
酯的合成方法研究
刘 聪
东北大学理学院高分子化学与物理
羧酸酯是一类重要的化工原料 ,它的用途相当广泛 ,可用作香料、溶剂、增塑剂及有机合成的中间体;同时在涂料、医药等工业中也具有重要的使用价值[1]。作为液晶化合物最基本和最重要的中心桥键之一,酯基的合成具有十分重要的意义。在过去很长一段时间里,酯的合成主要是采用一些经典的方法,如酸催化、酰氯法、酯交化法等;随着对各种新的催化剂和有机反应机理的研究,出现了一些新颖的合成方法,如Mitsunobu 反应、Steglich 酯化法、CAN 催化法、Me 3SiCl 催化法、DBU 催化法等等[2]。对这些新的合成方法进行研究,有助于在实验室推广采用更简单、更有效、更温和的方法合成羧酸酯,并进一步实用于工业化生产。 一、经典酯化反应 1、酯化反应机理:
羧酸与醇在催化剂作用下生成酯。例如:
CH 3COOH + HOC 2H 5
CH 3COOC 2H 5 + H 2O
H
酯化反应是可逆反应。为了提高酯的产率,可采取使一种原料过量(应从易得、 价廉、易回收等方面考虑),或反应过程中除去一种产物(如水或酯)。工业上生产乙酸乙酯采用乙酸过量,不断蒸出生成的乙酸乙酯和水的恒沸混合物(水6.1%,乙酸乙酯93.9%,恒沸点70.4℃),使平衡右移。同时不断加入乙酸和乙醇,实现连续化生产[3]。
羧酸的酯化反应随着羧酸和醇的结构以及反应条件的不同,可以按照不同的机理进行。酯化时,羧酸和醇之间脱水可以有两种不同的方式:
R C O
O H HO
R'
R C OH H O
O
R'
R ,R ’分别是烷基。(Ⅰ)是由羧酸中的羟基和醇中的氢结合成水分子,剩余部分结合成酯。由于羧酸分子去掉羟基后剩余的是酰基,故方式(Ⅰ)称为酰氧键断裂。(Ⅱ)是由羧酸中的氢和醇中的羟基结合成水,剩余部分结合成酯。由于醇
(Ⅰ) (Ⅱ)
去掉羟基后剩下烷基,故方式(Ⅱ)称为烷氧键断裂。
当用含有标记氧原子的醇(R 18OH)在酸催化作用下与羧酸进行酯化反应时,发现生成的水分子中不含18O ,标记氧原子保留在酯中,这说明酸催化酯化反应是按方式(Ⅰ)进行的。其反应机理可以表示如下:
R C O OH
H
R C OH OH
R C OH
OH
R C OH OH OH
R'
R C OH OH 2
R C OH R C OH R C O OR'
-H
这个机理可以概括如下:
R O OH
R'OH
+
-H
R C OH OH
R'O
R C O
OR' H 2O
+
叔醇的酯化反应经实验证明是按方式(Ⅱ)进行的:
R C O
OH +HO
CR'3
R C O
OCR'3
+
H 2O
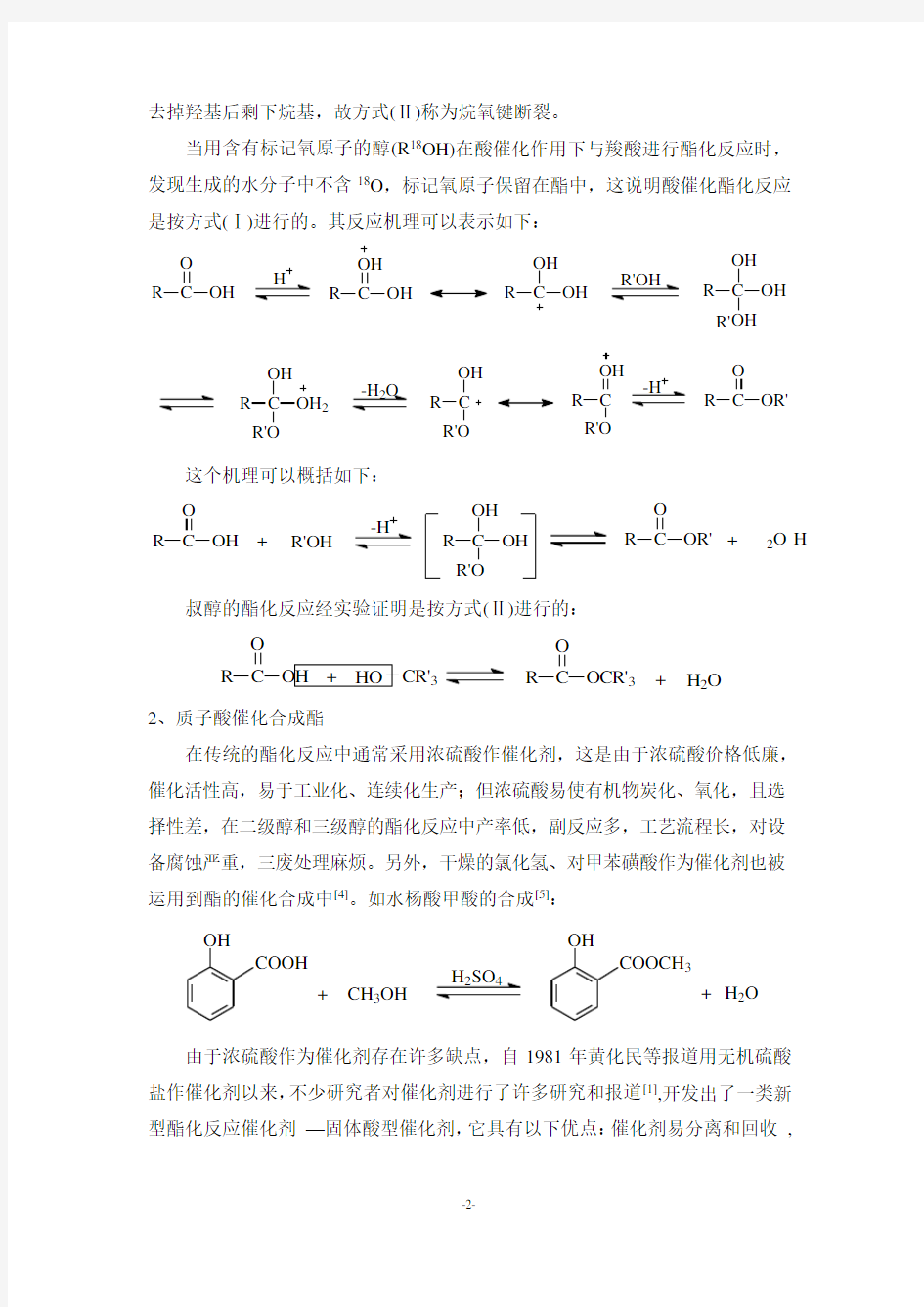
2、质子酸催化合成酯
在传统的酯化反应中通常采用浓硫酸作催化剂,这是由于浓硫酸价格低廉,催化活性高,易于工业化、连续化生产;但浓硫酸易使有机物炭化、氧化,且选择性差,在二级醇和三级醇的酯化反应中产率低,副反应多,工艺流程长,对设备腐蚀严重,三废处理麻烦。另外,干燥的氯化氢、对甲苯磺酸作为催化剂也被运用到酯的催化合成中[4]。如水杨酸甲酸的合成[5]:
OH
COOH
+
CH 3OH
OH
COOCH 3
+H 2O
由于浓硫酸作为催化剂存在许多缺点,自1981年黄化民等报道用无机硫酸盐作催化剂以来,不少研究者对催化剂进行了许多研究和报道[1],开发出了一类新型酯化反应催化剂 —固体酸型催化剂,它具有以下优点:催化剂易分离和回收 ,
不怕水,易再生可重复使用,它包括无机盐超强酸、分子筛、杂多酸、氧化物、高分子载体催化剂等。 3、酰氯酯化法
酰氯酯化法是合成羧酸酯应用最多的方法之一。该方法主要是先将有机酸转变为酰氯,酰氯再醇解得到相应的酯。酰化试剂有新制的二氯亚砜(SOCl 2) 、草酰氯(C 2O 2Cl 2)、光气(COCl 2)等。在实验室中,比较常用的是采用SOCl 2作为酰化试剂。SOCl 2酰氯酯法的优点是生成酰氯时的副产物是HCl 和SO 2,均为气体,有利于分离,且酰氯的产率较高,这将提高下一步反应的活性和产率。其缺点是制备酰氯时需对反应条件进行较严格的控制,如时间、温度等,不易除尽过量的SOCl 2,对设备的腐蚀较严重;而且酰氯需要现制现用,整体合成路线长[6]。
酰化和醇解过程中都生成大量氯化氢,因此在反应中加入氯化氢的去除剂,称为缚酸剂,如吡啶、三乙胺(TEA)、DMAP 、N,N ’-二甲苯胺等。酰氯遇水易发生分解,因此反应必须在无水条件下完成。
如10-十一烯酸(a)是合成不饱和酸胆甾醇酯常用的原料。李佩瑾等[7]以 SOCl 2为酰化试剂,N,N ’-二甲苯胺作为醇解反应的缚酸剂,75℃下回流8 h 得到了10-十一烯酸胆甾醇酯(b)。(b)是一种介晶单体,可与非介晶手性单体十一烯酸薄荷醇酯通过接枝共聚引入聚甲基含氢硅氧烷中,得到具有光化学活性的胆甾相液晶聚合物[8]。
CH 2=CH(CH 2)8COCl + HO Chol *
CH 2=CH(CH 2)8COO Chol *
(a)
(b)
CH 2=CH(CH 2)8COOH SOCl CH 2=CH(CH 2)8COCl +
4、酸酐酯化法
该方法一般用于双羧酸官能团物质与单羟基的醇或酚反应,制得单羧酸酯, 且保留一个羧酸,可进一步酯化或酰胺化。但由于酸酐化合物种类较少,限制了它的更进一步应用。
利用酸酐的反应特点,引入带羧酸官能团的柔性链,在合成氢键自组装超分子聚合体中具有特殊的应用。如沈永涛等[9] 以胆甾醇和丁二酸酐为原料合成了丁二酸单胆甾醇酯(AC),研究探讨了AC 自组装液晶特性。
O O +
2CH 2COO
(AC)
5、低级酯交换法
酯交换反应的实质是酯的醇解。将羧酸与低级醇(如甲醇或乙醇)制备成低级 酯(如甲酯或乙酯),然后与高级醇在酸性条件下进行醇解。该方法在某些特殊的反应中有着独特的作用,如羧酸的保护与脱保护。但由于该法合成路线较长,成本较高,使得其应用范围较小。在手性中心的合成中,需进行羧酸的保护,设想了下列方案:
OH
***
OCH 2CH 2COOCH 3
***
ClCH 2CH 2COOH +CH 3OH
ClCH 2CH 2COOCH 3ClCH 2CH 2COOCH 3
(2)OCH 2CH 2COOCH 3***
HO
COOH
+OCH 2CH 2COO
***
COOH
在该反应中,设想对羟基苯甲酸既作为反应物,又提供质子,促进反应的发
生(该方案第三步未验证)。 二、酯的合成新方法 1、Mitsunobu 反应
官能团的转化在有机合成化学中占了极其重要的地位,我们在合成中经常需要进行官能团转化,构建新的化学键,如 C —O ,C —N ,C —S ,C —C 等化学键。而Mitsunobu 反应[9]是在偶氮二碳酸二乙酯(DEAD)或者偶氮二碳酸二异丙酯
(DIAD)和三苯基膦作用下,醇类化合物和酸性化合物发生分子内或分子间脱水反应,形成C—O,C—N,C—S,C—C等键的反应。它最早是在1967年由Mitsunobu 等[10]发现。Mitsunobu 反应一般是在温和的中性条件下进行的,同时,如果是手性醇参加反应,醇羟基所连碳原子的绝对构型一般会发生翻转,因此,Mitsunobu反应广泛应用于各类天然产物的全合成或化合物的官能团转化,是一个应用范围较为广泛的反应。在这里我们仅仅把它作为一种羧酸缩合成酯的方法进行介绍。
DEAD or DIAD
ROOH+R'OH
ROOR'
3
近年来,对Mitsunobu 反应机理研究得比较多[11~13], 美国化学家V arasi等用31P NMR对Mitsunobu反应进行了仔细的研究。结合实验结果。他提出了如Scheme 1 所示的反应机理。他发现,酸性化合物在反应过程中加入的时间不同,反应的机理就有所不同,反应的第一步仍然是DEAD (1)和三苯基膦(2)进行加成,形成季鏻盐3,当 3 形成时酸存在于反应体系中,或者此时加入酸,季鏻盐3 就会马上发生质子化形成中间体5。此时加入醇,中间体5就会缓慢的形成鏻盐4,接着发生S N2取代反应并生成产物。当3 形成时,如反应体系中没有酸存在,此时加入醇,一半季鏻盐 3 会与醇发生反应,形成二烷氧基鏻盐6,在此时加入酸,剩下的季鏻盐会发生质子化生成5,而 6 也会很快和酸发生反应生成鏻盐4,同时释放出一半的醇去和中间体 5 发生反应。在此反应过程中,从5 到 4 的反应过程非常慢,从 6 到4的反应过程则非常快。
Scheme 1
2、Steglich酯化法(DCC-DMAP)
DCC-DMAP催化酸与醇直接在室温下反应[14],产率高,也能有效地合成具有空间位阻的酯。反应完毕,DCC(碳二酰亚胺)转化为脲不溶于溶剂被洗去[15]。研究发现1-甲基咪唑(MI)-DCC也是有效的催化体系。1-甲基咪唑的价格低,而且几乎无毒。
3、Me3SiCl催化法
Me3SiCl可以催化羧酸与醇反应生成酯,也可以合成具有空间阻碍的酯。可以加入吡啶或三乙胺中和反应生成的酸性副产物[16]。
4、DBU催化法
DBU催化羧酸与醇直接反应,可以促进有空间位阻的酸和氨基酸等与卤代烃反应生成酯[17]。
自60 年代DBU(1,8-二氮杂双环[5.4.0]-7-十一碳烯)问世以来,经过30多年的研究,已成为一种非常有用的试剂或催化剂,在消除、异构、缩合、酯化、环合、聚合等多种反应中得到应用[18,19]。其参与反应的特点是反应条件温和、副反应少,产物转化率高,产物选择性专一。尤其是对原料或生成物不稳定的反应,选用DBU 尤为适用。DBU 是一种有开发前途的试剂, 受到了广泛地重视。
DBU 是一个强碱性试剂,但它却是一个弱的亲核试剂, 易与质子结合而不易与碳原子结合, 因此DBU 的应用主要是作为强碱性试剂转移质子, 起到碱或催化剂的作用。DBU 参与反应的特点是, 一般需要等物质的量的DBU ,应用的反应也主要集中在有质子转移的一些反应, 如消除、异构、加成、酯化、醚化、酰胺化、重氮化等反应。
1978年,Ono[17]报道用DBU 催化羧酸、卤代烷进行酯化反应,得到高产率的酯。这种制备酯的方法有反应条件温和、无副反应等优点。一个代表性的例子是在DBU 作用下苯甲酸和碘乙烷反应1h 后得到苯甲酸乙酯,产率为95%。
另一个例子是在DM SO 中DBU 催化苄基溴、苯甲酸于30℃下反应10min ,几乎得到 100%的苯甲酸苄酯[ 20]。
利用上述方法还可以合成高分子量的聚酯。例如,在 THF 中,间苯二甲酸和间二溴甲基苯经DBU 作用反应1h ,得到粘度很高的聚合物,产率 90%[20]。而用其他有机碱(如三乙胺、吡啶等)催化均不能得到聚合物。
n HOOC
COOH
+Br
Br
DBU THF 1h
n n C
O O
O
C
O
DBU 可以催化聚合物进行酯化反应[21]。例如,聚甲基丙烯酸和对溴甲基硝基苯于DMSO 溶剂中反应30h ,得到聚甲基丙烯酸酯,聚甲基丙烯酸分子中羧基酯化率高达97%:
NO 3n BrCH 2
+
CH CH 3
CH COOH
( )
n DBU DMSO 30h
CH CH 3
CH ( )
n NO 3
CH 2
此外,在N-酰基咪唑存在下,用DBU 催化羧酸和叔丁醇反应也生成酯[22]。
RCOOH + t-BuOH
RCOOBu-t + H 2O
N N
CO 2
DBU
用该法制备的苯甲酸叔丁酯、肉桂酸叔丁酯和庚酸叔丁酯的产率分别为 91%、 64%和 68%,都没有副产物出现[22]。 5、羧酸盐与卤代烃反应法(相转移催化法)
羧酸盐与卤代烃反应由于其S N 2反应机理,卤代烃常为小分子,如:甲基, 乙基,烯丙基和苄基等。羧酸铯盐和季铵盐的反应效果较好,溶剂采用极性非质子溶剂[23](DMF ,DMSO)。
如邓书平等[24]由苯甲酸钠和氯化苄通过相转移催化法合成了苯甲酸苄酯,考察了几种廉价易得的季铵盐类相转移催化剂的亲核反应性能,筛选出性能较好的催化剂并优化了反应条件。
C6H5CH2Cl + C6H5COONa C6H5COOCH2C6H5 + NaCl
有机相水相有机相水相由苯甲酸钠和氯化苄通过相转移催化法合成苯甲酸苄酯,通过对合成成本的控制,筛选出四丁基溴化铵为合成苯甲酸苄酯反应的相转移催化剂,在n (苯甲酸钠):n(氯化苄)为1:1.3、反应温度为110℃、反应时间为2.5~3.0 h、催化剂用量为苯甲酸钠摩尔量的6 %、体系pH值为5~6时,苯甲酸苄酯的产率可达
83 %以上。
三、Steglich酯化法(DCC-DMAP)
在酯化反应中,最初的酰化催化剂为吡啶,但对于高位阻、低活性的底物,结果往往不理想。1967年Litvinenko和Kirichenko在研究间氯苯胺的苯甲酰化时发现,用4-二甲氨基吡啶(以下简称DMAP)代替吡啶时,反应速度提高104~105倍。1969年,Steglich和Hofle也发现DMAP和4-四氢吡咯基吡啶(PPY)对制备规模的酰化反应有着极强的催化作用。因此,人们对DMAP的制备方法和应用进行了广泛的研究[25~27], 发现DMAP 中给电子的二甲氨基与吡啶环的共轭作用,能强烈地激活环上的氮原子进行亲核取代反应,从而提高反应速度和产率。由于其显著的催化效果,被称为酰化反应的“超级催化剂”,并已在精细化工的诸多领域得到广泛应用。
DMAP,4-二甲氨基吡啶,用作酰化催化剂具有如下优点:
a) 反应速度快,与吡啶相比可提高反应速度104~105倍,因而大大缩短了反应时间;
b) 反应条件温和,许多反应可以在室温下进因而既降低能耗又易于控制;
c) 反应收率高,对于空间位阻大,反应活性低羟基化合物效果尤为显著;
d) 溶剂选择范围广,适用溶剂有苯、甲苯、甲苯、乙酸乙酯、己烷、四氢呋喃、氯仿、二氯甲烷、乙酸酐、吡啶、三乙胺、二甲亚砜等。
DCC,N,N’-二环己基碳二亚胺( dicyclohexyl-carbodiimide),是有机合成和医药制造工业常用的脱水剂,它可以使两个本来不能反应的分子,脱水形成化学
键。在我国主要用于丁胺卡那霉素和谷光甘肽等产品的生产。由于DCC分子中累积二烯结构使DCC具有很强的化学活性,不仅可以和羧酸、氢氰酸及硫化氢等许多酸性化合物起反应,也可以和其它如醇、胺及含活泼亚甲基等活泼氢一类化合物起反应[28]。
DCC-DMAP酯化法在合成具有空间位阻的羧酸酯的应用上具有不可比拟的优势,不仅反应条件温和,反应时间快,而且产率高。
1、反应机理
N,N'-二环己基碳二亚胺(DCC)主要用于多肽人工合成过程中氨基酸的缩合。在多肽合成,如Fmoc-固相合成中,是一个氨基酸的羧基与另一个氨基酸的氨基形成酰胺键。为了使羧基更容易接受亲核试剂的进攻,带负电荷的氧原子需首先被活化成一个较好的离去基团,DCC就发挥此作用。同样的,可用脂肪类或芳香类羧酸上羧基的氧原子作为亲核试剂进攻DCC分子中间的碳原子,从而使DCC结合在羧基上,形成一种活性酯结构,这使得醇或酚上的羟基的亲核进攻变得较容易进行。韦长梅等[29]在合成阿魏酸对硝基苯酚酯中,探讨了其反应机理,如下所示:
CH3 CH3O
O
(FA)(DCC)
CH3 CH3O
O
CH3 CH3O O
O NO2+NH NH
O
(DCU)
由反应机理可知。DCC 催化的酯化反应是通过DCC先与阿魏酸反应活化阿魏酸的羧基,再与羟基反应生成酯。
DMAP的应用更广泛,不仅可以同DCC一起用于反应中,充当缩合促进剂或催化剂,而且其本身也可催化酰氯酯化反应和酸酐酯化反应,对高位阻、低反应活性的底物有较高的活性,具有用量少、反应条件温和、操作简便、反应时间短、收率高、产品纯度高、没有副反应等优点。
DMAP在酰氯酯化反应中的催化机理表示如下:
N
N(CH 3)2
RCOOR'
+
+
N(CH 2CH 3)3HCl
233
+
RCOCl
N
N(CH 3)2
C
R '
O H
Cl R
S. Klemenc [30]研究了DMAP 在酸酐酯化反应中的机理,表示如下:
在本文中,重要研究DCC-DMAP 联用在酯化反应中的应用。 2、DDC-DMAP 催化合成酯的条件初探 1)、溶剂的选择
选择合适的溶剂对于有机合成特别是对于液体单体的合成有着重要的意义。首先要考查溶剂的毒性和成本,一般选用低毒或无毒、价廉易得的溶剂;然后考查羧酸和醇或酚在溶剂中的溶解性,同时还要考查后处理的难易程度。在液晶化合物中,通常由多个苯环、杂环或脂环构成其骨架,随着分子量的增加,环的个数也在不断增加,一般情况下使得化合物在溶剂中的溶解越来越困难,往往很难找到一种常见的单一溶剂使其溶解,这时就需要使用混合溶剂。
针对DCC-DMAP 反应体系,综合考查后,可选用以下溶剂,或两种溶剂的混合溶剂:
水溶性溶剂——THF ,丙酮,三乙胺,吡啶,乙酸乙酯,N,N-二甲基甲酰胺
(DMF),二甲基亚砜(DMSO);
非水溶性溶剂——二氯甲烷,三氯甲烷,1,2-二氯乙烷,甲苯,
溶剂种类的具体选择与所参加反应的原料有关。在实验前应查阅反应原料的理化性质或进行溶解性实验,大致掌握其溶解范围。如溶剂的溶解性能过强,会使少量的副产物溶解在其中,影响后处理及产物的纯度。如果THF、二氯甲烷在加入助溶剂(一般为吡啶)的情况下能将原料溶解,就不选用DMF和DMSO。通过大量实验发现,在使DMF或DMSO时,产物中会混入少量副产物DCU,分离不彻底。
溶剂的用量:一般以能够溶解反应物为标准,过多的溶剂不但造成浪费,也会使副产物DCU溶解其中,造成分离的困难;但过少的溶剂会造成产物产率的损失。
2)、原料用量和投料顺序
DMAP用量原则:羧酸质量的3%~5%。
投料的基本原则:DCC-DMAP溶解在溶剂中,向反应液中缓慢滴加。
投料顺序和原料的结构有着至关重要的关系。在实验室中,含有不同类官能团或官能团数目大于1的原料进行反应时,投料的顺序是不同的,大致可归纳为以下几种:
○1单羧酸化合物和单羟基化合物的反应
n(羧酸):n(羟基化合物):n(DCC) = 1.0 :1.0 :1.2。
在实际投料过程中,羧酸和羟基化合物的用量可以作一定程度的浮动,根据原料的成本和后处理的难易程度,可适当将其中一种原料过量,这样可以使反应进行得更充分。
这类反应的投料是最简单的。将羧酸溶解在溶剂中加入反应装置,将DCC-DMAP溶解后,缓慢滴加,搅拌0.5h,得到了羧酸活性产物,这时反应体系是混浊的,这是因为活性中间产物一般不溶于常见有机溶剂。最后加入含羟基的化合物。如果羟基化合物溶解性能不好,可加入助溶剂,待其完全溶解后,加入反应装置中。
○2单羧酸化合物和对羟基苯甲酸类化合物的反应
n(羧酸):(羟基化合物):(DCC) = 1.0:1.2~1.5:1.0。
一般情况下,对羟基苯甲酸类化合物适当过量,DCC不过量。
由于对羟基苯甲酸类化合物本身既有羟基又有羧基,根据反应机理,如果该化合物在接触到DCC-DMAP后可能发生本体的反应,生成二聚体或多聚体。因此,此类反应需要先将单羧酸化合物溶解在反应装置中,然后滴加DCC-DMAP,反应0.5h后,滴加对羟基苯甲酸类物质(滴加的原因是为了使得羟基能更充分接触DCC活化产物,使得反应朝着设计的方向发展)。对羟基苯甲酸类化合物适当过量,可以促使反应更快速、更彻底地进行。实验证明,此种投料方式基本能控制副反应的发生,产率在90%(按羧酸计算)左右。
○3单羧酸化合物和多羟基化合物的反应
n(羧酸):(羟基化合物):(DCC) = 1.0:2.5~3.0:1.0。
在实验室,经常遇到多羟基化合物,一般为双羟基化合物,如对苯二酚、对联苯酚等。实验要求一般是得到单酯化合物,保留一个羟基以进行下一步的合成。由于这类化合物在非极性溶剂中的溶解度不大,一般都选用水溶性溶剂。投料时,将多羟基化合物在反应装置中完全溶解,然后加羧酸化合物,剧烈搅拌1h,缓慢滴加DCC-DMAP溶液。由于羧酸和羟基化合物均完全溶解在溶剂中,根据分子在溶剂中的分布原理,羧酸在接触DCC-DMAP形成活性中间物后同羟基的接触几率是不改变的。利用上述原理,采用多羟基化合物在用量上远大于羧酸的投料方式,使得形成单酯的碰撞几率增加,这是分子动力学的优势;再考虑分子反应的热力学,根据反应能垒理论和空间位阻效应,生成双酯的几率被进一步降低。实验证明,长链羧酸基本上不会生成双酯,通过适当的后处理,可以得到纯度很高的产物,产率较高。
○4多羧基化合物和单羟基化合物的反应
n(羧酸):(羟基化合物):(DCC) = 2.0:1.0:1.0。
实验要求同样是制备单酯化合物,需保留一个羧基。投料时,先将羧酸溶于反应装置,缓慢滴加DCC-DMAP混合液,此时溶液变混浊;然后滴加加入单羟基化合物溶液。由于羧酸和DCC的用量关系,只有一半的羧酸官能团转化为羧酸活性产物,在此转化过程中,滴加速度和搅拌速度至关重要。一般情况下,需缓慢滴加(滴/3秒),快速搅拌(标准机械搅拌控制400)。这样可保证尽量多的羧酸活化反应只反生在一个羧酸官能团上,而另一个羧酸不参与反应。当然,这只
是一个几率的问题。最后得到的化合物当中,存在双酯,需要采取重结晶等方式提纯产品。单酯产率在50%以上(按羟基化合物计算)。
3)、反应温度
通过大量实验表明,常温25℃是反应的最佳时间。常温下,溶解性能不理想的原料,可适当加热反应,但温度不宜过高。在制备单体时,由于原料的反应活性较低,可适当提高反应温度。
4)、反应时间
一般反应24h。脂肪酸和醇的反应可缩短反应时间,一般3~12h,芳香羧酸和苯酚类物质的反应可延长时间,一般12~36h。在反应过程中,可采有TLC或红外监测的手段来考查反应完成情况。如采用红外监测,在等当量的DCC反应体系中,可将羧酸的特征吸收峰消失视为反应完成的指标。
5)、后处理
后处理根据所选用的溶剂种类的不同,大致有两种不同的处理方式:
○1水溶性溶剂
此类溶剂同水互溶,因此后处理相对简单。反应完毕后,加入5~10mL去离子水搅拌20min,这样可将未参与反应的DCC完全转化为DCU。停止搅拌,抽滤,尽量将不溶物除尽,用溶剂洗涤滤饼,合并溶液;将大部分溶剂旋转蒸发除去,浓缩液投入大量水中,沉淀析出。如果得到油状物,可加入冰水中,或冷冻。粗产品干燥,选用适当的溶剂重结晶。
○2非水溶性溶剂
反应完毕后,加入10~30mL去离子水搅拌20min;过滤,除尽不溶物;用溶剂洗涤滤饼,合并滤液。将滤液分别用1mol/LHCl溶液(x1)、2.5%Na2CO3溶液(x2)、去离子水洗涤(x3)。分液后,保留油层,无水MgSO4干燥2h,过滤,将滤液旋转蒸发,除去大部分溶剂,向浓缩液中加入大量甲醇,沉淀展开析出,静置一段时间后,过滤,得到粗产品,干燥,适当溶剂重结晶。
6)、DCC和DCU的红外特征峰
利用DCC的红外特征吸收峰判定反应的进行程度和原料是否处理完全。其结构中含有C=N,通常在2155-2100cm-1出峰。其红外谱图如下:
利用DCU 的红外特征吸收峰可以判定产物的纯度。 其特征结构是N-H ,C=O ,γ
N-H
通常在3540~3125cm -1出峰,γ
C=O 通常在
1690~1620 cm -1出峰,峰形较宽,通常出三个峰。其红外谱图如下:
下图是某个反应进行中,对反应液进行的红外测试。反应已经生成了酯基,但未反应完全,还有DCC 的特征峰。随着反应的进行DCC 的峰强度慢慢变小。值得注意的地方,即使反应完成后,反应液中也可能残留微量的DCC ,因此在反应结束后,需加入少量水将未反应完全的DCC 转化为DCU 。
477.64
538.54631.23787.39
818.22
842.15864.37891.56
949.57
1023.811046.55
1074.361090.631124.351146.451189.001239.861259.551298.56
1346.061359.571449.44
1629.76
2119.92
2659.00
2853.66
2930.57
3502.51
52 54
56
58 60 62 64
66
68 70
72
74
76
78 80 82
84
86%T
500
1000
1500 2000 2500
3000
3500
4000
cm-1
20
40
60
%T 50
60
70
%T 60
80
100
%T
500
1000 1500 2000 2500
3000 3500 4000 cm-1
2115cm -1
下图是某个反应产物的红外谱图,红外谱图中出现了DCU 的特征峰3330、1627cm -1,可能是由于所用溶剂过多或溶剂的溶解能力太强,将产品溶解的同时也溶解了部分产品。可以用溶解能力较弱的溶剂将产品重新溶解后过滤,因为DCU 一般不溶于有机溶剂。
下图是某个反应副产物的红外谱图,红外谱图中出现了目标产物酯基的特征峰1735、1718cm -1。这种产物和DCU 分离不好的原因:溶剂过少或溶剂溶解能力较弱,造成产物不能溶解在溶液中,析出,过滤时与DCU 混在一起,造成产率的损失。可选用一种溶解能力较强的溶剂洗涤DCU 滤饼。
%T
cm-1
1027.94
1174.96
1221.371297.57
1382.46
1467.78
1577.72
1626.82
1736.44
2120.60
2851.53
2934.76
3450.47
46 48 50 52
54
56 58
60
62 64 66 68
70
72
74
76 78 80
82 84 86 88%T
500
1000
1500
2000
2500
3000
3500
4000
cm-1
3、DMAP 催化酰氯法
在酰氯酯化过程中,需要加入缚酸剂,一般为吡啶、三乙胺等,既起到助溶、 催化的作用,又可中和反应生成的氯化氢。DMAP 作为一种高效的催化剂,在合成高位阻的羧酸酯中具有无可比拟的优势。一般情况下,将DMAP 作为催化剂,需要在反应体系中加入缚酸剂三乙胺。选择三乙胺的理由,是DMAP 作为碱性试剂,其碱性比三乙胺略弱(DMAP pKa=9.7,吡啶 pKa=5.29, 三乙胺 pKa=10.88),这样发生反应时生成的氯化氢优先和三乙胺结合,DMAP 可以循环催化。如果选择吡啶,氯化氢先和DMAP 反应,则起不到催化效果。
刘小平采用4-二甲胺基吡啶催化合成了美欧卡霉素[31]。
于三颈瓶中加入麦迪霉素20g (0.0246mol),醋酸乙酯60mL (0.613 mol),DMAP 适量,三乙胺适量,冰浴下,加乙酰氯10mL(0.140 mol),维持5~10℃,搅拌反应5h 左右,然后加热,温度低于60℃,搅拌10h 左右,冰水洗至酯层接
%T
cm-1
近中性,减压蒸出醋酸乙酯,得固体22.5g(0.0239mol),收率:97. 4%。
从反应过程可以看出,每酰化一分子,将释放三分子HCl ,HCl 如不及时从反应体系移走,将与DMAP 结合,使其失去催化活性,因此必须在反应体系中加入适量的缚酸剂,以保证DMAP 的催化效果,缚酸剂应该选用既溶于反应溶剂碱性又稍强于DMA P 的物质。根据试验,三乙胺(pKa= 10. 88)比较合适。 4、应用举例
1)、4-烯丙氧基苯甲酸-4`-十一烯酸对苯二酚双酯的合成 反应方程式:
COO CH 2=CHCH 2O
OH +
CH 2=CH(CH 2)8COOH
COO
CH 2=CHCH 2O
OOC(CH 2)8CH=CH 2
反应过程:
在250mL 三口烧瓶中加入2.7g(0.01mol) 4-烯丙氧基苯甲酸对苯二酚单酯,加入150mLCH 2Cl 2,加入适量的吡啶促使原料完全溶解;将 2.1g DCC(0.01)和0.1g DMAP(5%)溶解在20mL CH 2Cl 2中并投入三口烧瓶。将2.0g(约0.01mol) 十一烯酸加入10mL CH 2Cl 2中搅拌均匀,加入三口烧瓶中。搅拌,常温反应24h 。反应完毕后,加入20mL 去离子水,搅拌30min ,过滤,分液,弃去水层,保留CH 2Cl 2层。分别用1mol/L 盐酸溶液,5%Na 2CO 3溶液,去离子水洗涤CH 2Cl 2层。分液后将油层用无水MgSO 4干燥,旋转蒸发除去大部分溶剂,向浓缩液中加入甲醇,沉淀展开析出。过滤,干燥,乙醇结晶。产率69%,溶点61.3℃,清亮点78.4℃。
说明:这个反应投料顺序略有差异,是因为羟基化合物在CH 2Cl 2中的溶解能力较差,实验证明,此种投料方式得到的产品和用酰氯法得到的产品,理化参数均一致。
2)、十一烯酰氧基苯甲酰氧基苯甲酸 反应方程式:
CH(CH 2)8COO
COOH CH 2
COOH HO +CH(CH 2)8COO
COO
CH 2
COOH
DCC DMAP
反应过程:
将6.2g(0.02mol)4-十一烯酰氧基苯甲酸投入100mL三口烧瓶中,加入40mL THF,搅拌至完全溶解;将4.2g(0.02mol)DCC和0.2g(5%)DMAP溶解在20mLTHF 中,滴加入三口烧瓶中,搅拌0.5h;将3.5g(0.025mol)溶解于20mL CH2Cl2中,搅拌溶解完全,加入三口烧瓶中;常温反应24h。反应完毕,加入10mL去离子水,搅拌10分钟;静置过滤,得到白色沉淀;反复过滤,至滤纸上无白色固体;用THF洗涤滤饼,合并滤液,无水MgSO4干燥,旋转蒸发除去大部分溶剂,将浓缩液倾倒入大量水中,过滤,热水洗2~3次,过滤,干燥,乙醇重结晶,得白色粉末。产率70%。
3)、4-(2-丙烯酰氧基乙氧基)苯甲酸对联苯酚单酯的合成
反应方程式:
CH2=CHCOO(CH2)2O COOH+OH
HO
CH2=CHCOO(CH2)2O COO OH 反应过程:
将14.9g(0.08mol)对联苯酚投入装有机械搅拌和冷凝管的250mL三口烧瓶中,加入100mLTHF,搅拌至溶解完全。将4.8g (0.02mol)4-(2-丙烯酰氧基乙氧基)苯甲酸用40mLTHF溶解,加入三口烧瓶中,搅拌混合均匀。将4.2g (0.02mol) DCC和0.2g DMAP溶解后缓慢滴入三口烧瓶中。滴加完毕,常温反应24h。反应过程用IR监测,羧酸峰完全消失,可视为反应进行完全。反应完毕后,加入20mL去离子水搅拌20min,然后反复过滤除尽不溶物,旋转蒸发除去大部分溶剂,将浓缩液倾倒入大量水中,析出大量白色固体,过滤,热水洗2~3遍,过滤,用2%氢氧化钠碱液反复洗2~3遍,然后水洗至滤液无色。将所得粗产品加入去离子水中,调节pH=5~6,过滤,丙酮热过滤,保留滤液,冷却,析出固体物质,抽滤干燥得白色固体。
四、总结
结合实验室的实际情况,酰氯酯化法对于部分羧酸成酯还是具有一定的优势,如丙烯酸同对羟基苯甲酸反应,制备丙烯酰氯只需4~5h,且不用减压蒸馏除去
SOCl2(丙烯酸过量)。但总体来说,Steglich酯化法具有非常明显的优势,反应条件温和,反应时间适中,后处理简单,产率高,产品纯度较高(有些产物甚至不用重结晶就能达到纯度要求)等等。
当然,方法总是在摸索中不断完善和改进的,本文仅仅抛砖引玉,相信经过大家的努力,能够将酯化反应的各种方法引入实验室,大大扩展我们的合成手段和合成策略。文中很多想法还不成熟,由于时间的关系,很多都来不及验证和改正,希望在以后的时间里,错误之处能得到纠正,正确之处能得到采纳。
参考文献
1孙晓红. 羧酸酯化催化剂的研究进展与展望[J].化学与粘合,1998,4:230~233. 2武钦佩,李善茂.保护基化学[M].北京:化学工业出版社,2007,210~220.
3高鸿宾,有机化学[M].北京:高等教育出版社,1982,377~380.
4马勇林,吕鉴泉,曹颜琦,胡文祥.酯的催化合成[J]. 化学与粘合,2001,1:29~33
5程定海,山桂云,李述燕.水杨酸甲酯的合成研究[J].西北师范大学学报,2004,25(4):434~436.
6宋秀美,汪朝阳,毛郑州,罗玉芬,赵海军.胆甾醇酯的合成研究进展[J].广州化学,2008,33(1):59~66.
7李佩瑾,赵春山,李佳.胆甾相液晶的合成[J].化学工程师, 2005, 19 (1): 57-58.
8黄征青.对-10-十一烯羰氧基苯甲酸胆甾醇酯的合成[J].沈阳化工学院学报,2001,15(3): 186~188.
9Mitsunobu,O. Synthesis 1981, 1.
10Mitsunobu,O.; Yamada, M. Bull. Chem. Soc. Jpn.1967,40,2380.
11Crich,D.;Dyker,H.; Harris,R. J.J. Org.Chem.1989,54,257 and references therein. 12Varasi,M.;Walker,K.A.M.;Maddox,https://www.doczj.com/doc/5b6146680.html,.Chem.1987,52, 4235 and references therein.
13Hughes,D.L.;Reamer, R.A.;Bergan, J.J.;Grabowski,E.J.J.J.Am.Chem.Soc. 1988, 110, 6487 and references.
14(a) A. Hasner, V. Alexanian, Tetrahedron Lett. 1978, 46, 4475; (b) E. Haslam, Tetrahedron, 1980, 36, 2409; (c) A Williams, T. Ibranhim, Chem. Rev. 1981, 589;
嘧菌酯中间体合成新工艺及市场分析(下) 李强雷青菊 摘要:介绍了嘧菌酯原药市场行情及国内主流生产商,以及新中间体的合成工艺技术。 关键词:中间体合成工艺嘧菌酯市场分析杀菌剂 全球销量最大的杀菌剂嘧菌酯,2014年的销售额突破15亿美元,且需求量以15%左右的增幅持续增长。嘧菌酯原药及复配制剂均已过专利期,国内已有大量厂家进行生产。目前,国内原药产量约6000吨,其中88%销往国外。2016年,全球嘧菌酯原药产量有望突破1万吨。 一、工艺简介 目前国内现有和正在建设中的装置,合成技术都采用邻羟基苯乙酸、4,6-二羟基嘧啶、水杨酰胺为原料经过七步反应合成嘧菌酯,中间涉及20多种化学品,其中包括大量酸、碱和有机溶剂,反应及溶剂回收采用间歇操作,每吨嘧菌酯原药产生约50吨的废水。 二、嘧菌酯进入快速放量期 以甲氧基丙烯酸酯类杀菌剂王牌—嘧菌酯为代表的新一代杀菌剂,逐步显现出未来强劲的机会。通过研究某些已实现转移专利的杀菌剂进程来看,其过程可以分为三个阶段:首先是专利到期价格下降,经济劣势扭转; 产品逐渐被接受;第二阶段:中间体在国内逐步扩能,巨头(巴斯夫等)开始小规模的产能转移;相关上游产业链受益;第三阶段:价格企稳,大范围产能转移开始;国内厂商开始受益。目前,嘧菌酯刚刚进入第一阶段中期,即将迎来它的“快速放量期”。 嘧菌酯具有极其广泛的杀菌普,对几乎所有的真菌病害如白粉病,、锈
病、黑星病、霜霉病、稻瘟病等都有极好的活性,一般在保护性处理或病害发生早期使用,对作物种植影响甚微。 嘧菌酯可治疗作物疾病汇总 高效的杀菌效果与优异的经济效益是其胜出的核心因素。嘧菌酯具有良好的理化性质,能够制成各种制剂,包括:可湿性粉剂、水分散粒剂、悬浮剂等等。相关的田间药效试验表明在对马铃薯早疫病的测试中,其治疗效果优于代森锰锌;对黄瓜褐斑病的测试中显著优于百菌清。 三、嘧菌酯生产量情况 嘧菌酯产能集中度高,国内企业刚刚起步。目前全球嘧菌酯生产主要集中在先正达公司,总产能在8000吨。嘧菌酯2010年专利到期后,国内掀起了登记热潮,原药生产企业登记的有33家,但真正能够实现大规模生产的企业少之又少。 我国嘧菌酯主要生产企业 国内生产的嘧菌酯原药主要用于出口,出口国家包括乌拉圭、南非、
农 药 AGROCHEMICALS 第53卷第7期2014年7月Vol. 53, No. 7Jul. 2014 氟嘧菌酯合成方法述评 武恩明,孙 克,张敏恒 (沈阳化工研究院有限公司 新农药创制与开发国家重点实验室, 沈阳 110021) 摘要:总结了文献报道的氟嘧菌酯及其关键中间体的工艺路线和合成方法。 已知的氟嘧菌酯工艺合成路线 有2条,对涉及到的关键中间体的合成路线和合成方法进行了总结。 关键词:氟嘧菌酯;工艺路线;合成方法中图分类号:TQ450 文献标志码:A 文章编号:1006-0413(2014)07-0537-05 A Review of Synthetic Methods of Fluoxastrobin WU En-ming, SUN Ke, ZHANG Min-heng (State Key Laboratory of the Discovery and Development of Novel Pesticide, Shenyang Research Institute of Chemical Industry Co., Ltd., Shenyang 110021, China) Abstract: The process routes and synthetic methods of ? uoxastrobin and its intermediates reported in literatures were summarized. There are 2 main processes of ? uoxastrobin in prior art, the processes and synthetic methods of the key intermediates involved were also reported. Key words: Key words: ? uoxastrobin; process route; synthetic method 氟嘧菌酯(fluoxastrobin)是拜耳公司2004年开发上市的甲氧基丙烯酸酯类杀菌剂,该药对几乎所有真菌纲病害如锈病、颖祜病、网斑病、白粉病、霜霉病等数十种病害均有很好的活性,广泛应用于禾谷类作物、马铃薯、蔬菜和咖啡等作物。 与其他甲氧基丙烯酸酯类杀菌剂作用机理一样,氟嘧菌酯也是线粒体呼吸抑制剂。 2004年氟嘧菌酯首先在英国入市,2005年在荷兰登记上市,随后2006年在法国、2007年在墨西哥获准登记,2008年获得欧盟批准,2009年进入意大利市场,2011年进入阿根廷市场。 根据与拜耳公司协议,爱利思达生命科学公司在全球市场对氟嘧菌酯进行了进一步开发,2010年在美国获准登记,2012年在加拿大上市。 氟嘧菌酯入市后市场增长较快,2012年销售额达到1.65亿美元。 1 工艺路线 1.1 氟嘧菌酯工艺合成路线 已知的氟嘧菌酯工艺合成路线有2条,具体如下。路线1: 以关键中间体3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2 -二 嗪为原料,先与4,5,6-三氟嘧啶醚化后再与邻氯苯酚反应得到氟嘧菌酯[1-2]。 路线2: 4,5,6-三氟嘧啶先与邻氯苯酚醚化,然后再和中间体3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2-二 嗪醚化得到氟嘧菌酯[2]。 收稿日期:2014-05-17 作者简介:武恩明(1981—),男,工程师,硕士,主要从事农药合成研究。 E-mail :wuenming@https://www.doczj.com/doc/5b6146680.html, 。 1.2 中间体3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲 基]-5,6-二氢-1,4,2-二嗪的合成 氟嘧菌酯的合成存在3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2-二嗪和4,5,6-三氟嘧啶2个关键中间体,其中间体3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2-二嗪存在以下5种制备方法。 方法1: 以邻羟基苯乙酸甲酯为起始原料,先与3,4-二氢吡喃加成得到中间体2-四氢吡喃氧基苯乙酸甲酯,然后在叔丁醇中与叔丁醇钾和叔丁腈反应,得到的中间体在盐酸羟胺和1,2-二溴乙烷的作用下合环、离子交换树脂去保护后得到关键中间体[3]。 武恩明, 孙克, 张敏恒. 氟嘧菌酯合成方法述评[J]. 农药, 2014, 53(7): 537-541.
Synthesis of p -substituted tetraphenylporphyrins and corresponding ferric complexes with mixed-solvents method Zhicheng SUN 1,Yuanbin SHE (?)1,Rugang ZHONG 2 1Institute of Green Chemistry and Fine Chemicals,Beijing University of Technology,Beijing 100124,China 2College of Life Science &Bioengineering,Beijing University of Technology,Beijing 100124,China ?Higher Education Press and Springer-Verlag 2009 Abstract By using mixed-solvents method,?ve kinds of p -substituted tetraphenylporphyrin compounds [T(p -R)PPH 2,R =NO 2,Cl,CH 3,OCH 3,OH]were synthesized by the condensation of p -substituted benzaldehyde with pyrrole in mixed solvents (propionic acid,acetic acid and nitrobenzene),and corresponding ferric complexes [T(p -R)PPFe III Cl]were synthesized in dimethylformamide.The above free base porphyrins were obtained in 30%–50%yields,metalation yields were up to 90%and total yields of ferric complexes were 27%–50%.Effects of reactive conditions,solvents and oxidants on yields of free base porphyrins were investigated and the relevant mechanism was discussed.Structures of the above porphyrin complexes were characterized by ultraviolet-visible (UV-Vis),infrared (IR)and far infrared (FIR)spectroscopy. Keywords porphyrin,metalloporphyrin,mixed-solvents,synthesis,characterization 1Introduction Substituted tetraphenylporphyrin complexes with conju-gated macrocycles have been essential to the study of biomimetic chemistry in recent years [1–5].The porphyrin iron complexes are mostly used for the models of cytochrome P-450in which the dioxygen has been activated by metalloporphyrins under mild conditions [6,7].Based on that,the substituted metalloporphyrins present high catalytic activities and high selectivities in the catalytic oxidation of hydrocarbons without co-reducing reagents.So the catalytic effect of metalloporphyrins on the activity of inert C-H bonds has been given considerable attention [8]. However,the yields of substituted tetraphenylporphyrin complexes are lower and the cost of synthesis is still expensive,which have exceedingly restricted their current applications.Herein,the study on ef ?cient synthesis methods for improving the yields of metalloporphyrin complexes is obviously necessary. Chemists have developed a few synthetic methods to provide convenient access to synthesize substituent tetraphenylporphyrin complexes [9–11].The prevalent method of synthesis involves a mixed aldehyde condensa-tion with pyrrole via Adler method in re ?uxing propionic acid [12].Nevertheless,several limitations remain on the scope of synthetic porphyrin chemistry.One of these is the synthesis of porphyrins with only one solvent, e.g.,propionic acid or dimethylformamide,which brings the problems of a higher boiling point and inconsistent polarity [13].Therefore,the porphyrin complexes are often with low yields and the synthetic method is not universal for porphyrin complexes with various substituents. In this paper,a series of para -substituted tetraphenyl-porphyrin compounds and the ferric complexes [T(p -R)PPFe III Cl]were synthesized by using mixed-solvents method (Scheme 1).Different reaction conditions were investigated and the yields of porphyrin complexes were improved remarkably.This approach proved to be effective for the synthesis of a varity of metalloporphyrins. 2 Experimental 2.1 Reagent and instrument All chemicals were obtained commercially and used as received unless otherwise noted.Pyrrole was redistilled before use.Dichloromethane was dehydrated.Neutral Al 2O 3was baked at 100°C for 5h. Ultraviolet-visible (UV-Vis)spectra were obtained on HITACHI U-3010.Infrared (IR)spectra were obtained on Received September 18,2008;accepted November 10,2008E-mail:sheyb@https://www.doczj.com/doc/5b6146680.html, Front.Chem.Eng.China 2009,3(4):457–461DOI 10.1007/s11705-009-0169-6
提示:以下所有论文,将鼠标移至想要查看的标题上,按住Ctrl键并单击鼠标左键即可打开查看详细内容。也可咨询QQ:893628136 1. 电镀污泥中铜和镍的回收(字数:11869,页数:17 ) 2. 电渗析染料脱盐(字数:8183,页数:21 ) 3. 具有PULL-UP效应的合成革涂层配方及效应蜡的优化研究(字数:16506,页数:29 ) 4. 年产1500吨O,O—二乙氧基硫代磷酰氯车间设计(字数:12242,页数:34 ) 5. 年产1800吨氯乙酸的生产工艺设计(字数:10255,页数:32 ) 6. 汽液平衡测定及一致性模型的研究(字数:12042,页数:31 ) 7. 染料脱盐实验研究(字数:9594,页数:21 ) 8. 浙江万盛化工污水处理站设计(字数:11349,页数:27 ) 9. 异常数据的诊断、处理及化工应用(字数:12365,页数:32 ) 10. SBR法处理啤酒废水试验研究(字数:13062,页数:36 ) 11. 半水煤气生产过程计算及材料数据库程序编制(字数:8752,页数:28 ) 12. 吡硫醇锌检测方法的建立(字数:9522,页数:23 ) 13. 波长选择方法及其在近红外光谱数据中的应用(字数:20263,页数:37 ) 14. 长效防雾滴农膜的研制(字数:15278,页数:34 ) 15. 从杨梅叶中提取黄酮的研究(字数:10894,页数:24 ) 16. 催化合成水杨酸正戊酯的研究(字数:9079,页数:23 ) 17. 活性染料印花用增稠剂的研究(字数:11097,页数:23 ) 18. 婴幼儿谷基配方米粉酶法水解工艺研究(字数:5401,页数:16 ) 19. 含三催化中心手性配体的合成(字数:11985,页数:24 ) 20. 二步法合成梳形混凝土超塑化剂的研究(字数:12322,页数:24 ) 21. 茯苓多糖的氨基和羧甲基化研究(字数:10994,页数:23 ) 22. 活性蓝染料脱色研究(字数:19508,页数:37 ) 23. 梳形混凝土超塑化剂的合成(字数:12805,页数:24 ) 24. 油酸聚乙二醇加脂剂的制备工艺研究(字数:8328,页数:23 ) 25. 西他列汀中间体的合成工艺研究(字数:10460,页数:25 ) 26. 二步法合成聚羧酸盐高效减水剂的研究(字数:11802,页数:25 ) 27. 羟乙膦酸钠合成工艺的研究(字数:8890,页数:20 ) 28. 固相微萃取吸附水中有机污染物的研究(字数:8831,页数:22 ) 29. 有机硅阳离子乳液的制备及优化研究(字数:10447,页数:20 ) 30. 微波催化酯化反应SVM模型的优化(字数:10847,页数:24 ) 31. 三氟乙酰乙酸乙酯的制备(字数:12288,页数:26 ) 32. 基于判别分析的化工过程故障诊断方法及应用示例(字数:12586,页数:27 ) 33. 亚硫酸化蓖麻油的制备及其皮革加脂性能研究(字数:11946,页数:28 ) 34. 耐溶剂型水性聚氨酯的合成及其性能研究(字数:9592,页数:25 ) 35. 化物吸收耦合生物还原去除烟气中氮氧化物:电子穿梭体强化Fe(III)EDTA还原(字数:7180,页数:20 ) 36. 山药多糖提取工艺正交优化(字数:11265,页数:23 ) 37. 年产400吨亚氨基二苄甲酰氯的工艺设计(字数:12729,页数:40 ) 38. 污泥制取活性炭(字数:10615,页数:25 ) 39. 气相色谱-质谱法检测食品中多氯联苯的研究(字数:10726,页数:31 ) 40. 高维数据降维方法及化工应用(字数:12219,页数:30 ) 41. 响应面法优化菜籽油甾醇提取(字数:12991,页数:30 ) 42. 聚合物载药缓释微球药物释放动力学模拟(字数:10465,页数:24 ) 43. 水性聚氨酯皮革涂饰剂的合成及耐溶剂性能研究(字数:8865,页数:22 ) 44. 高维数据降维与建模在过程中的应用(字数:20751,页数:55 ) 45. 含二催化中心手性氨基醇配体的合成及其纯化(字数:12407,页数:27 ) 46. 溶胶凝胶制备纳米光催化剂及其应用的研究(字数:15784,页数:33 ) 47. 用半连续反应器和RAFT技术合成可控梯度聚合物(字数:11056,页数:31 ) 48. 水热合成法丙烷选择氧化催化剂的研究(字数:10815,页数:30 )
嘧菌酯生产方法简介 (八步法) 1、 氰化 在催化剂作用下,邻氯氯苄与氰化钠反应,生成邻氯氰苄 Cl CH 2Cl +NaCN Cl CH 2CN +NaCl 161 151.5 催A 2、 常压水解 邻氯氰苄在酸性条件下,常压水解生成邻氯苯乙酸 Cl CH 2CN +H 2O Cl CH 2COOH +NH 3 151.5 硫酸 170.5 2 3、 高压水解 在催化剂作用下,邻氯苯乙酸高压水解,生成邻羟基苯乙酸 +H 2O OH CH 2COOH +NaCl 催B 152 Cl CH 2COOH 170.5 NaOH 4、 闭环 在催化剂作用下,邻羟基苯乙酸分子内脱水,生成内酯 OH CH 2COOH +H 2O 催C 152 O O 134 5、 甲氧烯基化 以醋酸酐为溶剂,内酯与原甲酸三甲酯反应,生成苯并二醇
+HC(OCH 3)3 O O 134 +CH 3OH O O OCH 3 176 醋酐 6、 开环 苯并二醇与甲醇钠、二氯嘧啶反应,生成Z 型中间体 +CH 3ONa O O OCH 3 176 +N N Cl Cl 149+NaCl O N N Cl O O 320.5 7、 转位 在催化剂作用下,Z 型中间体高温转位成E 型中间体 催D O N N Cl O O Z 型320.5 高温 O N N Cl O O E 型320.5 8、 醚化 以碳酸钾作缚酸剂,在催化剂作用下,E 型中间体与水杨腈反应,生成嘧菌酯 催E +O N N Cl O O O E 型320.5 缚酸剂 O N N O O O O CN HO 119 嘧菌酯403.4
聚四氟乙烯合成方法 ——乳液合成法 此法属自由基聚合反应。在此法中. 使用的单体为气体四氟乙烯。具体方式如下:先向一个压力容器中加入一定量的水, 然后将自由基引发剂、乳化剂、 pH 值调节剂以及一些其它必要试剂以一定的顺序加入其中, 再将气体四氟乙烯单体通入反应器发生反应, 生成聚四氟乙烯颗粒。所用的表面活性剂一般为氟化型, 而引发剂一般使用水溶性过硫酸盐:但使用水溶性过硫酸盐作为引发剂时应注意一个原则:反应温度高于 50℃时,只单独使用此引发剂可以了;当温度在 5~ 50℃之间时, 需再加入一些还原剂, 如铁盐、硝酸盐和二硫酸钠等。此法所得的聚四氟乙烯颗粒尺寸一般较大。如 Bladel[3】合成的聚四氟乙烯颗粒尺寸在 50~150 nm 范围内, 平均粒子直径为 100 nm。因乳液合成法所获得的粒子一般是悬浮在溶液中, 此聚合过程并不是一个真正意义上的液相聚合反应, 有时把它称作悬浮聚合反应。乳化聚合反应具有高转化率、高反应速率以及可获得高分子量的聚四氟乙烯颗粒的优点。 膨胀聚四氟乙烯成型工艺 膨胀聚四氟乙烯的成型分两个阶段。第一阶段将 PTFE 散树脂与润滑助剂按 一定比例混合。放置一定时间后预成型, 然后将糊膏挤压成纵向排列纤维状的预成型品经干燥去除助剂; 第二阶段在低于聚四氟乙烯熔点的温度下进行高速拉伸, 再在高于熔点的温度下对处于拉伸状态的聚四氟乙烯半成品进行热定形,即可得到膨胀聚四氟乙烯制品。其工艺流程如下: (1混料将聚四氟乙烯树脂与助挤剂按一定质量比例, 混合均匀。选用分散树脂,它有良好的成纤性,粒子问的凝聚力低,分子链受到很小的剪切作用就会沿粒子长轴方向排列, 形成线形结晶。加入助挤剂可以增加颗粒问的粘连, 降低树脂颗粒间及树脂与容器间的摩擦力,提高加工性能。助挤剂通常可用石油醚、甲苯、丙酮、煤油、石蜡等。 (2预成型将混合料压制成与推压机膜腔相同形状的坯体。
【摘要】本文主要介绍了利用一种用1,4-二甲氧基苯作为反应的起始原料,用氢气作为还原剂,在金属钯复合催化剂的作用下反应直接生成产物对苯二酚。此工艺简单方便易行,副产物少,反应条件相对比较温和。本文对反应的催化剂的种类进行了帅选并且对催化剂的用量、反应温度、反应压力和反应时间进行了优化,最终优化的结果可以使得对苯二酚的产率达到90%。 【关键词】 1,4-二甲氧基苯对苯二酚氢气 对苯二酚是一个重要的有机化工原料,用途非常广泛。酚主要用于制取黑白显影剂、蒽醌染料和偶氮染料、合成气脱硫工艺的催化剂、橡胶和塑料的防老剂单体阻聚剂、食品及涂料清漆、橡胶和汽油的稳定剂和抗氧化剂、石油抗凝剂、洗涤剂的缓蚀剂、稳定剂和抗氧剂等,还用于化妆品的染发剂。 目前世界上生产对苯二酚的方法主要分为以下四种(1)苯胺氧化法;(2)对二异丙苯氧化法;(3)苯酚丙酮法;(4)苯酚羟基化法。 路线1:苯胺氧化法。 目前我国大部分生产厂家仍沿用苯胺氧化法,这是对苯二酚最早的生产方法,至今已有70多年的历史。该法反应过程为:在硫酸中(将138g的1,4-二甲氧基苯和5%不同的催化剂加入烧瓶中,往体系中加入氢气,在压力10mpa和120℃的温度下反应,取样分析对苯二酚的产率。结果如表3所示。 从上表可以看出一共四种催化剂,pd/sio2-al2o3和pd/al2o3的催化效果基本上没有什么差别,分别为81%和80%,但是在产率上都低于催化剂pd/caco3和pd/deloxan apii。pd/caco3 和pd/deloxan apii的催化效果都非常好。下面对催化剂的用量进行了一些实验,结果如表4所示。 从实验结果看出,随着催化剂用量的增加,产率得到了提高,但当用量达到5%的时候,再增加用量,产率基本上没有变化,使用6%pd/caco3为催化剂的产品最终产率为96%,使用6%pd/deloxan apii为催化剂的最终产率为93%。 2.4 反应时间对反应的影响 将138g的1,4-二甲氧基苯和5% pd/caco3催化剂加入烧瓶中,往体系中加入氢气,在压力10mpa和120℃的温度下反应,取样分析对苯二酚的产率,研究反应时间对产率的影响。结果如表5所示。 从上表可以看出反应时间在2小时以下,随着时间的推移对苯二酚的产率渐渐的提高,当反应时间大于2小时的时候,对苯二酚的产率基本没有什么变化,所以反应时间规定在2小时。 3 结语 本文比较了不同种类的催化剂对此反应的影响,确定以pd/caco3或pd/deloxan apii 为反应的催化剂,并且经过对pd/caco3和pd/deloxan apii的用量进行对比实验,确定pd/caco3 和pd/deloxan apii的用量比为5%,并对温度、压力和反应时间进行了对比。最终确定最佳工艺条件为:1. pd/caco3的用量比为5%;2.反应的温度为120℃;3.反应的压力为10mpa;4.反应的时间2小时。
步骤缺点备注 Rothemunde 法以荃类和吡咯为原料,以吡啶 和甲醇为溶剂。在封口的玻璃 管中反应,水浴90—95度下 反应30个小时。将反应液降 温后过滤,以吡啶洗涤反应管 和虑饼,合成虑液,再以百分 之五十乙酸萃取两次。最后将 醚液用饱和NAHSO3萃取三 次后,水洗至中性 反应时间长,反应条件苛刻,且要 求反应器密闭,底物浓度较低,后 处理非常麻烦,反应收率低 Adler-longo 法苯甲醛和新蒸的吡咯在丙酸 中回流30min。冷却至室温后 过滤,然后分别用甲醇和热水 洗涤滤饼,得到蓝紫色晶体, 最后真空干燥。 由于反应条件的限制,一些带敏感 基团或对酸敏感的取代苯甲醛不 能用作原料,同时带有强吸电基的 苯甲醛进行合成时产率特别低,而 且由于底物浓度大以及反应的温 度高,在反应过程中容易长生大量 的焦油,产物不容易纯化。 Lindsey法在室温下采用苯甲醛和吡咯 为原料,在氮气保护下,以二 氮甲烷为溶剂,三氟化硼乙醚 络合物为催化剂,生成卟啉 原,然后以二氯二氰基苯醌将 四苯基卟啉原氧化得到最终 产物四苯基卟啉,收率可达 20—30 优点:反应条件温和,不会产生焦油状的副产物,且产率较高,适合合成带有敏感基团或是空间位阻较大的卟啉。 缺点:此反应只能在比较稀的溶液中进行,且反应步骤相对较多。不仅原料较为昂贵,且反应过程需要无水及无氧操作 [2+2]法利用两分子的二吡咯甲烷缩 合成卟啉优点:可以方便的合成出各种带有不同取代基的不对称的卟啉,且产率比较高,具有较强的灵活性和区域选择性 缺点:合成过程中消耗会比较大且这类反应要在酸性条件下催化进行,而在该条件下容易使得二吡咯甲烷裂解,从而不利于反应的进行。同时,吡咯也容易进行自身缩合反应,且缩合产物难于分离。 微波激励法将吡咯和苯甲醛附于无机载 体硅胶上,利用载体的酸性催 化作用,在微波激励下合成四 苯基卟啉,反应10min后, 直接加入层吸柱进行吸分离, 得到四苯基卟啉,收率百分之 9.5 以二甲苯为溶剂,对硝基苯甲酸为催化剂,使苯甲醛吡咯在微波炉中反应20min,收率可达到百分之42.
第18卷第4期 佛山科学技术学院学报(自然科学版) Vol.18No.4 2000年12月 Journal of Foshan U niv ersity(Natural Science Edition)Dec.2000 文章编号:1008-0171(2000)04-0047-03 乙酸异戊酯的合成研究 关共凑,黄耀威,朱建辉 (佛山科学技术学院旅游与地理系,广东佛山528000) 摘要:研究了乙酸异戊酯的合成条件及工艺过程。用固体酸对氨基苯磺酸作催化剂,以异戊醇和冰乙酸直接酯化合成乙酸异戊酯。产物后处理简单,收率80%,催化剂可重复多次使用。 关键词:乙酸异戊酯;乙酸;异戊醇;对氨基苯磺酸 中图分类号:T Q413 文献标识码:A 乙酸异戊酯是重要的溶剂,也广泛用作食用果实香精。在我国,其合成主要是硫酸催化法[1]。近期有报道杂多酸催化,固体氯化物催化的方法[2],上述方法均存在着物料配比高,催化剂不能回收,废液污染环境,对设备腐蚀性强等缺点。采用对氨基苯磺酸作催化剂合成乙酸异戊酯[3],该反应为典型的可逆反应,随着反应的进行,产物浓度逐渐增大,逆向反应的趋势也逐渐加大,为促使反应向右进行,所以加入带水剂环已烷将生成的水从反应体系中分去。而有报道则认为醇、酯均能与水形成二元或三元共沸物,不需加入带水剂[4]。为了探讨清楚带水剂对实验的影响,本文对有无带水剂均进行实验比较。 1 实验方法 主要仪器有2WAJ型阿贝折射仪、Nicolet FT-IR550型红外光谱仪等。在装有回流冷凝管的圆底烧瓶中加入催化剂对氨基苯磺酸、异戊醇、冰醋酸和环已烷,装上分水器,加热回流反应至分水器中水层体积不再增加,此时反应已完成,停止加热。冷却后将反应物倒至分液漏斗,加入饱和NaHCO3,除去过量的乙酸,加无水M gSO4,干燥后,蒸馏收集136~142℃馏分,催化剂成固体沉结于反应瓶底,可重复使用。 2 结果与讨论 2.1 反应物配比对酯收率的影响 反应物乙酸与异戊醇的摩尔比对酯收率有一定的影响。实验反应条件均为对氨基苯磺 收稿日期:2000-09-16 作者简介:关共凑(1969-),女,广东开平人,佛山科学技术学院旅游地理系助理实验师,主要从事化学实验研究。
CoP-CMP的合成: 在50ml三口烧瓶中加入158.5mg对溴苯基钴卟啉、63.6mg1,4-苯二硼酸、 K 2CO 3 (221.1mg)溶液6ml、1,4-二氧六环30ml、Pd(PPh3) 4 18.5mg。冷冻到-70℃ 后通过三次15min-泵抽15min-通氮气-解冻除氧。然后在90℃下反应24小时。反应24小时后过滤,用去离子水、无水乙醇、THF、丙酮洗涤至滤液无色,再用甲醇、THF、丙酮索氏提取。最终得到0.1302g棕色粉末。 产物中的白色杂质比上次做的少很多,白色杂质可能是三口烧瓶的玻璃被碱腐蚀掉进产物里。由于对溴苯基钴卟啉在溶剂里溶解度低,产物很难洗干净。 对四硝基苯基卟啉(TNPP)的合成: 方法一:在装有冷凝管的250ml三口烧瓶中加入3.67g(24mmol)对硝基苯甲醛、3.9ml(41mmol)醋酸酐和100ml丙酸,加热至回流。10min内滴加1.6102g (48mmol)吡咯(溶于3ml丙酸),然后继续反应30min。反应混合物冷却至室温后过滤,得到的黑色固体用去离子水洗涤后40℃下真空干燥。得到的黑色固体里含有大量聚合物,这些聚合物易溶解于吡啶,而产物在冷的吡啶溶液里溶解度低。把黑色固体在50ml吡啶中回流1小时,冷却至室温后放在冰箱里过夜。混合物过滤后用丙酮洗涤固体至滤液无色,最后干燥得到0.8034g紫色产物。产率16.85%。 方法二:在装有冷凝管的100ml三口烧瓶中加入4g对硝基苯甲醛、7ml乳酸和25ml硝基苯,加热至130℃。20min内滴加12ml溶有1.71g吡咯的硝基苯溶液后继续反应2个小时。然后在60℃下加入20ml甲醇,搅拌30min。把反应物放在冰箱里过夜后抽滤得到紫黑色固体。然后把紫黑色固体在10ml吡啶中回流1个小时后放在冰箱里过夜。最后抽滤、用丙酮洗涤固体至滤液无色,最后干燥得到0.9735g紫色产物。产率18.5%。 UV-Vis(λ;nm;CHCl 3 溶剂):422,514,550,590,650。 对四氨基苯基卟啉(TAPP)的合成: 氮气保护下在装有冷凝管的250ml三口烧瓶中加入1.44g(108mmol)对四硝基苯基卟啉和HCl(12mol/l,70ml)。在75-80℃下加入14ml溶解有13.04 g SnCl 2·2H 2 O (58mmol)的浓盐酸至三口烧瓶中反应30min。然后在冰浴条件下加 入80ml浓氨水终止反应,在空气条件下继续搅拌1h。过滤收集固体,加入到140ml 氢氧化钠溶液(2%),剧烈搅拌30min。过滤收集固体产物,用水洗涤后干燥。再用氯仿索氏提取,收集滤液,旋转蒸发得到对四氨基苯基卟啉。 对四硝基苯基钴卟啉(TNPPCo)的合成: 氮气保护下在装有冷凝管的100ml三口烧瓶中加入200mg T(P-NO 2)PPH 2 和 100mlDMF,加热至回流后在1h内分4次加入300mg(1.5mmol)CoCl 2·6H 2 O。继 续反应1h。溶液由紫色逐渐变成红黑色,TNPP在DMF里溶解度差,上金属后溶解度变大。蒸馏回收50mlDMF。然后用冰水冷却,加入50ml去离子水,过滤收集产物。用去离子水反复洗涤后用少量乙醇洗涤。干燥后得到0.2050g产物。
氟嘧菌酯的合成方法 路线一合成路线: 以关键中间体3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2-二嗪为原料,先与4,5,6-三氟嘧啶醚化后再与邻氯苯酚反应得到氟嘧菌酯 路线一合成方法: 在0 ℃下将含47.2 g(0.2 mol)3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2-二嗪的1 L四氢呋喃溶液依次用29.3 g(0.22 mol)4,5,6-三氟嘧啶和6.0 g (0.2 mol)氢化钠(80%的植物油悬浮液)进行处理,反应物少量多次加入。反应混合物在0 ℃下继续搅拌3 h,随后不再进行冷却,继续搅拌过夜。残留物用乙酸乙酯消化并用水反复洗涤。有机相用硫酸钠干燥并减压浓缩,得到黏稠油状物,该油状物可缓慢形成结晶。得到熔点为98 ℃的68.7 g(理论值的98%) 3-{1-[2-(4,5-二氟嘧啶-6-基氧基)-苯基]-1-(甲氧亚氨基)-甲基}-5,6-二氢-1,4,2-二嗪。将124.1 g(0.333 mol) 3-{1-[2-(4,5-二氟嘧啶-6-基氧基)苯基]-1-(甲氧亚氨基)-甲基}-5,6-二氢-1,4,2-二嗪、42.8 g(0.333 mol)邻氯苯酚、46 g(0.333 mol)碳酸钾和3.3 g氯化亚铜于1 L二甲基甲酰胺的混合液中加热到100 ℃过夜;减压脱溶后冷却到20 ℃,加入乙酸乙酯和水,分出有机层,干燥、过滤、减压脱溶后得到148.2 g(97%)目的物。 路线二合成路线: 4,5,6-三氟嘧啶先与邻氯苯酚醚化,然后再和中间体3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2-二嗪醚化得到氟嘧菌酯 路线二合成方法: 在20 ℃,将136.8 g(0.56 mo1) 4-(2-氯苯氧基)-5,6-二氟嘧啶加入135.3 g(0.56 mol) 3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2-二嗪和197.6 g碳酸钾于460 mL乙腈混合溶液中,加完后温度升至31 ℃。并在50 ℃搅拌反应6 h,然后室温搅拌反应过夜,将所得混合液倒人2.3 L冰水中,搅拌反应5 h,过滤、水洗、干燥得到260 g (97.8%)目的物,熔点为75 ℃。 中间体3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2-二嗪的合成 氟嘧菌酯的合成存在3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2-二嗪和4,5,6-三氟嘧啶2个关键中间体,其中间体3-[1-(2-羟基苯基)-1-(甲氧亚氨基)-甲基]-5,6-二氢-1,4,2-二嗪存在以下5种制备方法
(10)申请公布号 CN 102199127 A (43)申请公布日 2011.09.28C N 102199127 A *CN102199127A* (21)申请号 201010130608.8 (22)申请日 2010.03.24 C07D 239/52(2006.01) (71)申请人淄博万昌科技股份有限公司 地址255068 山东省淄博市张店区朝阳路 18号 (72)发明人高庆昌 (74)专利代理机构青岛发思特专利商标代理有 限公司 37212 代理人巩同海 (54)发明名称 一种制备嘧菌酯的方法 (57)摘要 本发明涉及嘧菌酯的制备方法,按照苯并呋 喃-2(3H)-酮∶催化剂∶甲酸酯∶硫酸二甲酯= 1.0∶1.0~ 2.5∶1.0~10.0∶0.9~2.5的 比例,首先由苯并呋喃-2(3H)-酮与甲酸酯反应 生成中间体3-甲酰基苯并呋喃-2(3H)-酮,再使 用硫酸二甲酯甲氧基化,生成3-(α-甲氧基)-亚 甲基苯并呋喃-2(3H)-酮,后者再与4,6-二氯嘧 啶和2-氰基苯酚反应,生成嘧菌酯。本发明反应 工艺操作简单,原料成本低,“三废”生成少,环境 污染小,收率高。(51)Int.Cl. (19)中华人民共和国国家知识产权局(12)发明专利申请 权利要求书 1 页 说明书 4 页
1.一种制备嘧菌酯的方法,其特征在于首先由苯并呋喃-2(3H)-酮与甲酸酯在催化剂存在下进行甲酰化反应,生成3-甲酰基苯并呋喃-2(3H)-酮,后者与硫酸二甲酯进行甲氧化反应生成3-(α-甲氧基)-亚甲基苯并呋喃-2(3H)-酮,再与4,6-二氯嘧啶和2-氰基苯酚进行反应,生成嘧菌酯,其中: 反应物料的摩尔比为:苯并呋喃-2(3H)-酮∶催化剂∶甲酸酯∶硫酸二甲酯=1.0∶1.0~2.5∶1.0~10.0∶0.9~2.5; 甲酰化反应温度-10~45℃,反应时间2~24小时; 甲氧化反应温度0~60℃,反应时间1~4小时。 2.根据权利要求1所述的制备嘧菌酯的方法,其特征在于所述甲酸酯为甲酸甲酯或甲酸乙酯。 3.根据权利要求1所述的制备嘧菌酯的方法,其特征在于所述催化剂为氢化钠或者醇的钠盐或者钾盐。 4.根据权利要求3所述的制备嘧菌酯的方法,其特征在于所述醇的钠盐或者钾盐为甲醇钠/甲醇钾、乙醇钠/乙醇钾、丙醇钠/丙醇钾、异丙醇钠/异丙醇钾、丁醇钠/丁醇钾或者叔丁醇钠/叔丁醇钾。
比拉斯汀的合成工艺研究 发表时间:2017-10-30T17:35:09.573Z 来源:《医药前沿》2017年10月第29期作者:徐连德1 徐琪琪2 徐英明2 [导读] 比拉斯汀(Bilastine),中文化学名为2-[4-(2-(4-(1-(2-乙氧基乙基)苯并咪唑-2-基)哌啶-1-基)乙基)苯基]-2-甲基丙酸。(1沂水县第一中学山东临沂 276405) (2山东罗欣药业集团股份有限公司山东临沂 276017) 【摘要】α,α-二甲基-4-(2-溴乙酰基)苯乙酸甲酯[2]经过还原反应制得α,α-二甲基-4-(2-溴乙基)苯乙酸甲酯[3],3与1-(2-乙氧基-乙基)-2-哌啶-4-基-1H-苯并咪唑[4]发生烷基化反应,再经水解得到比拉斯汀[1],总收率约76%。 【关键词】比拉斯汀;组胺H1受体拮抗剂;合成 【中图分类号】R976 【文献标识码】A 【文章编号】2095-1752(2017)29-0353-01 比拉斯汀(Bilastine),中文化学名为2-[4-(2-(4-(1-(2-乙氧基乙基)苯并咪唑-2-基)哌啶-1-基)乙基)苯基]-2-甲基丙酸,是西班牙FAES制药公司开发的第2代组胺H1受体拮抗剂,2012年欧盟批准其用于治疗变应性鼻炎及慢性特发性荨麻疹[1]。本品安全性良好,无常用抗组胺药物存在的镇静作用及心脏毒性,口服给药吸收迅速,具有良好的耐受性、安全性和较高的生物利用度[2]。 已有文献报道了1的合成路线[3-5]。本文选择以下路线α,α-二甲基-4-(2-溴乙酰基)苯乙酸甲酯[2]经过还原反应制得α,α-二甲基-4-(2-溴乙基)苯乙酸甲酯[3],3与1-(2-乙氧基-乙基)-2-哌啶-4-基-1H-苯并咪唑[4]发生烷基化反应,再经水解得到比拉斯汀[1],并进行了工艺优化。 文献[5]报道了由2制备3的过程,用三乙基硅烷-三氟乙酸进行还原,反应时间长达72h,收率91%。本研究通过调整三乙基硅烷-三氟乙酸的用量,控制回流反应温度,缩短了反应时间,收率90%。文献[5]由3制备1的过程中,3依次与2-(4-哌啶基)-1H-苯并咪唑和2-氯乙基乙醚发生亲核取代反应后,水解得比拉斯汀,反应步骤长,且操作繁琐,且3在与2-(4-哌啶基)-1H-苯并咪唑发生亲核取代反应时,咪唑环上的氮-氢不可避免的会与哌啶基上的氮-氢进行竞争,生成副产物,影响收率和纯度。本研究在文献基础上进行了改进,将3直接与4进行烷基化反应,再进行水解,一锅法制备1,方法工艺简单,操作简便,收率及产品纯度均有较大幅度提高,总收率为76%,适合工业化生产。 图1 1的合成路线 Fig.1 Synthetic Route of 1 1.实验部分 1.1 α,α-二甲基-4-(2-溴乙基)苯乙酸甲酯[3]的制备 冰浴冷却下分别向反应瓶内加入20mL二氯甲烷、α,α-二甲基-4-(2-溴乙酰基)苯乙酸甲酯(30.0g,100.7mmol,采用文献方法[5]制得,纯度99.1%)、三氟乙酸(36mL,484.6mmol)、三乙基硅烷(18mL,112.7mmol)。冰浴下搅拌30min后,升温至60℃回流反应20h。反应结束,滴加饱和碳酸溶液(约150ml),加入乙酸乙酯(100ml*2)萃取,有机相浓缩干燥,得无色油状物(25.7g,90%)(文献:91%[5])。ESI-MS,m/z(%):307[M+Na]+,283[M-H]+。元素分析:C13H17BrO2,实测值(计算值)%:C54.96(55.00);H6.02(6.04);Br28.11(28.14);O11.27(11.27)。 1.2 比拉斯汀[1]的合成 在反应瓶中加入3(99.36g,0.35mol)和4(82.01g,0.3mol,购自:江苏弘和药物研发有限公司,纯度98%),搅拌下加入10ml聚乙二醇-400和45ml水,在冰水浴的冷却下慢慢加入混合碱(0.25molNaOH+0.1molNa2CO3),于40℃下快速搅拌3.5小时后放置,使反应液冷却至室温,加入3N丁二酸溶液2.1L,加热回流24小时,用10%氢氧化钠水溶液调至pH=7,用乙醚(450ml*2)萃取,旋出溶剂,得到固体1(116.83g,84%),mp291~293℃(文献:295-296[5])。纯度为99.8% [HPLC归一化法:同文献[5]。ESI-MS,m/z(%):487[M+Na]+,463[M-H]+。元素分析:C28H37N3O3,实测值(计算值)%:C72.50(72.54);H8.01(8.04);N9.07(9.06); O10.38(10.35)。 【参考文献】 [1] Corc6stegui R,Labeaga L,Inneririty A,et a1.Preclinical pharmacology of bilastine,a new selective histamine Hl receptor antagonist:receptor selectivity and in vitro anti-histaminic activity[J].Drugs R D,2005,6(6):371-384. [2] Carter NJ.Bilastine:in allergic rhinitis and urticaria [J].Drugs,2012,72(9):1257-1269. [3] Lee CH,Khoo JH,Kwon KC,eta1.Process for preparation of 2-methyl-2-phenylpropionic acid derivatives and novel intermediate compounds:WO,2009102155[P].2009-02-12. [4]王蕾,李科,王倩,等.2-(4-卤乙基)苯基-2-甲基丙酸酯的制备方法及合成比拉斯汀的方法:中国,102675101[P].2012-09-19. [5]孔昊,耿海明,梅玉丹,等.比拉斯汀的合成[J].中国医药工业杂志,2015,46(7):677-679.