
原核、真核表达载体构建 真核表达载体和原核表达载体的区别:主要是因为原核和真核表达系统所需的表达元件不同。 比如说启动子,终止子在两种表达系统中是不一样的。 带有真核表达元件的是真核载体,能在真核生物内表达; 带有原核表达元件的是原核载体,能在原核生物内表达。两者都具有的为穿梭载体。 ㈠原核表达载体指:能携带插入的外源核酸序列进入原核细胞中进行复制的载体。 原核表达载体调控原件 1.启动子 启动子是DNA链上一段能与RNA聚合酶结合并起始RNA合成的序列,它是基因表达不可缺少的重要调控序列。没有启动子,基因就不能转录。由于细菌RNA聚合酶不能识别真核基因的启动子,因此原核表达载体所用的启动子必须是原核启动子。原核启动子是由两段彼此分开且又高度保守的核苷酸序列组成,对mRNA的合成极为重要。在转录起始点上游5~10 bp处,有一段由6~8个碱基组成,富含A和T的区域,称为Pribnow 盒,又名TATA 盒或-10区。来源不同的启动子,Pribnow 盒的碱基顺序稍有变化。在距转录起始位点上游35 bp 处,有一段由10 bp组成的区域,称为-35区。转录时大肠杆菌RNA聚合酶识别并结合启动子。-35区与RNA聚合酶s亚基结合,-10区与RNA聚合酶的核心酶结合,在转录起始位点附近DNA被解旋形成单链,RNA聚合酶使第一和第二核苷酸形成磷酸二酯键,以后在RNA聚合酶作用下向前推进,形成新生的RNA 链。原核表达系统中通常使用的可调控的启动子有Lac(乳糖启动子)、Trp(色氨酸启动子)、Tac(乳糖和色氨酸的杂合启动子) 、lPL (l噬菌体的左向启动子)、T7噬菌体启动子等。 2. SD序列 1974年Shine和Dalgarno首先发现,在mRNA上有核糖体的结合位点,它们是起始密码子AUG和一段位于AUG上游3~10 bp处的由3~9 bp组成的序列。这段序列富含嘌呤核苷酸,刚好与16S rRNA 3¢末端的富含嘧啶的序列互补,是核糖体RNA的识别与结合位点。以后将此序列命名为Shine-Dalgarno序列,简称SD序列。它与起始密码子AUG之间的距离是影响mRNA转录、翻译成蛋白的重要因素之一,某些蛋白质与SD序列结合也会影响mRNA与核糖体的结合,从而影响蛋白质的翻译。另外,真核基因的第二个密码子必须紧接在ATG 之后,才能产生一个完整的蛋白质。 3.终止子 在一个基因的3¢末端或是一个操纵子的3'末端往往有特定的核苷酸序列,且具有终止转录功能,这一序列称之为转录终止子,简称终止子(terminator)。转录终止过程包括:RNA聚合酶停在DNA模板上不再前进,RNA的延伸也停止在终止信号上,完成转录的RNA从RNA聚合酶上释放出来。对RNA聚合酶起
如何阅读基因载体图谱 基因载体是基因工程的核心,也是基因治疗中强有力的生物工具,我们先来认识和阅读载体图谱吧。 一、载体分类及载体组成元件 载体分类 1、按属性分类:病毒载体和非病毒载体 病毒载体是一种常见的分子生物学工具,可将遗传物质带入细胞,原理是利用病毒具有传送其基因组进入目的细胞,进行感染的分子机制。可发生于完整活体或是细胞培养中。可应用于基础研究、基因疗法或疫苗。用于基因治疗和疫苗的病毒载体应具备以下基本条件: (1)携带外源基因并能包装成病毒颗粒; (2)介导外源基因的转移和表达; (3)对人体不致病; (4)在环境中不会引起增殖和传播。 非病毒载体一般是指质粒DNA。 2、按进入受体细胞的类型分类:原核载体、真核载体、穿梭载体(含原核和真核2个复制子,能在原核和真核细胞中复制,并可以在真核细胞中有效表达)。 3、按功能分类:克隆载体、表达载体 克隆载体:具有克隆载体的基本元件(Ori,Ampr,MCS等),可以携带DNA片段或外源基因进入受体细胞并克隆和大量扩增DNA片段(外源基因)的载体。 表达载体:克隆载体中加入一些与表达调控(具有转录/翻译所必需的DNA顺序)有关的元件即成为表达载体。 载体组成元件 1、复制起始位点Ori:即控制复制起始的位点。Ori的箭头指复制方向,其他元件标注的箭头多指转录方向(正向)。 2、抗生素抗性基因:可以便于加以检测,如Amp+ ,Kan+ (1)Ampr:水解β-内酰胺环,解除氨苄的毒性。
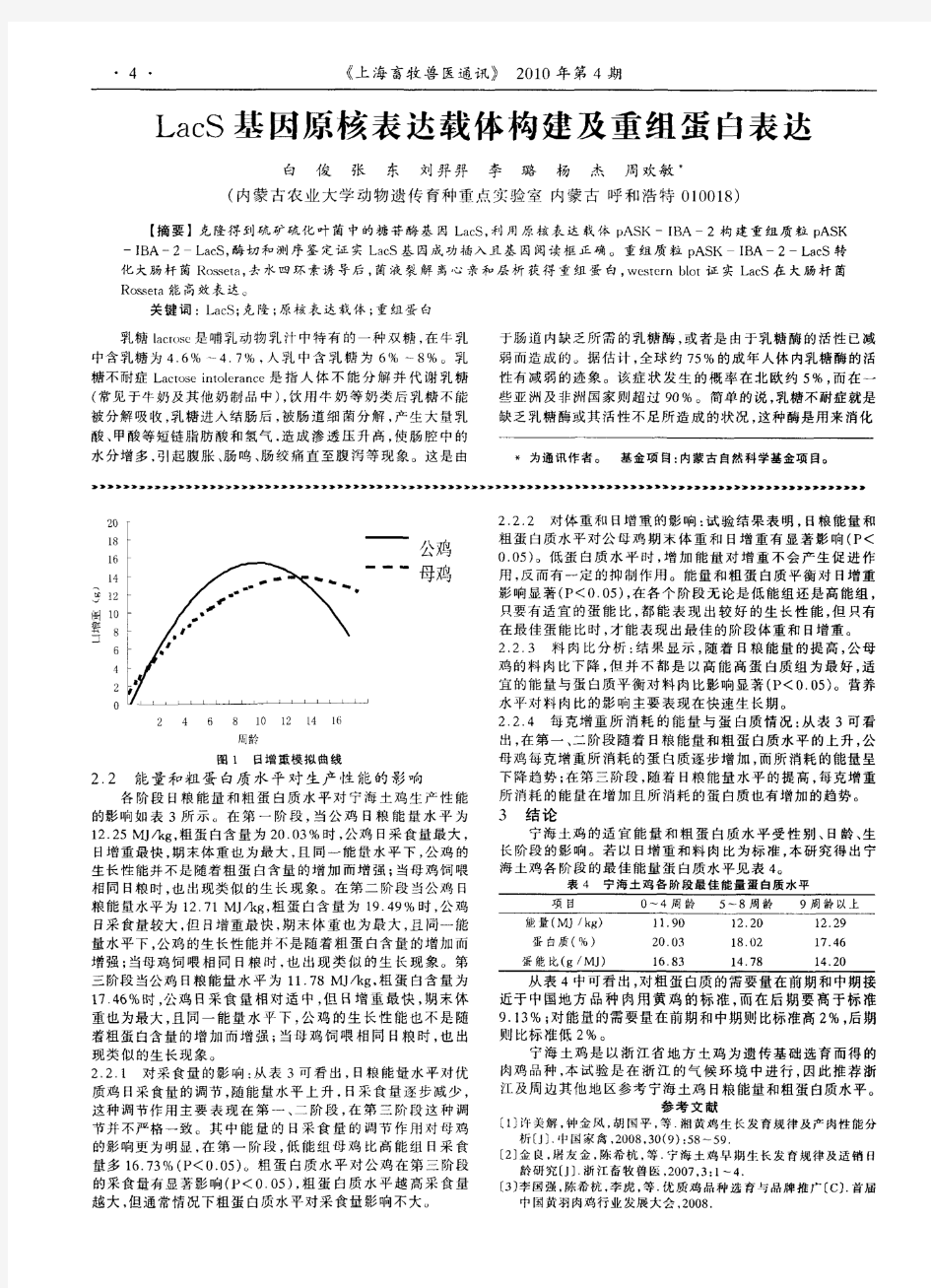
(2)tetr :可以阻止四环素进入细胞。 (3)camr:生成氯霉素羟乙酰基衍生物,使之失去毒性。 (4)neor(kanr):氨基糖苷磷酸转移酶,使G418(卡那霉素衍生物)失活。 (5)hygr:使潮霉素β失活。 3、多克隆位点:MCS克隆携带外源基因片段,它具有多个限制酶的单一切点,便于外源基因的插入。如果在这些位点外有外源基因的插入,会导致某种标志基因的失活,便于筛选。决定能不能放目的基因以及如何放置目的基因。还要再看外源DNA插入片段大小。质粒一般只能容纳小于10kb的外源DNA片段。一般来说,外源DNA片段越长,越难插入,越不稳定,转化效率越低。 4、P/E:启动子/增强子 5、Terms:终止信号 6、加poly(A)信号:可以起到稳定mRNA作用 示例阅读载体: pENTER载体 1)human ORF + pENTER载体 2) CMV启动子,T7启动子 3) ORF的C端融合了Flag和His tag 4) 多克隆位点,常用AsisI 和 MluI(人源基因上不常见的)
重组表达质粒的构建——原核表达载体选择质粒载体是重组蛋白表达的关键工具,其结构如下图。重组蛋白表达,我们首先要将基因插入到表达载体上,插入的位置为多克隆位点。质粒载体上有很多的功能元件,这些元件对于蛋白的表达都是至关重要的。尽管我们经过系统的分析和预测,但是仍有很多蛋白不能顺利表达、表达量很低或者表达状态不好。这个时候我们需要尝试构建不同的表达载体以期得到最好的效果,这些载体的主要区别是启动子和融合标签的差异。 蛋白表达优化主要工作也就是尝试构建不同融合表达标签,使用不同的宿主表达菌,测试不同的表达条件,筛选出最优表达体系。常用的融合标签有GST、MBP、Trx、6His、SUMO等,这些标签主要功能是促表达、促可溶、信号标记或助纯化。福因德生物可以提供以下系列载体以供科研表达研究。 1)促表达/促溶标签 2)信标标签
3)纯化标签 我们选择表达载体的时候不但要考虑蛋白怎么表达成功,更要考虑蛋白怎么纯化出来,纯化的问题主要是考虑纯化标签和酶切位点的选择,下表我们列举了常见的纯化标签和酶切位点。 4)酶切位点 以上为原核表达常用的标签和酶切位点,其性质也都作了简要的介绍,各专业网站或专业书籍已对此做详尽解释,科研工作者可根据具体实验设计方案,组合设计以上标签和酶切位点的使用。特别值得注意的是,选用和设计蛋白酶切位点的时候首要考虑的是序列内部有没有蛋白酶位点,同时要考虑酶切的效率和蛋白酶试剂成本。一般商业化载体,在标签蛋白与载体多克隆位点之间都设计有酶切位点。 标签可设计在N-端也可在C-端,设计在N-端的优势是,可通过标签高效翻译起始位点带动插入蛋白的表达,可溶性标签的高效表达更可促进蛋白的可溶性表达;同时,大部分的蛋白内切酶的切割位点在C-端,所以标签设计在N-端可将标签切割完全。 在设计标签序列与酶切位点的时候还要考虑N-端稳定性原则,也就是所谓宿主细胞的N-端规则(N-end rule),这个要避免;同时,还应该检查是否引入了可与别的蛋白相互作用的序列或者蛋白酶切位点。
原核表达载体的重要调控元件 启动子 启动子是DNA链上一段能与RNA聚合酶结合并起始RNA合成的序列,它是基因表达不可缺少的重要调控序列。没有启动子,基因就不能转录。由于细菌RNA聚合酶不能识别真核基因的启动子,因此原核表达载体所用的启动子必须是原核启动子。 原核启动子是由两段彼此分开且又高度保守的核苷酸序列组成,对mRNA的合成极为重要。在转录起始点上游5~10 bp处,有一段由6~8个碱基组成,富含A和T的区域,称为Pribnow 盒,又名TATA 盒或-10区。来源不同的启动子,Pribnow 盒的碱基顺序稍有变化。在距转录起始位点上游35 bp处,有一段由10 bp组成的区域,称为-35区。转录时大肠杆菌RNA聚合酶识别并结合启动子。-35区与RNA聚合酶s 亚基结合,-10区与RNA聚合酶的核心酶结合,在转录起始位点附近DNA被解旋形成单链,RNA聚合酶使第一和第二核苷酸形成磷酸二酯键,以后在RNA聚合酶作用下向前推进,形成新生的RNA链。 原核表达系统中通常使用的可调控的启动子有Lac(乳糖启动子)、Trp(色氨酸启动子)、Tac(乳糖和色氨酸的杂合启动子) 、lP L (l噬菌体的左向启动子)、T7噬菌体启动子等。 (1)Lac启动子:它来自大肠杆菌的乳糖操纵子,是DNA分子上一段有方向的核苷酸序列,由阻遏蛋白基因(LacI)、启动基因(P)、操纵基因(O)和编码3个与乳糖利用有关的酶的基因结构所组成。Lac启动子受分解代谢系统的正调控和阻遏物的负调控。正调控通过CAP(catabolite gene activation protein)因子和cAMP 来激活启动子,促使转录进行。负调控则是由调节基因产生LacZ阻遏蛋白,该阻遏蛋白能与操纵基因结合阻止转录。乳糖及某些类似物如IPTG可与阻遏蛋白形成复合物,使其构型改变,不能与O基因结合,从而解除这种阻遏,诱导转录发生。 (2)trp启动子:它来自大肠杆菌的色氨酸操纵子,其阻遏蛋白必须与色氨酸结合才有活性。当缺乏色氨酸时,该启动子开始转录。当色氨酸较丰富时,则停止转录。b-吲哚丙烯酸可竞争性抑制色氨酸与阻遏蛋白的结合,解除阻遏蛋白的活性,促使trp启动子转录。 (3)Tac启动子:Tac启动子是一组由Lac和trp启动子人工构建的杂合启动子,受Lac阻遏蛋白的负调节,它的启动能力比Lac和trp都强。其中Tac 1是由Trp启动子的-35区加上一个合成的46 bp DNA片段(包括Pribnow 盒)和Lac操纵基因构成,Tac 12是由Trp的启动子-35区和Lac启动子的-10区,加上Lac 操纵子中的操纵基因部分和SD序列融合而成。Tac启动子受IPTG的诱导。
VEGFA 基因重组真核表达载体的构建与表达 吴勇1,刘学刚2,郭嘉诚1,李慧敏1,丁周志1,王亚明1,宋伟3,徐家丽1,赵武1 (蚌埠医学院第一附属医院1.儿科;2.心胸外科;3.超声心动图,安徽蚌埠233004) 摘要:目的构建VEGFA 基因重组真核表达载体pEGFP-VEGFA ,并检测其在人胚胎肾细胞(HEK293T )中的表达。方法以人 脐血单核细胞的RNA 为模板,采用RT-PCR 技术扩增VEGFA 基因,并将其定向插入真核表达载体pEGFP-Nl 中,构建重组真核 表达载体pEGFP-VEGFA 。经限制性核酸内切酶BglII 和SalI 酶切和DNA 测序鉴定后,用脂质体法将pEGFP- VEGFA 转染HEK293T 细胞,转染后24h 和48h 采用荧光显微镜观察pEGFP- VEGFA 的表达。结果pEGFP-VEGFA 被双酶切为4697bp 和1251bp 两条条带,测序结果证实VEGFA 序列与GenBank 公布的VEGFA mRNA 序列完全一致。在经转染的HEK293T 细胞 内观察到较强的绿色荧光,表明pEGFP-VEGFA 成功转染HEK293T 细胞,并在其中得到了表达。结论成功构建重组真核表 达载体pEGFP-VEGFA ,为进一步研究VEGFA 基因的生物学功能奠定了基础。 关键词:血管内皮生长因子A ;基因;遗传载体 Construction and expression of recombinant eukaryotic expression vector of VEGFA gene WU Yong 1,LIU Xue-gang 2,GUO Jia-cheng 1,et al (Department of 1.Pediatrics ;2.Cardiothoracic Surgery ,The First Affiliated Hospital of Bengbu Medical College ,Bengbu 233004,China )Abstract :Objective To construct a recombinant eukaryotic expression vector pEGFP-VEGFA ,and to detect its expression in HEK293Tcells.Methods VEGFA gene was amplified from the total RNA of human umbilical cord blood monocytes by RT-PCR and then directionally inserted into the eukaryotic expression vector pEGFP-N1in order to construct the recombinant expression vector pEG-FP-VEGFA.The recombinant vector pEGFP-VEGFA was identified by restriction endonuclease BglII and SalI digestion analysis ,and se-quence analysis.The pEGFP-VEGFA was then transfected into the HEK293T cell by lipofectin reagent ,and its expression was detected by fluorescence microscopy at 24and 48h post-transfection.Results The recombinant vector pEGFP-VEGFA was digested into two bands of 4697and 1251bp.Sequencing results showed that the sequence of VEGFA in pEGFP-VEGFA was identical to GenBank VEG-FA accession number NM_001025366.Strong green fluorescence was observed in the HEK293T cells transfected with the pEGFP-VEG-FA ,which showed that the pEGFP-VEGFA had been successfully transfected into the HEK293T cells and acquired expression.Conclu-sion We successfully construct the recombinant eukaryotic expression vector pEGFP-VEGFA ,which lays a foundation to further study the biological function of VEGFA gene. Key words :vascular endothelial growth factor A ;gene ;genetic vectors 基金项目:安徽省自然科学基金面上项目(No 11040606M198) 作者简介:吴 勇,男,硕士研究生通信作者:赵武,男,博士,副教授,研究方向:小儿血管疾病的临床与基础研究,E-mail :zhaowuronghai@yahoo.com.cn · 54·安徽医药Anhui Medical and Pharmaceutical Journal 2013Jan ;17(1)
原核表达步骤
Chi l 原核表达基本试验步骤 将克隆化基因插入合适载体后导入大肠杆菌用于表达大量蛋白质的方法一般称为原核表达。这种方法在蛋白纯化、定位及功能分析等方面都有应用。大肠杆菌用于表达重组蛋白有以下特点:易于生长和控制;用于细菌培养的材料不及哺乳动物细胞系统的材料昂贵;有各种各样的大肠杆菌菌株及与之匹配的具各种特性的质粒可供选择。但是,在大肠杆菌中表达的蛋白由于缺少修饰和糖基化、磷酸化等翻译后加工,常形成包涵体而影响表达蛋白的生物学活性及构象。 表达载体在基因工程中具有十分重要的作用,原核表达载体通常为质粒,典型的表达载体应具有以下几种元件: (1)选择标志的编码序列; (2)可控转录的启动子; (3)转录调控序列(转录终止子,核糖体结合位点); (4)一个多限制酶切位点接头; (5)宿主体内自主复制的序列。 原核表达一般程序如下:获得目的基因-准备表达载体-将目的基因插入表达载体中(测序验证)-转化表达宿主菌-诱导靶蛋白的表达-表达蛋白的分析-扩增、纯化、进一步检测,其中包括: 一、试剂准备 (1)LB培养基。 (2)1M IPTG(异丙基硫代-β-D-半乳糖苷):2.38g IPTG溶于10ml ddH2O
中,0.22μm滤膜抽滤,-20℃保存。 CCY的IPTG是1M的,用时进行1000倍稀释。 二、操作步骤 (一)获得目的基因 1、通过PCR方法:以含目的基因的克隆质粒为模板,按基因序列设计一对引物(在上游和下游引物分别引入不同的酶切位点),PCR循环获得所需基因片段。 2、通过RT-PCR方法:用TRIzol法从细胞或组织中提取总RNA,以mRNA 为模板,逆转录形成cDNA第一链,以逆转录产物为模板进行PCR循环获得产物。 (二)构建重组表达载体 1、载体酶切:将表达质粒用限制性内切酶(同引物的酶切位点)进行双酶切,酶切产物行琼脂糖电泳后,用胶回收Kit或冻融法回收载体大片段。 2、PCR产物双酶切后回收,在T4DNA连接酶作用下连接入载体。我们用Soultion I连接。 (三)获得含重组表达质粒的表达菌种 1、将连接产物转化大肠杆菌BL21,根据重组载体的标志(抗Amp或蓝白斑)作筛选,挑取单斑,碱裂解法小量抽提质粒,双酶切初步鉴定。 2、测序验证目的基因的插入方向及阅读框架均正确,进入下步操作。否则应筛选更多克隆,重复亚克隆或亚克隆至不同酶切位点。 3、以此重组质粒DNA转化表达宿主菌的感受态细胞。
【建议】想和大家讨论讨论原核表达载体 原核表达载体,如pet系列,型号从小到大,那么多,往往让新手选择起来不知所措。所以希望和大家讨论讨论到底他们是怎么演变的,每个的优缺点,是不是号越大的就越好等新手们往往困惑不已的问题。 希望下面的讨论分系列进行,如pet系列、pgex系列等 这篇文章是我从网上找的关于pet载体的介绍,只要你耐心的看完,相信能有个基本的了解关于pet载体及应用。更详细的内容请高手进行补充 pET,原核表达金标准(转) pET 载体中,目标基因克隆到T7 噬菌体强转录和翻译信号控制之下,并通过在宿主细胞提供T7 RNA 聚合酶来诱导表达。Novagen 的pET 系统不断扩大,提供了用于表达的新技术和选择,目前共包括36 种载体类型、15 种不同宿主菌和设计用于有效检测和纯化目标蛋白的许多其它相关产品。 优点 ·是原核蛋白表达引用最多的系统 ·在任何大肠杆菌表达系统中,基础表达水平最低 ·真正的调节表达水平的“变阻器”控制 ·提供各种不同融合标签和表达系统配置 ·可溶性蛋白生产、二硫键形成、蛋白外运和多肽生产等专用载体和宿主菌 ·许多载体以LIC 载体试剂盒提供,用于迅速定向克隆PCR 产物 ·许多宿主菌株以感受态细胞形式提供,可立即用于转化 阳性pFORCE TM 克隆系统具有高效克隆PCR 产物、阳性选择重组体和高水平表达目标蛋白等特点。 pET 系统概述 pET 系统是在大肠杆菌中克隆和表达重组蛋白的最强大系统。根据最初由Studier 等开发的T7 启动子驱动系统,Novagen 的pET 系统已用于表达成千上万种不同蛋白。 控制基础表达水平 pET 系统提供6 种载体- 宿主菌组合,能够调节基础表达水平以优化目标基因的表达。没有单一策略或条件适用于所有目标蛋白,所以进行优化选择是必要的。 宿主菌株 质粒在非表达宿主菌中构建完成后,通常转化到一个带有T7 RNA 聚合酶基因的宿主菌(λ DE3 溶原菌)中表达目标蛋白。在λ DE3 溶原菌中,T7 RNA 聚合酶基因由lacUV5 启动子控制。未诱导时便有一定程度转录,因此适合于表达其产物对宿主细胞生长无毒害作用的一些基因。而宿主菌带有pLysS 和pLyE 时调控会更严紧。pLys 质粒编码T7 溶菌酶,
重组HER2真核表达载体的构建及其稳定转 染EMT6细胞株的筛选 作者:徐腾飞, 张文卿, 于红, 李丹 【摘要】目的: 构建人表皮生长因子受体(HER2)胞外区(1 896 bp)基因的真核表达质粒(pcDNA6/v5his HER2), 转染小鼠乳腺癌细胞(EMT6), 获得其稳定表达细胞株(EMT6/ HER2)。方法: 用PCR方法从含HER2全长基因的pcDNA3.1HER2质粒上扩增HER2胞外区基因序列; 经酶切、连接构建pcDNA6/v5his HER2; 转化大肠杆菌DH5α, 筛选阳性克隆, 对其进行酶切及测序鉴定; 以PEI法将pcDNA6/v5his HER2导入EMT6小鼠乳腺癌细胞, 经杀稻瘟菌素(Blasticidin)筛选1~2周, 获得抗性克隆EMT6/HER2; 用RT PCR检测EMT6/HER2中HER2 mRNA, 免疫组化法检测其HER2蛋白的表达。结果: PCR产物与预期片段大小一致; pcDNA6/v5his HER2经酶切、琼脂糖凝胶电泳后, 可见与PCR产物大小相同的片段; DNA测序结果显示, pcDNA6/v5his HER2 中HER2 基因序列无误, 读码框正确; 用RT PCR可在EMT6/HER2中检测到HER2 mRNA, 免疫组化法证实, EMT6/HER2中有HER2的阳性信号。结论: 成功地构建了HER2胞外区真核表达载体, 获得稳定表达HER2基因的小鼠乳腺癌EMT6细胞株, 为进一步研究HER2基因过表达与乳腺癌发生的关系及其基因治疗奠定基础。
【关键词】HER2; 真核表达; 转染 [Abstract]AIM: To construct an eukaryotic vector encoding extracellular domain of human epidermal growth factor receptors (HER2), pcDNA6/v5his HER2, and to screen HER2 positive clones from mouse breast cancer cell line EMT6. METHODS: The extracellular domain of HER2 was amplified from pcDNA3.1HER2 by PCR. pcDNA6/v5 his HER2 was prepared by inserting the fragment into the plasmid pcDNA6/v5his. Then the recombinant vector was identified by restriction enzyme and sequencing. Next, pcDNA6/v5his HER2 was transfected into the EMT6 cell line and the positive clones (EMT6/HER2) were screened with blasticidin. Finally, the expression of HER2 in EMT6/HER2 was detected by RT PCR and immunohistochemistry. RESULTS: The fragment of HER2 was amplified and pcDNA6/v5his HER2 was prepared successfully. No errors were found both in the sequence and ORF of the acquired fragment. The expected fragment of HER2 (1896 bp) was amplified from EMT6/HER2 by RT PCR and positive signals of HER2 were detected in EMT6/HER2 by immunohistochemistry. CONCLUSION: An eukaryotic plasmid encoding HER2 (pcDNA6/v5 his HER2) has been constructed and a cell line expressing HER2 stably has been prepared successfully.
基因的克隆、表达载体构建及功能验证(一般性方法) 一、基因克隆 ★事前三问 a.克隆这个基因干什么?它有什么功能? b.这个基因在哪种材料中扩增? c.材料需要怎么处理? ◎实验前准备工作 a.设计引物,准备材料, b.购置试剂:Taq酶、反转录试剂盒、凝胶回收试剂盒、质粒提取试剂盒、连接试 剂盒 c.实验试剂及用具:枪头、离心管、培养皿、滤纸灭菌;Amp+ 、Kan+等抗生素准 备 ※基本流程 提取和纯化RNA—cDNA第一条链合成—PCR—凝胶电泳—胶回收—连接—转化—涂平板—挑单菌落—摇菌—提质粒—测序 1.总RNA的提取、纯化及cDNA第一链合成 1.1叶片、根总RNA的提取 Trizol是一种高效的总RNA抽提试剂,内含异硫氰酸胍等物质,能迅速裂解植物细胞,抑制细胞释放出的核酸酶,所提取的RNA完整性好且纯度高,以利于下一步的实验。 1)实验前准备 预先配制0.1%的DEPC水(ddH2O中含0.1%DEPC,V/V,37 ℃过夜处理12 h),高温灭菌后,用DEPC水配制75%乙醇,研钵、量筒、试剂瓶等需200℃灭菌至少4 h,所用枪头和枪盒均去RNA酶处理(直接购买)。 2)Trizol 法(小麦)叶片或根的总RNA实验步骤如下: (1)提前在1.5 ml离心管中加入1 mlTrizol,然后将200 mg样品液氮中研磨成白色粉末,
移入管内,用力摇15 s,在15-30℃温育5 min,使核酸蛋白复合物完全分离。 (2)4℃,12000g离心10min,取上清,离心得到的沉淀中包括细胞外膜、多糖、高分子量DNA,上清中含有RNA。 (3)吸取上清液加0.2 ml氯仿,盖好盖,用力摇15 s,15~30 ℃温育2~3 min。(4)在≤12000g,4℃离心10 min,样品分为三层:底层为黄色有机相,上层为无色水相和一个中间层,RNA主要在水相中,水相体积约为所用TRIzol试剂的60%。 (5)将上层水相转移到新的1.5 ml离心管中,加2倍体积的无水乙醇沉淀RNA,室温静止30 min。 (6)在≤12000g,4℃离心10 min,离心前看不出RNA沉淀,离心后在管侧和管底出现胶状沉淀。 (7)用≥1 ml的75%乙醇洗RNA,涡旋振荡样品,在≤7500g,4℃离心5 min,弃上清。(8)室温放置干燥或真空抽干RNA沉淀,大约晾5-10分钟,加无RNase的水100μl用枪头吸几次,55~60℃温育10 min使RNA溶解。 (9)配制以下体系: 10×DNase buffer 5 μl DNase I (RNase-free)(40 μg/μl) 1 μl RNasin Inhibitor(40 μg/μl) 1 μl Total RNA 70 μg 加去RNase水至总体积为50 μl (10)37 ℃水浴1h,加DEPC处理的水至总体积为100 μl,加入等体积氯仿抽提一次。(11)取上清,加入10 μl的3 mol/L NaAC溶液,200 μl的无水乙醇,-80 ℃沉淀30 min。 (12)2~8 ℃,12000g离心10 min,弃清液,干燥后取50μl无RNase的水溶解RNA。3)RNA的质量及纯度检测 (1)电泳检测取2ul RNA 与1 ul 10×Loading buffer上样缓冲液混合均匀在1% 的琼脂糖凝胶上电泳,在紫外灯下观察RNA 条带并记录实验结果。 (2)分光光度计RNA纯度检测 取1ul RNA液,以DEPC水为空白对照,测定A260/ A280 比值,估测RNA质 量。 4)cDNA第一条链的合成 按照以下体系将提取的总RNA反转录成第一链cDNA: 1)在Eppendorf管中配制下列混合液:
标签:真核细胞酵母表达系统细胞表达载体真核表达系统昆虫表达系统动物表达系统 摘要 : 原核表达系统是常被用来研究基因功能的成熟系统,由于原核表达系统具有包涵体蛋白不易纯化、蛋白修饰不完整等缺陷,人们也开始利用真核细胞表达系统来研究基因。 原核表达系统是常被用来研究基因功能的成熟系统,由于原核表达系统具有包涵体蛋白不易纯化、蛋白修饰不完整等缺陷,人们也开始利用真核细胞表达系统来研究基因。 自上世纪70年代基因工程技术诞生以来,基因表达技术已渗透到生命科学研究的各个领域。并随着人类基因组计划实施的进行,在技术方法上得到了很大发展,时至今日已取得令人瞩目的成就。随着人类基因组计划的完成,越来越多的基因被发现,其中多数基因功能不明。利用表达系统在哺乳动物细胞内表达目的基因是研究基因功能及其相互作用的重要手段。 在各种表达系统中,最早被采用进行研究的是原核表达系统,这也是目前掌握最为成熟的表达系统。该项技术的主要方法是将已克隆入目的基因DNA段的载体(一般为质粒)转化细菌(通常选用的是大肠杆菌),通过iptg诱导并最终纯化获得所需的目的蛋白。其优点在于能够在较短时间内获得基因表达产物,而且所需的成本相对比较低廉。但与此同时原核表达系统还存在许多难以克服的缺点:如通常使用的表达系统无法对表达时间及表达水平进行调控,有些基因的持续表达可能会对宿主细胞产生毒害作用,过量表达可能导致非生理反应,目的蛋白常以包涵体形式表达,导致产物纯化困难;而且原核表达系统翻译后加工修饰体系不完善,表达产物的生物活性较低。 为克服上述不足,许多学者将原核基因调控系统引入真核基因调控领域,其优点是: ①根据原核生物蛋白与靶DNA间作用的高度特异性设计,而靶DNA与真核基因调控序列基本无同源性,故不存在基因的非特异性激活或抑制; ②能诱导基因高效表达,可达105倍,为其他系统所不及; ③能严格调控基因表达,即不仅可控制基因表达的“开关”,还可人为地调控基因表达量。 因此,利用真核表达系统来表达目的蛋白越来越受到重视。目前,基因工程研究中常用的真核表达系统有酵母表达系统、昆虫细胞表达系统和哺乳动物细胞表达系统。 1.酵母表达系统 最早应用于基因工程的酵母是酿酒酵母,后来人们又相继开发了裂殖酵母、克鲁维酸酵母、甲醇酵母等,其中,甲醇酵母表达系统是目前应用最广泛的酵母表达系统。目前甲醇酵母主要有H Polymorpha,Candida Bodini,Pichia Pastris3种。以Pichia Pastoris 应用最多。
载体,质粒,基因表达载体3者有何不同? 载体是一种运输工具,细胞膜上的蛋白质也是一种载体,其识别很单一,也因此有了选择透过性。 质粒是一种小型环状DNA,广泛存在于微生物中,一般的基因工程中,质粒被用来当做目的基因的运载体,也是一种运输工具。 基因表达载体是目的基因和运载体连接后的产物。 基因表达载体和重组质粒的区别 基因表达载体的构建(即目的基因与运载体结合)是实施基因工程的第二步,也是基因工程的核心。 将目的基因与运载体结合的过程,实际上是不同来源的DNA重新组合的过程。如果以质粒作为运载体, 首先要用一定的限制酶切割质粒,使质粒出现一个缺口,露出黏性末端。然后用同一种限制酶切断目的基因,使其产生相同的黏性末端(部分限制性内切酶可切割出平末端,拥有相同效果)。将切下的目的基因的片段插入质粒的切口处,首先碱基互补配对结合,两个黏性末端吻合在一起,碱基之间形成氢键,再加入适量DNA连接酶,催化两条DNA链之间形成磷酸二酯键,从而将相邻的脱氧核糖核酸连接起来,形成一个重组DNA分子。这个重组的DNA分子也叫重组质粒。 就是这样。简单的说,重组质粒是基因表达载体的一种。 生物学上marker是什么意思 从文字上说,标记(Marker),染色体上一个可以被识别的区域(比如限制性内切酶的酶切点,基因的位置等)。标记的遗传能够被检测出来。标记可以是染色体上有表达功能的部分(比如基因),也可以是没有编码蛋白质功能但遗传特性能够被检测出来的部分 从实验上说,marker有两种,一种是基因marker,一种是蛋白质marker。两种marker分别是进行琼脂糖凝胶电泳和SDS-PAGE(蛋白质电泳)所用的标准参照物,用以指示目的基因或者蛋白的大小。 一般常称呼的marker都是指基因marker,即DNA marker DNA Marker 作为一种分子标记(Marker),构建分子图谱。分子标记克隆在质粒上,可以繁殖及保存。 DNA Marker 是分子量不同的DNA片段,主要用途就是DNA 分子凝胶电泳时,加样用做对比来检测琼脂糖凝胶是否有问题。 现在常用的DNA MARKER有两种,一种是病毒等DNA经过酶切获得的,分子量大小有零有整,另外一种是固定数值的,比如100BP,200BP等。 两种MARKER都不难制作,对于第一种,稍微难一些,主要是要获得病毒等大的基因组片段,然后用适当的酶切,切割完全以后,就能得到相应的图谱。第二中实际上很简单,如果你手头有任何一个载体,而且它的序列完全清楚,那么可以采用PCR的方法获得一系列大小不同的片段,比如上游引物可以使用一个,然后用数数的方法确定下游100BP处的下游引物,扩增出的就是100BP的片段,200BP处的引物就是200BP的片段,依此类推,可以获得一系列不同大小的片段,扩增后,把它们放到一起,就获得了自制的MARKER了
实验一载体与目的基因的连接与转化以及 重组DNA的提取与酶切鉴定 一、实验目的 1.CaCl2法制备感受态细胞 2.目的基因与载体连接(c-myc+pSV2;粘端连接) 3.重组质粒转化大肠杆菌并筛选转化体(HB101;Amp r) 4.质粒DNA的小量快速制备 5.质粒DNA的限制性内切酶酶切 6.DNA的琼脂糖凝胶电泳 二、实验原理 通过粘端连接法将具有相同粘性末端的DNA分子连接在一起,通过碱基配对氢键形成一个相对稳定的结构,利用连接酶发挥间断修复的功能,从而获得重组的DNA分子。 受体细胞经处理后(电击或CaCl2等处理),细胞膜通透性发生变化,从而使外源的载体分子通过感受态细胞,并使受体细胞获得新的稳定遗传的性状,该过程称为转化。由于本实验种pSV带有抗氨苄青霉素的基因,因而转化后的细胞在含氨苄青霉素的平板上培养可以筛选出转化成功的受体细胞。 分离质粒DNA的步骤包括:培养细菌使质粒扩增、收集和裂解细菌以及分离和纯化质粒DNA。SDS可以使细胞壁裂解,碱变性抽提质粒DNA的原理是利用染色体DNA与质粒DNA的变性复性的差异达到分离目的,当pH>12.6时,染色体DNA氢键断裂,双螺旋结构解开而变性,质粒DNA由于超螺旋共价闭合环状结构,两条互补链不会完全分离。当采用pH 4.8的NaAc高盐缓冲液调节pH至中性时,质粒DNA恢复原有的构型,而染色体DNA则不能复性而缠绕形成网状结构。通过离心可将染色体DNA及大分子RNA、蛋白质等去除。 三、实验器材和试剂 1.器材 恒温摇床、电热恒温培养箱、电热恒温水浴、台式离心机、低温离心机、涡旋振荡器、移液枪及枪头、1.5 ml离心管、制冰机、三角推棒、酒精灯、细菌培
Chi l 原核表达基本试验步骤 将克隆化基因插入合适载体后导入大肠杆菌用于表达大量蛋白质的方法一般称为原核表达。这种方法在蛋白纯化、定位及功能分析等方面都有应用。大肠杆菌用于表达重组蛋白有以下特点:易于生长和控制;用于细菌培养的材料不及哺乳动物细胞系统的材料昂贵;有各种各样的大肠杆菌菌株及与之匹配的具各种特性的质粒可供选择。但是,在大肠杆菌中表达的蛋白由于缺少修饰和糖基化、磷酸化等翻译后加工,常形成包涵体而影响表达蛋白的生物学活性及构象。 表达载体在基因工程中具有十分重要的作用,原核表达载体通常为质粒,典型的表达载体应具有以下几种元件: (1)选择标志的编码序列; (2)可控转录的启动子; (3)转录调控序列(转录终止子,核糖体结合位点); (4)一个多限制酶切位点接头; (5)宿主体内自主复制的序列。 原核表达一般程序如下:获得目的基因-准备表达载体-将目的基因插入表达载体中(测序验证)-转化表达宿主菌-诱导靶蛋白的表达-表达蛋白的分析-扩增、纯化、进一步检测,其中包括: 一、试剂准备 (1)LB培养基。 (2)1M IPTG(异丙基硫代-β-D-半乳糖苷):2.38g IPTG溶于10ml ddH2O
中,0.22μm滤膜抽滤,-20℃保存。 CCY的IPTG是1M的,用时进行1000倍稀释。 二、操作步骤 (一)获得目的基因 1、通过PCR方法:以含目的基因的克隆质粒为模板,按基因序列设计一对引物(在上游和下游引物分别引入不同的酶切位点),PCR循环获得所需基因片段。 2、通过RT-PCR方法:用TRIzol法从细胞或组织中提取总RNA,以mRNA 为模板,逆转录形成cDNA第一链,以逆转录产物为模板进行PCR循环获得产物。 (二)构建重组表达载体 1、载体酶切:将表达质粒用限制性内切酶(同引物的酶切位点)进行双酶切,酶切产物行琼脂糖电泳后,用胶回收Kit或冻融法回收载体大片段。 2、PCR产物双酶切后回收,在T4DNA连接酶作用下连接入载体。我们用Soultion I连接。 (三)获得含重组表达质粒的表达菌种 1、将连接产物转化大肠杆菌BL21,根据重组载体的标志(抗Amp或蓝白斑)作筛选,挑取单斑,碱裂解法小量抽提质粒,双酶切初步鉴定。 2、测序验证目的基因的插入方向及阅读框架均正确,进入下步操作。否则应筛选更多克隆,重复亚克隆或亚克隆至不同酶切位点。 3、以此重组质粒DNA转化表达宿主菌的感受态细胞。
常用的哺乳动物细胞表达载体和组成成分 有SV40病毒表达载体、痘病毒表达载体、逆转录病毒表达载体,常见的哺乳动物表达载体的组成成分有:原核DNA序列、启动子、增强子、拼接信号、终止信号和多聚腺苷酸化信号、筛选标记及真核病毒序列等。 (1)原核DNA序列:为了能在大肠杆菌中增殖,得到大量能转染哺乳动物细胞的重组DNA,哺乳动物表达载体中通常有一段原核序列,包括一个能在大肠杆菌中自身复制的复制子,便于挑选含重组DNA细菌的抗生素抗性基因,以及便于把真核序列插入载体的少数单一限制性酶切位点。当具备这些序列以后,外源的真核基因序列可由单一酶切位点插入载体中,形成的重组DNA可在大肠杆菌中增殖,经抗生素筛选后进行DNA提取,即可得到大量的所需的哺乳动物细胞表达载体。 (2)启动子:真核生物的启动子区域位于TATA区上游100bp到230bp之间,TAT区位于转录起始点上游25-30bp处。启动子的转录效率因细胞而异。因此需根据宿主细胞类型选择不同的启动子。 (3)增强子:增强子是使启动子的基因转录效率显著提高的一类顺式作用元件,有多个独立核苷酸序列组成。它们的作用通常不具有方向性,在位于转录起始点的下游或离启动子很远时仍有活性。许多增强子只能在特定的组织或细胞中起作用,即具有组织细胞的特异性,因此在构建真核表达载体的时候,应根据宿主细胞来选择增强子。 (4)剪接信号:真核基因由许多内含子和外显子组成。被转录成mRNA前体以后,需通过剪除内含子、连接外显子才能成为成熟的mRNA。一般mRNA拼接需要的基本序列位于内含子的5’和3’末端,因此,改变拼接位点5’和3’末端两侧的外显子序列可能会影响邻近拼接位点的使用效率,在替换外显子时应注意。 (5)终止信号和多聚腺苷化的信号:转录的终止信号常常位于多聚腺苷化位点下游的一端长度为几百个核苷酸碱基的DNA区域内。多聚腺苷化需要两种序列:位于腺苷化位点下游的GU丰富区或U丰富区和位于腺苷化位点上游11-30个核苷酸处的一个有6个核苷酸碱基组成并高度保守的AAUAAA序列。为了保证目的mRNA能有效地多聚腺苷化,真核表达载体上必须包括多聚腺苷化下游的一段序列。最常用的方法是用SV40的一段237bp长的BamHⅠ- BclI 限制性片段,含有多聚腺苷化的信号。另一种的方法是将全长cDNA与已组装在表达载体上一个顺式作用因子的部分片段融合,提供多聚腺苷化的信号。 (6)遗传标记:从成千上万个哺乳细胞中,检测出极少数的含DNA重组体的转染细胞,并鉴定已导入外源DNA是哺乳动物细胞基因表达系统的一个关键内
真核细胞常见表达载体 真核细胞, 表达载体 1、pCMVp-NEO-BAN载体 特点:该真核细胞表达载体分子量为6600碱基对,主要由CMVp启动子、兔β-球蛋白基因内含子、聚腺嘌呤、氨青霉素抗性基因和抗neo基因以及pBR322骨架构成,在大多数真核细胞内都能高水平稳定地表达外源目的基因。更重要的是,由于该真核细胞表达载体中抗neo基因存在,转染细胞后,用G418筛选,可建立稳定的、高表达目的基因的细胞株。 插入外源基因的克隆位点包括Sal1、BamH1和EcoR1位点。注意在此载体中有二个EcoR1位点存在。 2、pEGFP, 增强型绦色荧光蛋白表达载体(Enhanced Fluorecent Protein V ector) 特点: pEGFP表达载体中含有绿色荧光蛋白,在PCMV启动子驱动下,在真核细胞中高水平表达。载体骨架中的SV40origin使该载体在任何表达SV40 T抗原的真核细胞内进行复制。Neo抗性盒由SV40早期启动子、Tn5的neomycin/kanamycin抗性基因以及HSV-TK基因的聚腺嘌呤信号组成,能应用G418筛选稳定转染的真核细胞株。此外,载体中的pUC origin 能保证该载体在大肠杆菌中的复制,而位于此表达盒上游的细菌启动子能驱动kanamycin抗性基因在大肠杆菌中的表达。 用途: 该表达载体EGFP上游有Nde1、Eco47111和Age1克隆位点,将外源基因扦入这些位点,将合成外源基因和EGFP的融合基因。借此可确定外源基因在细胞内的表达和/或组织中的定位。 亦可用于检测克隆的启动子活性(取代CMV启动子,Acet1-Nhe1)。 3、pEGFT-Actin, 增强型绿色荧光蛋白/人肌动蛋白表达载体 特点:pEGFP-Actin表达载体中含有绿色荧光蛋白和人胞浆β-肌动蛋白基因,在PCMV启动子驱动下,在真核细胞中高水平表达。载体骨架中的SV40origin使该载体在任何表达SV40 T抗原的真核细胞内进行复制。Neo抗性盒由SV40早期启动子、Tn5的neomycin/kanamycin 抗性基因以及HSV-TK基因的聚腺嘌呤信号组成,能应用G418筛选稳定转染的真核细胞株。此外,载体中的pUC origin 能保证该载体在大肠杆菌中的复制,而位于此表达盒上游的细菌启动子能驱动kanamycin抗性基因在大肠杆菌中的表达。 用途: pEGFP-Actin载体在真核细胞表达EGFP-Actin融合蛋白,该蛋白能整合到胞内正在生的肌动蛋白,因而在活细胞和固定细胞中观察到细胞内含肌动蛋白的亚细胞结构。 4、pSV2表达载体 特点:该表达质粒是以病责SV40启动子驱动在真核细胞目的基因进行表达的,克隆位点为Hind111。SV40启动子具有组织/细胞的选择特异性。此载体不含neo基因,故不能用来筛选、建立稳定的表达细胞株。 5、CMV4 表达载体 特点:该真核细胞表达载体由CMV启动子驱动,多克隆区域酶切位点选择性较多。含有氨苄青霉素抗性基因和生长基因片段以及SV40复制原点和fl单链复制原点。但值得注意的是,该表达载体不含有neo基因,转染細胞后不能用G418筛选稳定的表达细胞株。 其他常用克隆Vector: pBluscript II KS DNA 15 ug pUC18 DNA 25 ug pUC19 DNA 25 ug 说明: pBluescript II kS、pUC18 &Puc19载体适合于DNA片段的克隆、DNA测序和对外源基因进行表达等。这些载体由于在lacZ基因中含有多克隆位点,当外源DNA片段扦入,转化lacZ基因缺乏细胞,并在含有IPTG和X-gal的培养基上培养时,含有外源DNA载体的细胞