式(a,b)参与共振,CH3与带正电荷的碳相连,CH3有给电子效应,可以中和部分正电荷,使正碳离子稳定,杂化产生的活性中间体也比较稳定。亲电试剂进攻间位时,没有比较稳定的极限式,没有CH3与带正电荷的碳相连的极限式参与杂化。因此,甲基是邻、对位定位基。 ②如苯环上的取代基为 -F、-Cl、-Br、-I、-CH2Cl、-CH匉CHNO2等时,则具有这些取代基的苯的亲电取代反应性较苯低,即这些基使苯环钝化。邻位和对位钝化程度较间位小,有利于形成邻位和对位的
取代基定位效应解析 一、定位基分类与定位效应解析: 苯环上已有的取代基叫做定位取代基。 1、邻对位定位取代基 ①概念:当苯环上已带有这类定位取代基时,再引入的其它基团主要进入它的邻位或对位,而且第二个取代基的进入一般比没有这个取代基(即苯)时容易,或者说这个取代基使苯环活化。 ②特征:这类取代基中直接连于苯环上的原子多数具有未共用电子对,并不含有双键或三键。 ③定位取代效应按下列次序而渐减: -N(CH3)2,-NH2,-OH,-OCH3,-NHCOCH3,-R,(Cl,Br,I) 二甲氨基氨基羟基甲氧基乙酰氨基烷基卤素 2、间位定位取代基 ①定义:当苯环上己有在这类定位取代基时,再引入的其它基团主要进入它的间位,而且第二个取代基的进入比苯要难,或者说这个取代基使苯环钝化。 ②特征:取代基中直接与苯环相连的原子,有的带有正电荷,有的含有双键或三键。 ③定位效应按下列次序而渐减: -N+(CH3)3,-NO2,-CN,-SO3H,-CHO,-COOH 三甲铵基硝基氰基磺酸基醛基羧基 3、取代定位规律并不是绝对的。实际上在生成邻位及对位产物的同时,也有少量间位产物生成。在生成间位产物的同时,也有少量的邻位和对位产物生成。 4、苯环的取代定位规律的解释 当苯环上连有定位取代基时,苯环上电子云密度的分布就发生变化。这种影响可沿着苯环的共轭链传递。因此共轭链上就出现电子云密度较大和电子云密度较小的交替现象,从而使它表现出定位效应。 ①邻对位定位取代基的定位效应: 邻对位定位取代基除卤素外,其它的多是斥电子的基团,能使定位取代基的邻对位的碳原子的电子云密度增高,所以亲电试剂容易进攻这两个位置的碳原子。 卤素和苯环相连时,与苯酚羟基相似,也有方向相反的吸电子诱导和共轭两种效应。但在此情况下,诱导效应占优势,使苯环上电子云密度降低,苯环钝化,故亲电取代反应比苯难。但共轭使间位电子云密度降低的程度比邻对位更明显,所以取代反应主要在邻对位进行。
苯环上原有的取代基对新导入取代基有影响,这种影响包括反应活性和进入位置两个方面。通常,苯环上原有的第一取代基称为定位基,从大量实验事实的分析总结中发现,定位基的定位作用遵循一定的规律,这一规律称为苯环上亲电取代反应定位规律(又称定位规则)。下面分别讨论定位基的类型;定位规则的理论解释;二元取代苯的定位规律;定位规律的应用。 (一)定位基的类型 1.邻、对位定位基。这类定位基的结构特征是定位基中与苯环直接相连的原子不含不饱和键(芳烃基例外),不带正电荷,且多数具有未共用电子对。常见的邻、对位定位基及其反应活性(相对苯而言)如下: 强致活基团:―NH2(―NHR,―NR2),―OH 中致活基团:―OCH3(―OR),―NHCOCH3(-NHCOR) 弱致活基团:―ph(―Ar),―CH3(-R) 弱致钝基团:―F,―Cl,―Br,―I 这类定位基多数使亲电取代反应较苯容易进行,但卤素例外。 2.间位定位基。这类定位基的结构特征是定位基中与苯环直接相连的原子一般都含有不饱和键(-CX3例外)或带正电荷。常见的间位定位基及其定位效应从强到弱顺序如下:―N+H3,―N+R3,―NO2,―CF3,―CCl3,―CN,―SO3H,―COH,―COR,―COOH,―COOR,―CONH2等。 这类定位基属致钝基团,通常使苯环上亲电取代反应较苯难进行,且排在越前面的定位基,定位效应越强,反应也越难进行。 (二)定位规则的理论解释 苯环上的取代反应是亲电取代反应。因此,从反应活性的角度分析,凡有助于提高苯环上电子云密度的基团,就能使苯环活化,反应活性提高;反之,凡是使环上电子云密度降低的基团,就能使苯环钝化,反应活性降低。从反应位置的角度分析,当苯环上没有取代基时,环上六个碳原子的电子云密度是均等的;但当苯环上有取代基时,由于取代基的电子效应沿着苯环共轭体系传递。在环上出现了出现了电子云密度的疏密交替分布现象。第二个取代基总是进入苯环上电子云密度相对较大的部位,从而使这些碳原子上的取代物占了多数。现以―CH3,―OH,―Cl,―NO2为代表加以说明。 1.甲基(―CH3)。甲基具有正的诱导效应(+I),是供电子基;此外,甲基的 C-H键的σ电子可与苯环的п电子发生σ,п-超共轭效应。其结果均可使苯环上的电子云密度增大,特别是甲基的邻、对位增加的更多。 因此,甲苯比苯易发生亲电取代反应,而且主要发生在邻、对位上。 2.酚羟基(-OH)。从诱导效应看,氧的电负性大于碳,存在负的诱导效应(-I),但氧上的未共用电子对可与苯环上的п电子产生给电子的p,п-共轭效应(+C)。在反应时,动态的共轭效应占主导地位,总的结果是使苯环上电子云密度提高,而不是降低,而且邻、对位增加的较多。 所以,苯酚的亲电取代反应比苯容易进行,且第二个取代基主要进入酚羟基的邻、对位。 3.氯原子(―Cl)。氯原子的电负性较大,是吸电子基,存在负的诱导效应(-I)。但同时,氯原子的未共用电子对,同样可以与苯环上的п电子产生给电子的p,п-共轭效应(+C)。但与酚羟基不同的是氯原子的+C不足以抵消-I,总的结果是使苯环上电子云密度降低,且间位降低较多,邻、对位降低的较少,量子化学的计算也表明同样的结果。 (+)表示电子云密度比苯小
取代基效应 张巧燕 赤峰学院化学化工学院,赤峰024000 摘要:取代基不同而对分子性质产生的影响。取代基效应可以分为两大类。一类是电子效应,包括诱导效应、共轭效应和场效应。电子效应是通过键的极性传递所表现的分子中原子或基团间的相互影响,取代基通过影响分子中电子云的分布而起作用。另一类是空间效应,是由于取代基的大小和形状引起分子中特殊的张力或阻力的一种效应,空间效应也对分子的反应性产生一定影响。本文主要讨论电子效应。 关键字:诱导效应共轭效应场效应空间效应 取代基效 应 电子效应 空间效应(位阻效应,物理的相互作用 ) 诱导效应(σ,π) 共轭效应(π-π, p-π)超共轭效应(σ- π,σ- p ) 场效应空间传递 一、电子效应 电子效应是通过键的极性传递所表现的分子中原子或基团间的相互影响,取代基通过影响分子中电子云的分布而起作用。 有机化学中的电子效应有诱导效应(inductive effect)、共轭效应(conjugation)、超共轭效应(hyperconjugation)和场效应(field effect)。 1、诱导效应(inductive effect) 1.1共价键的极性与静态诱导效应 (1)共价键的极性(polarity): 成键原子间因电负性不同而使电子云不对称分布,电子云偏向电负性较高的原子一边,从而使共价键有了极性,称为极性共价键或极性键。 (2)诱导效应(inductive effect): 由于原子或基团电负性的影响沿着分子中的键传导,引起分子中电子云按一定方向转移或键的极性通过键链依次诱导传递的效应称为诱导效应或I效应。 (3)静态诱导效应(Is): 存在于未发生反应的分子中的诱导效应(电负性决定)
芳环上取代基定位效应的研究 摘要: 通过苯环上芳氢化学位移的变化, 判断苯环上取代基的定位效应及苯环亲电取代反应的强弱. 关键词: 化学位移; 定位效应; 亲电取代反应 1 取代基的定位分类 人们通过大量实验, 归纳出苯环亲电取代定位效应根据新基导入的位置和反应的难易分成3 类, 见表1. 第1 类, 邻、对位定位基. 主要使反应易于进行, 并使新导入的基团进入苯环的邻位或对位; 第2 类, 间位定位基. 主要使反应难于进行, 并使新导入基进入苯环的间位. 第3类定位基, 既使反应较难进行, 又使新基团导入邻位或对位. 原有取代基X 主要决定反应的难易 与产物异构体的量, X 的电子效应解答 了定位效应问题. 实验表明, 第1 类定 位基团活化苯环的顺序大体为[ 1] ) O- > ) NR2 > ) NH2 > ) OH > ) OR > ) NHCOR> ) OCOR; 第2 类定位基钝化苯环, 强弱顺 序依次是[ 1] ) N+ R3 > ) NO2> ) CN > ) SO3H> ) C= O > ) COOH;第3 类定位基, 卤苯进行亲电取代反应速度是[ 1] C6H5F > C6H5Cl U C6H5Br > C6H5I. 芳烃亲电取代反应的定位效应是很复杂的, 定位效应的解释, 主要是经验性的[ 1] , 说不上什么/ 规律0或/ 法则0. 因为定位效应不仅与定位基的电子效应和主体效应有着密切的关系, 还与亲电试剂的种类、性质及反应条件有关. 现代物理实验方法的应用, 推动了有机化学的飞速发展. 利用红外光谱、拉曼光谱、偶极效应、核磁共振吸收及电子光谱等方法可以知道有关分子内原子振动频率、键能的大小、分子的电荷密度分布以及电子状态等有关物质的动态结构. 对芳环定位基的定位效应的解释, 应用核磁共振谱、芳氢化学位移变化进行研究, 发现定位基对芳环亲电取代反应速度以及新引入基团的位置与定位基对芳氢化学位移的变化有着紧密的联系, 可以通过芳氢化学位移的变化, 判断定位基定位效应. 2 化学位移的变化判断取代基的定位效应 核磁共振可在有机化合物中, 依不同化学环境的质子共振信号出现在不同的
取代基效应对有机化合物性质的影响摘要:有机化学中,取代基是取代有机化合物中氢原子的基团。烷烃、烯烃、炔烃、芳香烃等均可以除去 一个或几个-H而形成称作取代基的基团(如-CH?甲基,-C2H5乙基,-CH2CH2-亚乙基)。含氧的取代基有-OH 羟基,-COOH羧基,-CHO醛基,>C=0羰基等。取代基往往是决定有机化合物性质的官能团,也是命名化合 物时要特殊考虑的地方(如图)。不同的取代基会导致不同的效应,如诱导效应、共振效应、电子效应及立 体效应等,从而使不同的化合物产生不同的性质。 关键词:取代基理论;有机化合物 取代基效应是分子中某些基团或原子所引起的电子效应和空间效应的总称。电子效应,包括诱导效 应,共轭效应,场效应和极化效应。由于一个键产生的极性将影响到分子的其余部分,而电子的转移既可 以通过静电诱导方式沿分子链或空间传递,也可在共轭体系中由轨道离域或电子离域产生,前者就是我 们通常所说的电子的诱导效应,而后者就是共轭效应。而场效应与诱导效应紧密相联.常常在分子中同 时出现,而且多数情况下两者的作用方向一致,很难把它们明确区分开来。对于极化效应,也是通过取 代基分子本身的极化作用,对化合物分子本身的电子云产生影响,所以电子效应是通过影响分子中电子 云的分布来影响分子的性质的。而空间效应,是由于取代基的大小或形状引起分子中特殊的张力或阻力 的一种效应。空间效应在有机化学中相当普遍,当分子内的原子或基团处于范德华半径不许可的范围时 产生的排斥作用,或两个分子相互接近时由于基团之间的非键作用所引起的化学效应,都是空间效应的 具体表现,所以空间效应是通过几何结构来影响化合物分子的性质的。 取代基效应的影响涉及到有机化学的很多方面,包括有机化合物的物理性质、酸碱性、反应活性,以 及有机反应的类型、速度、平衡、位置及产物等。本文拟从取代基效应对化合物的物理性质和反应性质两 方面的影响进行分析。 1 取代基效应对有机化合物物理性质的影响 1.1 取代基对有机化合物酸碱性的影响一般来讲,负诱导效应(原子或原子团吸引电子 的能力大于氢原子,通常用-I表示)将增强化合物的酸性,而正诱导效应(原子或原子团吸电子的能力小于氢原子,通常用+I表示)将减小化合物的酸性。 -I:CN>F>Cl>Br>I>CH3O>C6H5>CH2=CH>H +I:(CH3)3C->CH3CH2->-CH3>H 从共轭效应的影响来看,一般π-π共轭、p -π共轭能增强化合物的酸性,而减小了化合物的碱性强度。如苯酚由于存在p-π共轭,而增大了O-H键的极性具有一定的酸性,比普通醇的酸性强。三硝基苯酚由于具有共轭效应和诱导效应的协同作用,所以具有强酸性,其酸度接近于无机的强酸。烯醇式的1,3-二酮也因为p-π共轭而具有微弱的酸性。再如羧酸中存在电负性大的取代基时,其酸性增强,因为吸电子作用对羧基负离子有一定稳定作用,从而促进了羧基的电离。酸性增大程度决定于取代基与-COOH 的作用大小,后者与基团的吸电子能力及两者距离有关。与之对应,胺类的碱性(供电子能力)会因取代基的供电子作用而得到提高。在实际应用中,可以通过测定取代酸和胺类的酸碱性来确定取代基的供电子或吸电子作用大小。但同时也应注意空间效应对酸碱性的影响。 1.2 取代基对有机化合物荧光性能的影响理论研究表明,对于不同的发光母体,同类取代基所处的位 置不同所表达的荧光强弱的变化规律也不相同。例如苯环上-Me取代的卟啉化合物和-OMe取代的卟啉 化合物的荧光数据表明:对于苯环上-Me取代的卟啉化合物,间位取代卟啉的荧光强度最大,对位其次,邻位最小;对于苯环上-OMe取代的卟啉化合物,间位取代卟啉的荧光强度最大,邻位其次,对位最小。此结果说明,即使对于相同的发光母体,不同取代基位置的变化,其荧光强弱的变化规律也有所不同[5]。同样,不同性质的取代基也会对荧光强度产生影响。例如卟啉的苯环上供电子取代基(如-NH2、-OMe、-Me)及吸电子取代基(如-NO2、-Cl)均使卟啉荧光发射波长比未取代的苯基卟啉有一定程度的红移,由于取 代基的孤对电子或者σ电子参与卟啉分子的共轭大π 键,增大了共轭体系,使卟啉大环上的电子跃迁能 级降低所致。然而,供电子取代基比吸电子取代基对卟啉荧光发射波长的影响更明显。卟啉的苯环上供电 子取代基使卟啉的荧光强度增加,而吸电子取代基却使荧光强度明显降低,因为吸电子取代基中的n电子跃迁到π*键上属于禁阻跃迁,产生的激发态分子数较少,同时,吸电子取代基使S1→T1的系间跨 越程度增加,S1→S0放出光子的数量大大减少,致使荧光减弱。其中-NO2吸电子能力强,对荧光的抑 制最明显。 2 取代基效应对有机化合物反应性质的影响 2.1 取代基对反应类型的影响由于烯烃碳碳双键的电子云密度大且暴露,易于发生亲电加成反应。但当烯 烃的双键碳上连有多个强吸电子原子或基团时,其电子云密度大大降低,因此较难进行亲电加成反应,
同学们,大家好。今天要讲的是定位效应的解释。 通过上节课的学习,我们已经知道,有些基团会使苯环的亲电取代反应活性增大,称为活化基;有些基团会使苯的亲电取代反应活性减小,称为钝化基;苯环上的基团还会影响取代位置,根据定位效果分为邻对位定位基和间位定位基。苯环上原有基团为什么会影响亲电取代活性和取代位置呢?今天我们就来分析并解释这一问题。 大家都知道,苯亲电取代时,亲电试剂靠近苯环生成σ-络合物是整个反应的决速步骤。同样,取代苯反应的决速步骤也生成σ-络合物,如图,决速步骤中苯与亲电试剂的成键能力与苯环上电子密度有关。若原有基团是供电子基,苯环电子密度增大,容易受到亲电试剂的进攻,则亲电取代活性增大,该基团就是活化基。若原有基团是吸电子基,会使苯环电子密度减小,吸引亲电试剂的能力减小,则反应活性减小,该基团是钝化基。因此判断一个基团是活化基还是钝化基,只需要分析基团与苯环间的电子效应(包括诱导效应和共轭效应)来确定该基团是供电子基还是吸电子基即可。那么如何分析判断一个基团是邻对位定位基还是间位定位基呢?从反应式可以看出,决速步骤中生成了三种σ-络合物:邻位、间位、对位,这三个平行反应的相对速度决定了最终产物的多少,即决定了取代位置。 这三个平行反应的相对速度可以从两个角度比较。一方面可以从反应物中邻、间、对三个位置上碳的电子密度相对大小分析。基团与苯环间的电子效应使邻间对位碳上电子密度不同,电子密度大的碳自然容易受到亲电试剂的进攻而表现出较大的反应活性。另一方面也可
以从三个σ-络合物的稳定性比较。σ-络合物越稳定,能量越低,生成时经历的过渡态能量越低,反应的活化能越小,反应速度快,相应的σ-络合物生成的就多。通过以上讲解,大家脑海中要有这样几个概念:第一,分析基团与苯环间的电子效应可以判断基团是供电子基还是吸电子基,从而来确定基团使苯环活化还是钝化;第二,分析基团与苯环间的电子效应可以比较邻间对位碳的电子密度大小,以此判断基团的定位效果;第三,分析σ-络合物的稳定性也可以判断基团的定位效果。也就是说不论是对活性的影响还是对定位效果的影响都和电子效应有关。 我们知道,甲基是活化基,又是邻对位定位基。下面就以甲基为例,通过分析甲基与苯的电子效应解释甲基为什么是活化基?为什么是邻对位定位基?甲基碳是sp3杂化,苯环碳是sp2杂化,因此苯环碳电负性大,电子由甲基向苯环偏移,甲基表现出供电子的诱导效应;甲基中α-碳氢键与苯环发生σ-π超共轭效应,电子由甲基转向苯环,甲基表现出供电子的共轭效应。供电子的诱导和供电子的共轭使甲基成为供电子基,使苯环上电子密度增大,亲电取代活性增大,因此甲基是活化基。 甲基为什么是邻对位定位基呢?甲基与苯环间的供电子诱导效应在苯环中传递时,如图所示,会使邻位、对位碳带部分负电性,即邻对位碳的电子密度大,间位碳电子密度小。事实上甲苯中电子密度分布确实如此,如图所示,邻位碳电子密度是1.017,对位是1.011,而间位只有0.999。邻对位电子密度大容易受亲电试剂进攻而被取代,
一、定位基定位效应 苯环上已有的取代基叫做定位取代基。 1、邻对位定位取代基 ①概念:当苯环上已带有这类定位取代基时,再引入的其它基团主要进入它的邻位或对位,而且第二个取代基的进入一般比没有这个取代基(即苯)时容易,或者说这个取代基使苯环活化。 ②特征:这类取代基中直接连于苯环上的原子多数具有未共用电子对,并不含有双键或三键。 ③定位取代效应按下列次序而渐减: -N(CH3)2 , -NH2 , -OH , -OCH3 , -NHCOCH3 , -R , (Cl,Br,I) 二甲氨基氨基羟基甲氧基乙酰氨基烷基卤素 2、间位定位取代基 ①定义:当苯环上己有在这类定位取代基时,再引入的其它基团主要进入它的间位,而且第二个取代基的进入比苯要难,或者说这个取代基使苯环钝化。 ②特征:取代基中直接与苯环相连的原子,有的带有正电荷,有的含有双键或三键。 ③定位效应按下列次序而渐减: -N+(CH3)3 , -NO2 , -CN , -SO3H , -CHO , -COOH 三甲铵基硝基氰基磺酸基醛基羧基 3、取代定位规律并不是绝对的。实际上在生成邻位及对位产物的同时,也有少量间位产物生成。在生成间位产物的同时,也有少量的邻位和对位产物生成。 4、苯环的取代定位规律的解释 当苯环上连有定位取代基时,苯环上电子云密度的分布就发生变化。这种影响可沿着苯环的共轭链传递。因此共轭链上就出现电子云密度较大和电子云密度较小的交替现象,从而使它表现出定位效应。 ①邻对位定位取代基的定位效应: 邻对位定位取代基除卤素外,其它的多是斥电子的基团,能使定位取代基的邻对位的碳原子的电子云密度增高,所以亲电试剂容易进攻这两个位置的碳原子。 卤素和苯环相连时,与苯酚羟基相似,也有方向相反的吸电子诱导和共轭两种效应。但在此情况下,诱导效应占优势,使苯环上电子云密度降低,苯环钝化,故亲电取代反应比苯难。但共轭使间位电子云密度降低的程度比邻对位更明显,所以取代反应主要在邻对位进行。 ②间位定位基的定位效应: 这类定位取代基是吸电子的基团,使苯环上的电子云移向这些基团,因此苯环上的电子云密度降低。这样,对苯环起了钝化作用,所以较苯难于进行亲电取代反应。 ③共振理论对定位效应的解释 邻对位中间体均有一种稳定的共振式(邻对位定位基的影响)。 在间位定位基的影响下,在三个可能的碳正离子中间体中,邻对位共振式中正电荷是在连有吸电子基的碳上,它使碳正离子中间体更不稳定。所以间位碳正离子中间体是最有利的。 二、二取代苯的定位规律 如果苯环上已经有了两个取代基,当引入第三个取代基时,影响第三个取代基进入的位置的因素较多。定性地说,两个取代基对反应活性的影响有加和性。 1.苯环上已有两个邻对位定位取代基或两个间位定位取代基,当这两个定位取代基的定位方向有矛盾时,第三个取代基进入的位置,主要由定位作用较强的一个来决定。 2.苯环上己有一个邻对位定位取代基和一个间位定位取代基,且二者的定位方向相反,这时主要由邻对位定位取代基来决定第三个取代基进入的位置。 3.两个定位取代基在苯环的1位和3位时,由于空间位阻的关系,第三个取代基在2位发生取代反应的比例较小。 参考资料:有机化学高等教育出版社
工业技术兰!竺::!二::型:!:!:!型鲨垦翌垦垦囡取代基效应对有机化合物性质的影响 赵艳丽周淑晶李锦莲 (佳木斯大学化学与药学院化学系黑龙江154007) 摘要:取代基效应是有机结构理论的重要组成部分。取代基效应对有机化合物的物理性质、化学性质和反应活性等都有着重要影响,了解有机化学中的取代基效应及其在有机化学中的作用方式,对于学习有机化学及应用有机化学知识都至关重要。 关键词:取代基理论有机化合物 中图分类号:062文献标识码:A文章编号:1673—0534(2006)08(a)一0063—01 1引言 取代基效应是分子中某些基团或原子所引起的电子效应和空间效应的总称。电子效应,包括诱导效应,共轭效应,场效应和极化效应。由于一个键产生的极性将影响到分子的其余部分,而电子的转移既可以通过静电诱导方式沿分子链或空间传递,也可在共轭体系中由轨道离域或电子离域产生,前者就是我们通常所说的电子的诱导效应,而后者就是共轭效应。而场效应与诱导效应紧密相联.常常在分子中同时出现,而且多数情况下两者的作用方向一致,很难把它们明确区分开来。对于极化效应,也是通过取代基分子本身的极化作用,对化合物分子本身的电子云产生影响,所以电子效应是通过影响分子中电子云的分布来影响分子的性质的。而空间效应,是由于取代基的大小或形状引起分子中特殊的张力或阻力的一种效应。空间效应在有机化学中相当普遍,当分子内的原子或基团处于范德华半径不许可的范围时产生的排斥作用,或两个分子相互接近时由于基团之间的非键作用所引起的化学效应,都是空间效应的具体表现,所以空间效应是通过几何结构来影响化合物分子的性质的。 取代基效应的影响涉及到有机化学的很多方面,包括有机化合物的物理性质、酸碱性、反应活性,以及有机反应的类型、速度、平衡、位置及产物等。本文拟从取代基效应对化合物的物理性质和反应性质两方面的影响进行分析。 2取代基效应对有机化合物物理性质的影响 2.1取代基对有机化合物酸碱性的影响一般来讲,负诱导效应(原子或原子团吸引电子的能力大于氢原子,通常用I表示)将增强化合物的酸性,而正诱导效应(原子或原子团吸电子的能力小于氢原子,通常用+I表示)将减小化合物的酸性。 一I:CN>F>Cl>Br>I>CH30>C6H5>CH.=CH>H +I:(CH3)3C一>CH3CH2>CH3>H 从共轭效应的影响来看,一般n—n共轭、p一丌共轭能增强化合物的酸性,而减小了化合物的碱性强度。如苯酚由于存在p丌共轭,而增大了0H键的极性具有一定的酸性,比普通醇的酸性强。三硝基苯酚由于具有共轭效应和诱导效应的协同作用,所以具有强酸性,其酸度接近于无机的强酸。烯醇式的l,3一二酮也因为pn共轭而具有微弱的酸性。再如羧酸中存在电负性大的取代基时,其酸性增强,因为吸电子作用对羧基负离子有一定稳定作用,从而促进了羧基 的电离。酸性增大程度决定于取代基与一 C00H的作用大小,后者与基团的吸电子能 力及两者距离有关。与之对应,胺类的碱性 (供电子能力)会因取代基的供电子作用而得到 提高。在实际应用中,可以通过测定取代酸 和胺类的酸碱性来确定取代基的供电子或吸 电子作用大小。但同时也应注意空间效应对 酸碱性的影响。 2.2取代基对有机化合物荧光性能的影响 理论研究表明,对于不同的发光母体, 同类取代基所处的位置不同所表达的荧光强 弱的变化规律也不相同。例如苯环上Me取 代的卟啉化合物和一0Me取代的卟啉化合物 的荧光数据表明:对于苯环上Me取代的卟 啉化合物,间位取代卟啉的荧光强度最大,对 位其次,邻位最小;对于苯环上OMe取代 的卟啉化合物,间位取代卟啉的荧光强度最 大,邻位其次,对位最小。此结果说明,即使 对于相同的发光母体,不同取代基位置的变 化,其荧光强弱的变化规律也有所不同”]。同 样,不同性质的取代基也会对荧光强度产生 影响。例如卟啉的苯环上供电子取代基(如一 NH,、一OMe、一Me)及吸电子取代基(如一 N0,、一C1)均使卟啉荧光发射波长比未取代 的苯基卟啉有一定程度的红移,由于取代基 的孤对电子或者。电子参与卟啉分子的共轭 大n键,增大了共轭体系,使卟啉大环上的电 子跃迁能级降低所致。然而,供电子取代基比 吸电子取代基对卟啉荧光发射波长的影响更 明显。卟啉的苯环上供电子取代基使卟啉的 荧光强度增加,而吸电子取代基却使荧光强 度明显降低,因为吸电子取代基中的n电子跃 迁到n+键上属于禁阻跃迁,产生的激发态分 子数较少,同时,吸电子取代基使S1一T1的 系间跨越程度增加,S1一S0放出光子的数量 大大减少,致使荧光减弱。其中N02吸电子 能力强,对荧光的抑制最明显。 3取代基效应对有机化合物反应性质的 影响 3.1取代基对反应类型的影响 由于烯烃碳碳双键的电子云密度大且暴 露,易于发生亲电加成反应。但当烯烃的双键 碳上连有多个强吸电子原子或基团时,其电 子云密度大大降低,因此较难进行亲电加成 反应,而多为采取亲核加成反应历程,如F,C =CF,、(NC),C=C(CN),等。同样,芳 环一般易于进行亲电取代反应,但若有多个 强吸电子取代基(如N0,等)处于离去基团 (如卤素、一S0,C。H,等)的邻、对位时,就 会发生亲核取代反应。空间效应亦能对反应 类型产生影响,如D碳上无支链的伯卤代烷 与亲核性强的试剂主要起SN,反应;而D碳 上有支链时则容易进行消除反应,因B碳上 的烃基阻碍了试剂从背面接近Ⅸ碳原子。应 该说明的是,官能团本身的反应类型亦受控 于电子效应。例如,与碳碳双键不同,碳氧双 键(羰基)易于进行亲核加成反应,由于氧原子 电负性大,其周围的电子云密度很大,在 >C+O中氧为相对稳定的八隅体结构, 而碳缺电子易被亲核试剂进攻。 3.2取代基对反应活性和速度的影响 碳碳双键上的取代基决定了烯烃的反应 活性与速度,带有供电子基团能增大双键的 电子云密度,使亲电加成反应速度加快;反 之,吸电子基团或原子会降低亲电加成反应 的速度。取代基对苯环的亲电取代反应是活 化还是钝化,要看其存在是使苯环的电子云 密度增大还是减少,或对中间碳正离子的稳 定性起增加还是降低的作用。醛、酮的结构对 羰基亲核加成反应活性的影响是电子效应和 空间效应(包括环状物的角张力)综合作用的结 果。 3.3取代基对自由基热力学稳定性的影响 自由基的热力学稳定性通常用键的均裂 键能(BDE)来表征的。有机物的均裂键解离能 (也称键裂能)被定义为AB—A?+B?,即 在标准状况下,化合键均裂前后各物种焓的 变化值““。通过一些科研小组所得的研究结 果我们得知:对于以碳原子为中心芳环体系的 自由基,不论连有吸电子基团还是供电子基 团,碳自由基的稳定性均有所提高,BDE减 小。 4结语 综上所述,有机化学中的取代基效应对 化合物的物理化学性质和反应性质以及谱学 等方面盼l生质都有明显的影响,所以正确掌握 和运用取代基效应,对于有机化学的学习以及 对化学试剂的结构和性质的研究都具有重要 的作用。 参考文献 [1]曹晨忠.有机化学中的取代机效应【M】.北 京:科学出版社,2003. [2】刘传银.高等函授学报,2005,18:45 [3]高志农.大学化学,2003,18:50. [4]张华山,王红,赵媛媛.分子探针与检 测试剂M】.北京:科学出版社,2002.科技咨询导报ScienceandTechnoIogyConsu…ngHeraId63 万方数据
龙源期刊网 https://www.doczj.com/doc/5f2742996.html, 有关芳环上取代基定位效应的教学探索 作者:何芝洲樊亚鸣陈国术刘浩怀艾小红陈怡莎 来源:《科技视界》2015年第05期 【摘要】芳环上取代基的定位效应是芳烃一章教学中的重点和难点之一。本文结合多年 的教学实践和探索,总结出一套简便判断取代基定位效应的方法,即通过分析取代基与芳环直接相连原子的正负性来判断取代基的定位效应。运用该方法,学生能轻松掌握芳烃取代基的定位规律,教学效果显著。 【关键词】芳环;亲电取代;定位效应;教学探索 Teaching Exploration for Orientation Effect of Substituents on Aromatic Ring HE Zhi-zhou FAN Ya-ming CHEN Guo-shu LIU Hao-huai AI Xiao-hong Chen Yi-sha (School of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou Guangdong 510006, China) 【Abstract】The orientation effect of substituents on aryl ring is one of teaching emphasis and difficulty in aromatic hydrocarbon chapter. In this paper, an easy way was put forward to judge the orientation effect of substituents. Firstly, to analyze the positive/negative of the carbon directly connected to aromatic ring, and then confirm the positive/negative of each carbon on aromatic ring according to the charge principle of similar poles repel and opposite poles attract. Using this way,the student can easily understand the orientation rules t of substituents. 【Key words】Aromatic ring; Electrophilic substitution; Orientation effect; Teaching exploration 芳烃的亲电取代反应是有机化学教学的一个重要内容,是在学习了烯烃的亲电加成反应后接触到的又一重要的有机反应类型。学习了烯烃的亲电加成反应后,学生对本章的亲电取代反应机理比较容易理解,但对取代基的定位效应却难以理解和掌握。在现有的各种有机化学教材[1-2]和参考资料[3-4]中对该知识点都是从电子效应、空间效应和中间体的稳定性等方面进行阐述。学生在学习过程中,往往一团雾水,基本上是通过死记硬背来记住各种基团的定位规律,时间一长很容易忘记,且在遇上没有见过的基团时,更是一片茫然。笔者在多年教学实践和探索中,总结出了一套比较简便易掌握的判断基团定位效应的方法,即通过分析取代基中与苯环直接相连原子的正负性来判断基团的定位效应。 在具体的教学中,笔者采用比喻、对比等教学方法,通过分析芳烃的结构特征,从学生所熟悉的知识点出发,引导、鼓励学生一步步分析问题,得出结论。
取代基效应对有机化合物性质的影响 摘要:取代基效应是有机结构理论的重要组成部分。取代基效应对有机化合物的物理性质,化学性质和反应活性等都有着重要影响,了解有机化学中的取代基效应及其在有机化学中的作用方式,对于学习有机化学及应用有机化学知识都至关重要。 关键词:取代基理论有机化合物 取代基效应对有机分子性质的影响是多方面的,归纳起来,可以分为两个方面:(1)电子效应,包括诱导效应,共轭效应和场效应。电子效应是通过影响分子中电子云的分布来影响分子的性质。(2)空间效应,是由于取代基的大小或形状引起分子中特殊的张力或阻力的一种效应。空间效应是通过几何结构来影响化合物分子的性质的。 1 取代基效应对有机化合物物理性质的影响 1.1 取代基对有机化合物酸碱性的影响 一般来讲,负诱导效应(原子或原子团吸引电子的能力大于氢原子,通常用-I表示)将增强化合物的酸性,而正诱导效应(原子或原子团吸电子能力小于氢原子,通常用+I表示)将增强化合物的碱性。 -I:CN>F>Cl>Br>I>CH3O>C6H5>CH2=CH>H +I:(CH3)3C->CH3CH2->-CH3>H 如:CCl3COOH,CHCl2COOH,CH2ClCOOH和CH3COOH的pKa分别为0.08,1.29,2.31和4.76。而HCOOH,CH3COOH,CH(CH3)2COOH和(CH3)3CCOOH的pKa则为3.74,4.76,4.92和4.97。 从共轭效应的影响来看,一般∏-∏共轭,p-∏共轭能增强化合物的酸性,而减小了化合物的碱性强度。如苯酚由于存在p-∏共轭,而增大了O-H键的极性具有一定的酸性,比普通醇的酸性强。三硝基苯酚由于具有共轭效应和诱导效应的协同作用,所以具有强酸性,其酸度接近于无机的强酸。烯醇式的1,3-二酮也因为p-∏共轭而具有微弱的酸性。再如羧酸中存在电负性大的取代基时,其酸性增强,因为吸电子作用对羧基负离子有一定稳定作用,从而促进了羧基的电离。酸性增大程度决定于取代基与-COOH的作用大小,后者与基团的吸电子能力及两者距离有关。与之对应,胺类的碱性(供电子能力)会因取代基的供电子作用而得到提高。在实际应用中,可以通过测定取代酸和胺类的酸碱性来确定取代基的供电子或吸电子作用大小。但同时也应注意空间效应对酸碱性的影响。 1.2取代基对有机化合物荧光性能的影响 理论研究表明,对于不同的发光母体,同类取代基所处的位置不同所表达的荧光强弱的变化规律也不相同。如苯环上-Me取代的卟啉化合物,间位取代卟啉的荧光强度最大,对位次之,邻位最小。对于苯环上-OMe取代的卟啉化合物,间位取代卟啉的荧光强度最大,邻位次之,对位最小,说明即使对于相同的发光母体,不同取代基位置的变化,荧光强弱也有变化。同样,不同性质的取代基也会对荧光强弱有影响。 1.3取代基效应对化合物紫外和红外吸收光谱性质的影响 有机化合物具有最大吸收波长是该化合物的主要特征,并由该化合物所具有的发色团及其他多种因素所决定。发色团之间的共轭效应能使其发生显著红移,这是因为C-H的σ电子与共轭体系的∏电子发生一定程度的共轭,扩大了共轭范围,从而使吸收峰向长波长方向移动。某些饱和原子团在近紫外区并不“发色”,但当他们与发色团相连或共轭时,能是发色团红移。这些基团有-OH,-OR,-NR2,-SR,-X等。由于这些基团都含有p孤对电子,他们与发色团的∏电子发生共轭,是发色团的吸收峰红移。 有机化合物的红外吸收光谱也受化合物的取代基的影响。由于诱导效应会影响到特征吸收的极性和力常数, 所以会导致吸收频率发生变化。以C = O 为例, 卤素原子的吸电子作用