
为活细胞研究设计光学显微系统时,首要考虑的是检测器的敏感度(对信号乃至噪音的检测),图像获取的速度,以及在此基础上标本的可行性。对于固定细胞的成像,曝光时间及光强度相对来说都很高,这时可能会造成光漂白;然而对于活细胞成像,上述光的影响必须去除。几乎在所有情况下,活细胞显微镜都会在尽可能高的图像质量与尽可能好的细胞活性之间取得一个平衡。对于此类实验,时间及空间上的分辨率需要设定在能满足实验要求的水平上,而不是给予过度的光照或设定过多采样时间点。
基本上,一个理想的活细胞成像系统必需有足够的敏感度,来满足在弱荧光条件下仍能得到高图片质量;同时,系统也必需足够快,以记录整个动态过程。另外,这个系统还需要有足够高的分辨率以捕捉样品细节,并且能够准确的实时测量每个微小的光强变化。然而不幸的是,要改善上述的任意一条都需建立在牺牲其它性能的基础上。因此现在还不能够设计出一个可以满足所有要求的活细胞成像体系。研究人员现在只能在尽量减低不重要的信息的遗失的同时,尽可能的获得最优的重要参数。这样,显微镜的配置最终取决于成像的要求,对于样品在实验期间活性的要求,进行标记的难度水平,以及仪器的可用性等实际因素。
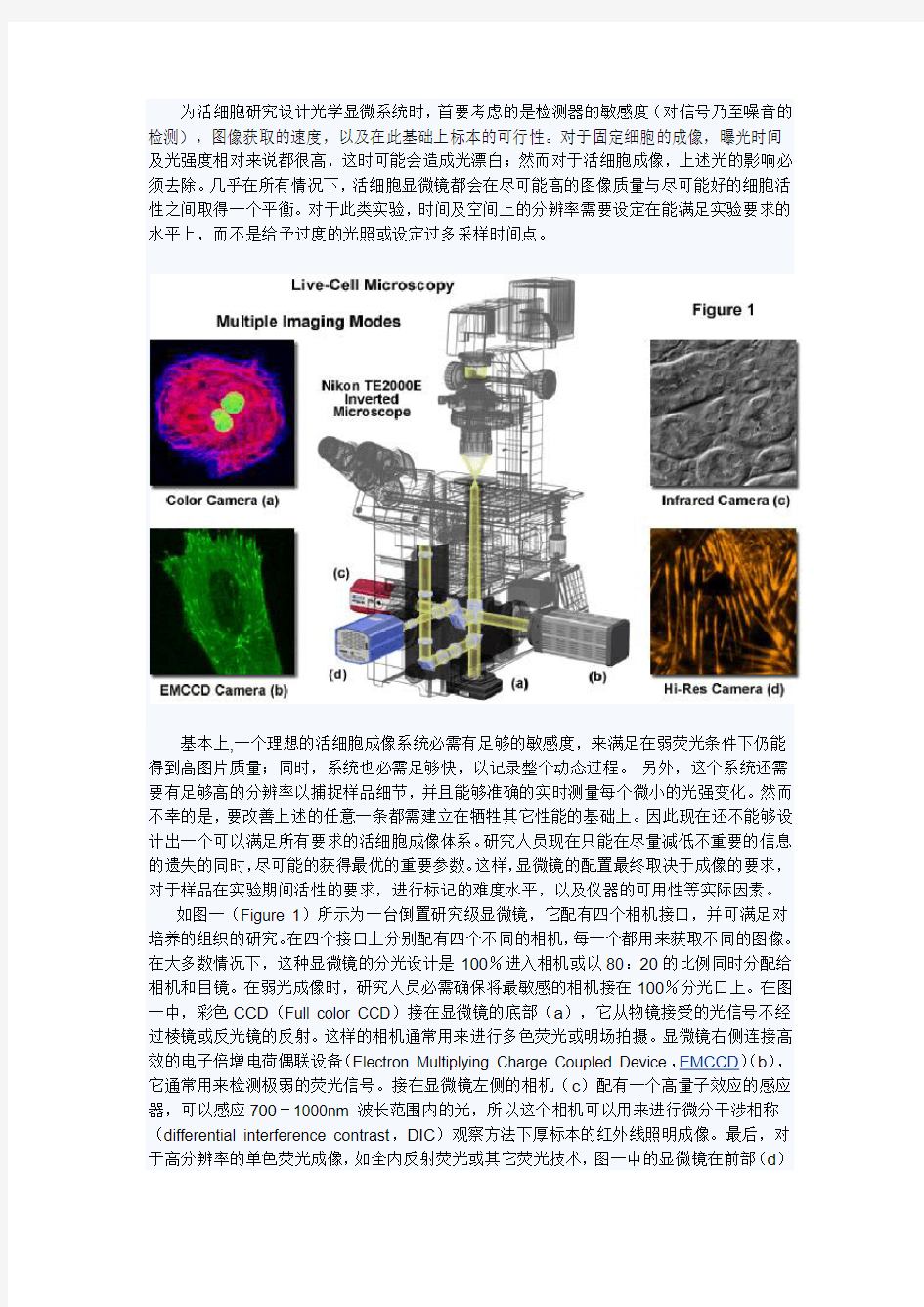
如图一(Figure 1)所示为一台倒置研究级显微镜,它配有四个相机接口,并可满足对培养的组织的研究。在四个接口上分别配有四个不同的相机,每一个都用来获取不同的图像。在大多数情况下,这种显微镜的分光设计是100%进入相机或以80:20的比例同时分配给相机和目镜。在弱光成像时,研究人员必需确保将最敏感的相机接在100%分光口上。在图一中,彩色CCD(Full color CCD)接在显微镜的底部(a),它从物镜接受的光信号不经过棱镜或反光镜的反射。这样的相机通常用来进行多色荧光或明场拍摄。显微镜右侧连接高效的电子倍增电荷偶联设备(Electron Multiplying Charge Coupled Device,EMCCD)(b),它通常用来检测极弱的荧光信号。接在显微镜左侧的相机(c)配有一个高量子效应的感应器,可以感应700-1000nm 波长范围内的光,所以这个相机可以用来进行微分干涉相称(differential interference contrast,DIC)观察方法下厚标本的红外线照明成像。最后,对于高分辨率的单色荧光成像,如全内反射荧光或其它荧光技术,图一中的显微镜在前部(d)
配备了Peltier-cool 相机。这个相机最小像素点为6μm,适合于此类成像的需求。如图一中的配置非常昂贵,但是它能满足用户所有的成像要求。
进行活细胞成像的光学显微镜需要宽光谱增称模块。大多数的研究者使用荧光显微镜,通常还会配合一种或多种透射光技术。其中荧光技术包括了传统的宽视场落射荧光,激光扫描共聚焦,真空碟片共聚焦,场扫描共聚焦,多光子,全内反射(TIRF),荧光关联光谱,荧光寿命成像(FILM),光活化,镭射陷阱等技术。相应的成像技术有光谱成像,多色成像,共定位,时间序列,荧光光漂白恢复,(FRAP,FLIP,FLAP),荧光共振能量转移(FRET),荧光散斑显微镜,膜片钳技术等,以上这些技术通常要配合一种或多种基础成像技术。当荧光与明场,DIC,霍夫曼调制相称(HMC),相差等技术相结合时,荧光技术作为探针,可用于定位.对于绝大多数的活细胞成像来说,显微镜的配置都需要满足一些特定的需要以
保证试验能够成功进行。除了显微镜与数字成像系统,活细胞成像还需要考虑到两个限制因素——一个是保持细胞的活性,另一个是在北京噪音及自发荧光的基础上尽可能的提高信号强度。
提高活细胞成像的信噪比
活细胞成像与固定细胞成像之间的基本区别是,后者的成像没有过多的限制,如合适视野的选定,曝光时间的设定,电子益增的设定,读出率及,偏移值等。这样,染色的固定样品的成像就可以充分利用相机的全部动态设定。因此,固定成像可以得到理想的信噪比。但不幸的是,由于细胞活性对照明的严格要求,活细胞成像的情况又与固定细胞的成像有所不同。在活细胞成像时,样品的灰度往往比背景高不了多少。在这种情况下,成像首要考虑的是检测系统的暗电流及噪音值。经济型的相机通常噪音都会比较高,导致背景混乱并掩盖样品信号,在读出速率增加时这种影响更为严重。所以,几乎所有的活细胞成像设备质量的衡量标准都是检测器的好坏。
数字图像的噪音主要由四方面造成,检测器,照明系统,Poisson 噪音或shot 噪音(由于光子通量的随机性造成),以及弥散光。在大多数情况下,通过选择好的检测器或优化照明条件可以降低系统噪音。事实上,任何一种光子检测器在记录的时候都会产生某种形式上的噪音。扫描光聚焦及多光子显微镜中的光电倍增管(PMT)会产生杂电子。同样的,用于明场,全内反射,针孔碟片共聚焦,场扫描显微镜的CCD也会生成背景暗电流。对此,CCD 及光电倍增设备也做了一系列的改进,比如制冷设备,它会使仪器工作时保持在低温状态,从而降低暗电流的水平。然而,在读取光信号并将模拟信号转换为数字信号的时候,特别是CCD这种仪器,也会产生噪音,即read noise,制冷数码相机产生的主要噪音。
Figure 2所示为影响活细胞成像质量的数字相机的读出速度与敏感度的相互依存关系。The specimen is a culture of human 图中样品为人的宫颈癌细胞(HeLa 细胞系),展示mKO (monomeric Kusabira Orange)荧光蛋白与人的α珠蛋白融合,图片在宽视场荧光下单色相机拍摄,照明中添加TRITC滤片,读出速度为10或1.25 MHz。Figures 2(a) & 2(b)所示,在关闭CCD 光闸的条件下,低读出速度下的噪音要明显高于高读出速度下的噪音。但是,如果将CCD 调节到全动态范围的话,这方面的影响就不明显了,如Figures 2(c) & 2(d) 所示;然而,在低光照条件下,低读出速度(Figure 2(f))的图片质量要明显的优于高读出速度(Figure 2(e)。 Figures 2(e) & 2(f) 的照明强度大约是 Figures 2(c) & 2(d) 的十分之一。如将照明强度再降低5倍效果会更加明显,如Figures 2(g) & 2(h),低读出速度下的图片至少还能够表达出需要的效果(Figure 2(h)),然而高读出速度下的成像质量则很不理想(Figure 2(g))。
一个成功的数字成像实验,要求样品的空间照明模式是连续的,即整个视野内的照明是连续一致的,每次成像间的图片也是连续一致的。这取决于照明光源,使用钨卤素灯照明,样品的光学性质在显微镜下通常都是连续的(被称为光学一致性)。然而使用激光扫描显微镜的照明装置对某一特定的点进行扫描,则每一次的扫描结果都不一样。另外,使用汞灯(the mercury plasma arc-discharge lamp)作为宽视野荧光显微镜的照明光源,图片结果会在光谱上体现为特定的分离的波长峰值。相比之下氙灯(Xenon lamps)在可见光谱下的结果就相当连续,÷但是在它在紫外区的结果不佳(这一光谱范围通常不用于活细胞成像)。
对于金属卤素灯这样的照明光源,它的特性介于汞灯与氙灯之间,也许是性能最佳的光源。金属卤素灯能够提供从紫外到红外间连续的光谱,此外,保有汞灯的光强。除了调整照明光源,为了达到照明一致的效果,还可以在灯箱及显微镜的照明输入端口之间添加一个光导纤维(或液体光导)。在视场中的照明梯度可以通过计算来矫正(flat-field algorithms)。灯泡有时在输出的亮度会与平均值有10%的偏离,这时需要注意的是它不能够通过光学纤维来矫正,而要使用稳定的电源。
对光子通量的测量是统计学的处理,在检测信号时会产生偏差。所谓的Poisson 或 shot noise(如前所述),它体现在测量N个光子时,出现的偏差与N的平方根成正比,即信噪比。所以,增加信号光子的数量N可直接提高信噪比及图像对比度。提高信号最简单的方
法是手机更多的光子,通常做法是增加曝光时间或提高激发照明的强度,但是这些做法会增加光毒性并降低细胞活性。其次,另一个更有用的方法是加强检测器的敏感度。
为了解决活细胞成像低光照的问题,现在的数字CCD相机检测器更加敏感,这是通过面元划分(binning)来实现的(如Figures 3 & 8所示)。一般CCD读取像素点会将横向上的像素值转移到只读寄存器上,然后进行顺序分析。在binning 处理时,相互临近的一群光信号会被合在一起检测(2 x 2 或 4 x 4)为一个信号值进行输出。这通过将多行像素转移到一系列只读寄存器(比如两行进行 2 x 2 binning ,四行进行4 x 4 binning)中然后多行同时读取来实现的。在2 x 2 binning 情况下,空间分辨率会降低一半,信号强度增加4倍,信噪比增加2倍。很明显,虽然降低了空间分辨率,但对于弱信号强度的提高是很显著的。
Figure 3显示的是将临近的像素结合在一起并不断的增加binning 水平,对数字图像的空间分辨率及最终尺寸的影响。样品为印度麂鹿皮肤组织的纤维原细胞,它表达mCherry 荧光蛋白(红色伪彩)及人的β肌动蛋白,位于应力纤维组成的丝状细胞骨架网络中。通常在进行荧光蛋白标记时,表现出很好的共定位的细胞常常表达很低水平的融合蛋白,其发出的信号极微弱。如果不用binning处理(Figure 3(a)),在不产生光毒性及光漂白的曝光时间下得到的信号弱到无法辨别。Binning 2 x 2 渐进到 4 x 4增加了信号水平,但降低了空间分辨率(Figures 3(b) and 3(c))。在binning 8 x 8 像素下的图像(Figure 3(d))最亮。但分辨率降低极多。另外很重的一点是,在binning 处理时,增加合并像素点,图像尺寸按比例缩小例如,不进行binning 处理(1 x 1)的图像(Figure 3)的原始大小为1360 x 1024(pixels),在(2 x 2), (4 x 4), 及 (8 x 8) 处理条件下的图像分辨率分别为680 x 512, 340 x 256, 及 170
x 128(pixels)。如上所述,这些图像的尺寸被缩小了。同样的,一个512 x 512 的数码相机在进行上述比例处理时,图像的大小将分别缩小为256 x 256, 128 x 128, and 64 x 64(pixels)。
Binning 处理的重点是读出噪音产生在binning 处理后,所以虽然读取了几个像素的集合,但是每个binning处理后的像素只读取一次。相对的,像 2 x 2 这样的像素集合,如果先收集信号再进行平均计算,那么由于每个点都会产生噪音,这样的结果会比binning差。因此,虽然binning处理会降低空间分辨率,但是这样的处理可以显著提高空间分辨率,这在光敏细胞的试验中是非常有意义的,因为在这样的试验中,光照水平非常的低,而且曝光时间和很短。在活细胞成像中最重要的参数通常是检测若荧光信号的能力,而不是空间分辨率,所以,通过binning处理提高敏感度及缩短曝光时间是成像的重点,不需太多考虑空间分辨率。除binning 处理外,许多高效相机通过小范围拍摄或只拍摄感性的区域来缩短曝光时间,这种拍摄方法可以在保有空间分辨率的同时减少曝光对细胞的损害。
克服常规高效相机的缺点
为了满足极低照明下高速成像的需求,开发商们推出了一种有效的放大弱信号的相机,EMCCD。这种EMCCD通过芯片上益增寄存器进行信号放大,敏感度能够达到单光子检测级别,同时不会像传统CCD 一样降低量子效率和空间分辨率。EMCCD的一个显著特性是,通过芯片上专门的外延串行寄存器,在硅亚结构内发生碰撞电离过程中进行增益。(Figure 4). 由光子激发出的电荷要高于读出噪音,这即使在高帧频下也能实现,并且对于现在的任何CCD 都使用,包括背照芯片CCD(back-illuminated CCD)。
EMCCD的优越性在于由于放大过程直接发生在CCD的结构内,它要比其它的强化CCD(intensified CCDs)便宜一些。对于单分子成像,全内反射,针孔碟片场扫描共聚焦离子通量检测实时3D试验等弱信号的测定,EMCDD要比其它针对弱光的感光原件好很多。另外,如果使用传统的荧光成像技术,EMCCD极高的敏感性可以在弱光及/或低激发强度下得到高信号强度,这样,可以降低光毒性及荧光的光漂白。
显微镜的光学系统
用于活细胞成像的显微镜的结构必需足够坚固,或者是铝制的,同时,也要足够稳固防震。而显微镜外露的光学表面及聚光镜,灯箱,物镜转盘等装置必需保持洁净,存放显微镜的房间要保持低湿度的无尘状态。在活细胞成像的日常使用时,必需保证所有的光学通路及光圈在同一轴线上,并应用克勒照明。存放显微镜的房间也有相应要求,室内不要安装顶灯,以减少直接进入目镜及样品处的光线。
电子数字相机通常通过专用的接口接在显微镜的一个或几个相机接口上。研究级的显微镜通常有多个接口同时连接几个相机(如 Figure 1)。这对于需要使用不同手段得到最佳图像效果的显微成像是十分有用的。在显微镜机身内部有一系列反光镜,分光镜,棱镜分别被安置在恰当的位置以将光信号反射到不同的相机接口及目镜中。需要注意的是,由于部分光进目镜而减少了进入相机接口的光线,相机成像的信噪比会降低。因此,在配置显微镜时,应该将若荧光照相的分光口的分光率设置为100%。
物镜负责收集由样品发出的光线,并将其传到显微镜的光学组件中,因此,物镜的选择是至关重要的。在光路中的光学组件,如反光镜,分光镜,聚光透镜,滤光镜,棱镜等会严重的降低信噪比,所以,这种效应必需降低。在荧光成像时,应当使用高数值孔径的物镜,因为它能够尽可能多的收集由样品发射出的光线。数值孔径(Numerical aperture)体现了其棱镜能够从单一点光源收集光线的能力,因此,对于活细胞成像,数值孔径是最重要的考虑因素。其数值标注在物镜筒上,大小从0.25(低放大倍数,如10x干镜)到1.20(水镜)及1.45(油镜,40x - 100x范围)不等。数值孔径的数学表达式如下:
Numerical Aperture (数值孔径,NA) = Refractive Index (折射率,η) × Lens Acceptance Angle (镜口角的正弦值,sin(θ))
因此,物镜对光的收集能力与物镜前透镜及样品间介质的折射率有关,与镜口角的正弦值有关。数值孔径为物镜分辨率数学表达式的一个因数。根据瑞利判据(Rayleigh criterion,一种保守的估计),分辨率等于照明光波长与一个常数(1.61)的乘积与数值孔径的商。另外,数值孔径还决定了图像的亮度,图像亮度与数值孔径的四次方成正比,与放大倍数的二次方成反比。所以,稍微提高数值孔径都能够显著地提高信号强度。因此活细胞成像要求使用数值孔径最高的物镜及高折射率的浸入物(油或水)。
如上,图像的亮度与物镜放大倍数的二次方成反比,因此弱信号成像最好使用低倍数的物镜。比如说,在100x数值孔径1.4的物镜下观察样品,如果这个时候信噪比成为影响图片质量的主要因素,那么将物镜转换到60x或40x,则同样的数值孔径下图片的亮度会得到显著的提高。事实上,将60x/1.4与100x/1.4的物镜进行对比,对同一张荧光图像成像,前者要比后者亮三倍。观测强度也是衡量物镜透光能力的一个因素。在某些试验中,为了达到要求的放大率,通常要将放大与binning 处理借个起来。
在多重标记的固定细胞试验中(有时也能是活细胞试验),具有色差及平场矫正的物镜是最理想的。然而不幸的是,复消色差物镜和平场复消色差物镜中相对于消色差物镜和萤石物镜(低精确度物镜)来说,不可避免的添加了很多光学器件(透镜)。高矫正的物镜不会生成假象,可应用于透射光技术,如相差,DIC等。然而,矫正的结果降低了光透过率,这对固定细胞的成像不会有很大的影响,但是对活细胞成像来说却不然。阐述这种现象最好的例子是,一个40x/1.4的平场复消色差的物镜,理论上要比同类100x物镜亮六倍,但实际上,前者的亮度只有后者的四倍左右。但无论如何,40x及60x的物镜在光学分辨率极限下,仍能提供最亮的图片。
如Figure 5 所示,为就图像分辨率及亮度方面,物镜数值孔径的重要性。样品为非洲绿猴的肾脏上皮细胞培养物 (CV-1 line),用表达特异结合线粒体的EGFP 的质粒转染该培养物,在荧光显微镜下可以观测到线粒体在细胞中的网络分布。
Figure 5 为平场萤石物镜下相同视野的图片(数值孔径1.3,放大倍数由40x到100x 不等)。虽然图中的拍摄条件及矫正手段相同,但40x下的线粒体要比其它条件的亮(Figure 5(a),图片大小为经调整)。相比之下,60x及100x下的图片逐渐变暗,100x下的线粒体几乎不可见 (Figures 5(b) & 5(c)).这样的物镜仍然可用,但是必需放大检测器的增益值,这样一来信噪比就会降低,图片质量也相应下降。需要注意的是,由于数值孔径相同,从Figures 5(d)到5(f)的分辨率是一样的。
以上的例子说明在选择活细胞成像的物镜时,如果样品标记荧光,则不要盲目的追求放大倍数。对共聚焦显微镜的40x或60x物镜来说来说,在成像时进行放大后的结果和100x 物镜的成像效果相同(Figures 5(d) and 5(e))。由于具有相同的数值孔径,两个物镜的分辨率是一样的。但这种说法并不是说60x或100x 的物镜没有任何作用。事实上,观察小样品通常要使用高倍数的物镜,比如宽视场显微镜下观察过氧化物酶体和分泌颗粒。由于图像大小通常与检测器的尺寸有关,所以,光学成像最终的放大率取决于CCD的参数(像素点大小,中间变倍等)。因此,物镜的选择通常要考虑光学显微镜的配置,以及试验的特殊要求。
信噪比比分辨率的影响更大的时候,最好要选择矫正度低的物镜,这样的物镜中光学原件更少,相对的光的透过率会显著提高。同样的,相差物镜中的光学器件也会减少光的透过率。这种水平的减少在透射光成像时不会有影响,但是在荧光成像时大概会减少15 %-20%的光透率。因此,在进行活细胞成像时,如果试验用的荧光标记较弱,则不能用相差物镜,除非实验步骤方面有特殊的要求。同样的,当试验要求将荧光图片同DIC 图片进行叠加时,在荧光成像前要将物镜下方的偏光装置拿开,因为它大概会减少30%的透光率。
光路中的任何缺陷都会直接影响最终成像的信噪比。对于活细胞成像来说必需消除高数值孔径物镜的球差。通常引起球差的因素有培养液及物镜浸入物等。球差是由于光沿光轴不均匀分布造成的,会导致成像变形。由于成像样品通常较大,球差还会影响信噪比。这个问题在对厚组织成像时要比薄层细胞成像时严重很多,由于水的折射率与培养基的折射率接
近,通常使用水镜来消除这个问题。另外一种方法是矫正因培养基或盖玻片导致的折射率的改变。由于折射率会受温度影响发生变化,所以,在室温下与在37摄氏度下的浸入物及盖玻片厚度需要做适当调整。最新的物镜可通过矫正环来消除温度引起的折射率的改变。
如 Figure 6 所示,为显微镜光学系统(a)及数码相机检测系统(b)对活细胞成像的影响。图中样品为鼠袋鼠肾脏的一个上皮细胞(PtK2 line),该细胞表达EGFP,EGFP与
H2B组蛋白结合定位于细胞核。观察方法为宽视场落射荧光。如图,左图右下方及右图左下方为分辨率最佳的两张。在活细胞显微成像方面,光学系统中物镜的数值孔径及光源的波长决定了图像对比度与物理分辨率。在左图中,随着照明强度的增加,图像中的噪音明显减少;随着光学分辨率的提高,细胞核中的细节变得更加清晰。
在右图中,图像质量随着灰度及空间分辨率改变。增大像素尺寸,图像的细节变得模糊不清,图像几乎无法辨认;而降低灰度,会改变图像外形,并仅呈现出具有高对比度的信息。以上两种因素共同改变图像质量,同样,在活细胞成像试验中以上因素需要根据试验需要进行配置。
在活细胞成像试验中还应用了各种滤色片及反光镜,它们的作用是调整从照明器经显微镜到检测器中光的波长。在透射光系统中这些增称光学组件包括起偏器,棱镜(DIC),聚光镜环,相差环等。在透射光成像系统中,如明视野,DIC,相差等,光的强度要比荧光大得多,所以,为了提高信噪比,需要在光路中添加一些组件。其中首要考虑的是过滤金属卤素灯的紫外及红外波段的光以保护样品。红外线产生的热效应会损害细胞活性,并会降低红外敏感CCD 成像的信噪比。紫外线很少出现在透射光源中,为了阻止这些波长范围的光,只需添加绿光片或红光片即可。红外及可见光滤光片都会显著降低光强,所以在成像时需要增加曝光时间及增益以保证适宜的信噪比。
在荧光成像中,为了保证激发光及发射光的波长在适宜的范围内,会在光路中添加滤光片及半透半反滤光片。在相对光强较低的荧光成像中,选择滤光片首要考虑的是它是否能提供适宜的信噪比。大多数情况下,光会不分波长大量的通过光学组件以至于掩盖了荧光染料发出的信号,这时带通滤色片就显得非常重要。通常这中窄波段的滤色片会限制绝大部分光透过,所以,为了得到满意的图片,研究人员必需选择适宜带宽的滤色片。
现在,市场上有很多厂商提供特制的带通滤色片及半透半反滤色片组合。在为活细胞成像选择滤片组时,为了得到好的信噪比,所选的组合要最接近荧光子的光谱性质。例如,选择标准的多色荧光成像FITC 滤色片组专门为进行YFP成像时,就不需要为了提高信噪比而滤掉部分YFP 范围内的光(Figure 7(a))。在这种情况下,选择符合YFP 吸收及发射波段的带通激发光滤色片及长通发射光滤色片会得到最佳的效果。
再举一个两个荧光蛋白衍生物波段的选择实例(Figure 7)。图中上部为YFP 的变种Venus 的激发及吸收光谱,它配合FITC 滤片组及宽波段发射光光谱以达到最好的信号收集效果。发射光滤色片的选择与检测器收集的信号直接相关,而上述组合显然不如宽带通YFP 组和收集的信号多。同样的,TRITC 滤片组(Figure 7(c))在mCherry 荧光蛋白的激发波段不如Texas Red 滤片组(Figure 7(d))效率高。另外,Texas Red 滤片组的发射光波段更宽,允许检测器收集更多的信号。所以,在为活细胞成像选择荧光基团时需要配合滤片组合一起考虑,以保证得到最佳的激发及发射信号。
现在有更多的带宽组合可供多色荧光标记同时成像。另外还需要考虑的是在显微镜光学组件中添加中性密度滤色片来降低激发及发射强度——这在活细胞成像中是重要的,如同阻挡或减少紫外,红外线的细胞光毒性。这些滤色片通常放在显微镜光源接口处,并可以随时取出以增加光强。通过显微镜的光强及数值孔径可以通过聚光镜或落射照明器的孔径光阑及视场光阑进行调节。
检测器几何形状与光学分辨率间的匹配
由于样品有检测器成像,图像的空间分辨率取决于光学系统的分辨率。因为CCD 的检测器有一列光电二极管组成,每个都有固定的尺寸,所以,每个像素的几何学特性限制了图像的最终的分辨率。为了达到显微镜的分辨率,检测器的尺寸要符合Nyquist 抽样标准——2.5-3 pixels/Airy disk unit。举个例子,对于光学分辨极限250 nm,图像最终的像素大小应接近80-100 nm。对活细胞成像来说,信噪比要比空间分辨率更重要,所以最佳的成像
方式为2 pixels/Airy disk,这种方式不会对图像质量造成明显损害。降低每个像素与Airy Unit 的比例会增加图像的亮度,并且可以使用更大的CCD 光电二极管,以累积更多的光电子。
Figure 8(a) 中描述的是应用高数值孔径物镜进行活细胞成像时Airy disk 与CCD 像素列阵的投射关系。列阵中每个像素大小为6μm。100x/N.A.1.4 的物镜投影在CCD表面上的像直径为20μm(Table 1),因此,可以达到Nyquist抽样标准(3.3 pixels/Airy unit)。在这样的采样频率下,有充足的空白可以进行2x2 的binning 处理。相对的相同数值孔径下
60x 物镜的投影直径为12 μm,刚好低于Nyquist 的下限。另外,即使将数值孔径降到1.3,40x 物镜投影直径仍只有8.4μm,需要连接放大接口才能达到Nyquist 分辨率标准。光电二极管列阵的像素尺寸可大可小以匹配显微镜的空间分辨率,而无需添加中间变倍体;或者可以配置一个特制的CCD 接口,这样的接口中包含了一系列棱镜系统。
Figure 8(b) 中显示的为2x2 binning处理的原理图,列阵中包含16个像素。曝光后,CCD 收集的光电子将进行两次平行迁移,然后,串行寄存器上发生两次迁移,这样一个像素上将收集4个像素(2x2 binning)的光电子,并将其传输到输出节点进行放大。4x4 binning 的处理方式与此相同,在处理过程中收集列阵中16 个像素信号并在输出前进行放大,这样一来可以显著地增强信号,同时降低读出噪音。如前面讨论过的,binning 处理是活细胞成像中一个绝佳的弱信号收集方式,这种处理可以降低对低荧光表达量细胞的毒害。
实际上,由1.4数值孔径的物镜成的像,最终大小介于100-120 nm 之间。将这样大小的信号一合适的物理尺寸转移到CCD 光电二极管中,需要考虑物镜的放大倍数。例如,要达到60x/1.4 物镜的最大分辨率,光电二极管不能大于6μm——大小取决于放大倍数及分辨率。然而,对100x/1.4 的物镜来说,光电二极管最大可至10μm。因此可以很明显的看出,只有当CCD 的配合了适当的物镜,数字分辨率才会与光学分辨率相匹配。10μm光电二极管与100x 物镜组合生成的图片像素大小为100 nm;当与60x物镜组合时生成的像素大小为167 nm,这时;图片的分辨率会低一些。相比之下,6μm光电二极管与60x 的物镜配合则是最佳的,但是当与100x物镜组合时,则会取样过度。这种情况下的取样过度会降低图像
亮度,从而无法改善图像分辨率。
从前面的讨论看来,似乎想要得到理想的分辨率就得为不同的物镜配不同的CCD。实际上,这种方法一点都不实际,所以,需要找到一个折衷的办法。在有些情况下,这个问题可以通过在CCD 与显微镜之间添加一个耦合器来消除物镜放大倍数造成的影响。例如,
0.6x 的接口可以将100x 物镜的投影大小降低到60x 物镜的效果。通过售后经销商可以得到各种耦合器,可放大或缩小CCD 光电二极管上投影的大小。以上的建议中需要注意的是,这些组件添加或移除相对来说非常容易,最好不要把CCD 从显微镜接口处拿下来,以防灰尘或颗粒进入显微镜机体内或相机成像传感器上。清洁传感器是件非常困难的事,尤其是要彻底清除那上面的每一个灰尘及颗粒。
与显微镜光学分辨率相匹配的像素大小
物镜(数值孔径)
分辨率
(μm)
投影尺寸
(μm)
要求像素尺
寸
(μm)
1x (0.04) 6.9 6.9 3.5 2x (0.06) 4.6 9.2 4.6 2x (0.10) 2.8 5.6 2.8 4x (0.10) 2.8 11.2 5.6 4x (0.12) 2.3 9.2 4.6 4x (0.20) 1.4 5.6 2.8 10x (0.25) 1.1 11.0 5.5 10x (0.30) 0.92 9.2 4.6 10x (0.45) 0.61 6.1 3.0 20x (0.40) 0.69 13.8 6.9 20x (0.50) 0.55 11.0 5.5 20x (0.75) 0.37 7.4 3.7 40x (0.65) 0.42 16.8 8.4 40x (0.75) 0.37 14.8 7.4 40x (0.95) 0.29 11.6 5.8 40x (1.00) 0.28 11.2 5.6 40x (1.30) 0.21 8.4 4.2 60x (0.80) 0.34 20.4 10.2 60x (0.85) 0.32 19.2 9.6 60x (0.95) 0.29 17.4 8.7 60x (1.40) 0.20 12.0 6.0 100x
(0.90)
0.31 31.0 15.5
100x
(1.25)
0.22 22.0 11.0
100x
(1.30)
0.21 21.0 10.5
100x
(1.40)
0.20 20.0 10.0
Table 1
现在许多为荧光显微镜设计的CCD 的光电二极管大小在5-16μm范围内,并可进行binning 处理。如上讨论,通过binning 处理,几个光电二极管上的信息倍整合到一个单元中,从而有效的增大像素尺寸。6-8μm大小的光电二极管可匹配60x 的物镜,经过2x2 binning 处理,可匹配100x 物镜。这中情况下,使用高透过性的物镜,可使100x 物镜下成像更加明亮。这样,100x 物镜下,bin 2 下的图片几乎是60x 物镜下,bin 1 图片亮度的二倍,而分辨率近似或高于bin 1。当要求更高的敏感度时,使用60x 物镜进行2x2 binning 处理可以增加图像亮度,这时对于活细胞成像来说,分辨率上的损失在可接受范围内。
在一些实验中,例如大群细胞的同时成像,获得视野内更多的信息要比空间分辨率重要。在没有中间变倍体的情况下,CCD 的矩形传感器无法获取目镜视野内所有的信息,通常只有30-80% 左右,大小取决于芯片尺寸。如果要获取更大的图片,需要添加一个具缩小功能的耦合器(如前所述)。通常0.6x 的透镜就可以使整个视野内的图片投影到芯片上,但同时会降低一些分辨率。如果要保有图像分辨率,可以通过x-y扫描获取多张图片然后用软件进行拼接。
光学显微镜上使用近紫外光源来获得最高分辨率。近紫外后,分辨力的顺序是蓝,绿,红。在大多数情况下,研究人员使用金属卤素灯产生的宽光谱进行照明,但是在活细胞成像中,照明光谱通常被滤色片控制在很窄的波段。可见光谱中部波长大约为550 nm,人眼最敏感的绿色。Table 1 中的分辨率就是在这个波长下测定的。所以在不同波长下,这些值是不同的。数值孔径也是重要因素,如表中所列,放大倍数相同的条件下,数值孔径越高,分辨率越高。
总结
也许在活细胞成像中最关键的就是在获得最高光学分辨率的同时平衡试验中信号强度(降低放大倍数),分辨率(提高放大倍数)及细胞活性间的关系。研究人员还必需选择尽量少的透镜来配合光学放大及检测器像素尺寸。配合不同物镜(放大倍数及数值孔径)的各种类型的中间变倍体都可以购买得到,以满足弱光及高分辨率试验的要求。在选择物镜放大倍数时,必需严格遵守Nyquist 分辨率标准。每个像素不得大于分辨率极限的一半;由于显微镜中衍射极限,工作距离通常不能小于50 nm。
活细胞成像用标准物镜通常为10x 及40x 干镜,及40x, 60x,100x 油镜或水镜。经济的低放大倍数的干镜通常用来进行初步检测,比如找到细胞的位置,因此过高不需要光学矫正。然而浸润镜头则需要高数值孔径(油镜1.3-1,4,水镜1.2),及高光透过性。由于图片采集与视场中部,多以高平常物镜并没有多大意义,而同时这样的物镜昂贵并且光透性比其它物镜低。所有用于荧光成像的物镜都需要用荧光珠检验其点扩散函数。
无论落射荧光还是透射光(DIC 及相差),单色冷CCD 是成像的最佳选择,这包括培养中的活细胞。在选择相机时需要考虑的是量子效率,噪音水平(暗电流及读出噪音),像素大小(及满肼容量),及扫描频率。为了减少光对样品的损害,当选择高敏感度的相机,即高量子效率,低噪音,大像素尺寸,和低扫描频率。因此,敏感度还需与分辨率(高分辨率,低像素尺寸)及显像率(高扫描频率)之间平衡。
低扫描CCD 的帧速率通常不高,因此,除非样品的亮度极高,在低曝光条件下信噪比会很低。这些因素都限制了CCD 在高速成像中的应用(30帧/s),并是样品成像变得令人挫败。另外还需避免光漂白及光毒性,一些产品在拍摄时可以在细胞活性与亮度之间取得平衡。处于以上考虑,相机应无光闸,具帧转移或插值传感器,这样除了低速数字信号扫描外,
可以持续地在录像速率下拍摄图像。在荧光探针亮度很低或量子产量很低等极弱光条件下,如果要求高速成像,可选择增益型或电子扩增型CCD。
GE超高分辨活细胞成像系统 利用活细胞成像工作站进行细胞和基因的功能研究,是生物医学研究的最新趋势。固定细胞观察仅能提供固定瞬间细胞的静态信息,无法反映细胞在正常生理生化条件下的状态。活细胞观察,对处于正常生理状况下的细胞进行全程扫描和记录,获得其连续、全面、动态过程由于其显示的正常细胞动态的活动过程,很容易发现和确定细胞间相互作用和信号传导的过程,以及在活细胞水平上的生物分子间的相互作用,不仅可以解决长期以来悬而未解的问题,更为未来的研究提出新的问题,指出新的方向。 一、活细胞成像系统原理 目前主流的活细胞成像系统从原理上可以分为两大类: 基于宽场反卷积技术 基于共聚焦技术 两种技术作为目前最流行的活细胞成像技术,均可以实现在维持细胞存活的情况下,快速获取单一焦平面的信号,在具体性能上则各有擅长。 宽场反卷积技术 对光线进行反卷积运算是光学成像领域的成熟技术,最早由美国国家航空航天局开发并成为观察微弱天体信号的标准技术。去卷积和共聚焦技术是光学显微镜领域获得单一焦平面光线的两大主流技术(J.M.Murray, live cell imaging, 2010)。通过将非焦平面的光线还原至焦平面上,大大提高了样品信号的强度以及图像的信噪比。由于去卷积技术设计到大量的后期运算,因此在高性能计算机发明以前,一直受制于运算能力,没有得到大规模的推广。随着近年来计算机性能的大幅提升和价格的下降,去卷积技术逐渐成为光学显微镜的主流技术。一个点光源经过显微镜的光路,由于镜片对光线的衍射和散射,最终呈现在观察者面前的是一个模糊的点,所以点光源变成模糊的点的过程即为卷积。反卷积就是把模糊的点还原成点光源的过程。 以API 公司的DeltaVision系统为例,其反卷积过程经历以下几步: 1)首先通过无数的计算和实验,得到点光源经过显微镜物镜后变模糊的规律,建立模型。 2)选择完美的物镜,保证样品信号经过物镜后变模糊的规律符合步骤一中得到的模型。 3)将通过显微镜光路的所有的光信号进行收集,因为点光源经过显微镜光路后会变成一个空 间中的倒圆锥形,所以在收集信号的时候需要很准确的记录信号的Z 轴信息。 4)对收集到的所有光信号按照步骤一中的模型进行还原,最终将模糊的点还原成清晰的点, 客观反映它在空间的位置和强度。 目前去卷积技术越来越广泛地应用于生物学图像的研究中。 共聚焦技术 共聚焦显微镜它采用点光源(point lightsource) 照射标本,在焦平面上形成了一个轮廓分明 的小的光点(light spot ) ,该点被照射后发出的荧光被物镜收集,并沿原照射光路回送到探测器。探测器前方有一个针孔(pinhole) ,几何尺寸可调。这样,来自焦平面的光,可以会聚在探 测针孔范围之内,而其它来自焦平面上方或下方的散射光,都被挡在探测针孔之外而不能成象。 光束扫描器又分为单光束、多光束或狭缝扫描器几种。其中单光束扫描获得的图像质量最好, 狭缝扫描器虽然产生图像的速率很高(可达实时水平) ,但其图像信噪比低于单光束扫描,这是 因为从狭缝长轴来的漫射光不能被有效遮挡。多光束扫描如碟片式共聚焦是由电动马达驱动
实验目的 为了获得大量的质粒作为克隆载体,我们需要将购买来的质粒通过转化的方法导入细菌的细胞内。制备出感受态的细胞,我们就可以方便的将需克隆的质粒导入其中,使其繁殖,就能获得大量质粒。本次实验的目的就是增加质粒的数量。同时,我们能掌握大肠杆菌感受态细胞的制备及转化的基本原理和实验技术方法。 实验原理及过程 实验步骤 1挑取一个JM109单菌落于3ml LB(无抗生素)培养基中,37℃,180rpm培养过夜。 [让菌复苏,进入对数期的早中期。此时更容易制得感受态细胞。] 2取过夜培养物加入到40mL LB(无抗生素)培养基中,37℃250rpm大约小时。 [减少达到所需亮的均所需繁殖的世代数,以减少菌的变异。] 3取培养物置于40mL的灭菌离心管中,冰浴10min,4000rpm4℃离心 10min,弃上清。(将所有上清倒光,可以将离心管倒过来) [使细菌的生长停止,代谢减慢。因为这时已经没有培养基了,如果细菌仍有很高的代谢效率,则有大量细菌死亡。] 4菌体重悬于10mL100mmol/L冰冷CaCl2中,轻吹,4℃30min,4000rpm 离心5min弃上清。 [细菌处于0度,CaCl2低渗溶液中,菌细胞膨胀成球形,外源DNA形成抗DNA酶的羟基-钙磷酸盐混合物黏附于细胞表面(羟基来源于细胞壁上的糖,磷酸来源于DNA),经短时间420C热激处理,促进细胞吸收DNA复合物。(一
定要轻吹,这时细胞壁打开,十分脆弱)]5菌体重悬于1mL 100mmol/L冰冷的CaCl2中,轻吹,用于转化。 [除去所有的LB以及细胞破损后释放出来的细胞内含物。防止细胞分裂,以致大量细胞死亡。(一定要轻吹,这时细胞壁打开,十分脆弱)] 6取感受态细胞100uL,加入2uLpET-21a质粒,冰浴30 min受体菌:感受态细胞100uL,加入2 uL无菌双蒸水阴性对照:LCaCl2溶液100uL,加入2 uL阴性对照 [第一个阴性对照是为了验证LB中的抗生素有活性;第二个阴性对照是为了验证CaCl2溶液和pET-21a质粒未被污染。] [冰浴是为了降低细胞代谢率,防止细胞大量死亡。] 7热激42度,90s 【在低温处理后,热激,可以使细胞壁热胀冷缩,再低温,使细胞壁上黏附的质粒进入细胞体内。】 8冰浴2 min 9加入800 uL无抗生素的LB,37℃复苏1h 【用LB复苏细胞,使细胞恢复活力,从而使细胞中的质粒表达抗抗生素的基因,只有这样才能使转化成功的细菌在有抗生 素的LB平板上生长。】 10取100ul细胞直接在含50ug/mLCarbenicillin(羧苄西林)的LB培养基上铺平板,将平皿放置37 oC温箱30min至液体被吸收。倒置平皿37 oC培养过夜,出现菌落。 【这些菌落为转化成功的菌落,而对照组应该没有菌落。在目的菌落的旁边有可能出现卫星菌落,这是因为亩的菌落的抗性基因似的菌落周围的抗生素失效,长出了转化为成功的菌落。用羧苄西林因其对酸稳定、不易被细菌降解,所以可以降低卫星菌落的数量。
1:DH5a菌株 DH5a是一种常用于质粒克隆的菌株。E.coliDH5a在使用pUC系列质粒载体转化时,可与载体编码的β-半乳糖苷酶氨基端实现α-互补。可用于蓝白斑筛 选鉴别重组菌株。 基因型:F-,φ80dlacZΔM15,Δ(lacZYA-argF)U169,deoR,recA1,endA1,hsdR17(rk-,mk+),phoA,supE44,λ-,thi-1,gyrA96,relA1 2:BL21(DE3) 菌株 BL21菌株用于高效表达克隆于含有噬菌体T7启动子的表达载体(如pET系列)的基因。T7噬菌体RNA聚合酶位于λ噬菌体DE3区,该区整合于BL21 的染色体上。该菌适合表达非毒性蛋白。 基因型:F-,ompT, hsdS(rBB-mB-),gal,dcm(DE3) 3:BL21(DE3) pLysS菌株 BL21(DE3) pLysS菌株含有质粒pLysS,因此具有氯霉素抗性。PLysS含有 表达T7溶菌酶的基因,能够降低目的基因的背景表达水平,但不干扰目的蛋 白的表达。该菌适合表达毒性蛋白和非毒性蛋白。 基因型:F-,ompT hsdS(rBB-mB-),gal,dcm(DE3,pLysS ,Camr 4:JM109菌株 JM109菌株在使用pUC系列质粒载体进行DNA转化或用M13phage载体进行转染时,由于载体DNA产生的LacZa多肽和JM09编码的LacZΔM15进行α -互补,从而显示β-半乳糖苷酶活性,由此很容易鉴别重组体菌株 基因型:recA1,endA1,gyrA96,thi-1,hsdR17,supE44,relA1,Δ(lac -proAB)/F’[traD36,proAB+,lacIq,lacZΔM15] 5:TOP10菌株 TOP10菌株适用于高效的DNA克隆和质粒扩增,能保证高拷贝质粒的稳定遗传。 基因型:F- ,mcrAΔ(mrr-hsd RMS-mcrBC),φ80,lacZΔM15,△lacⅩ74,recA1 ,araΔ139Δ(ara-leu)7697, galU,galK ,rps, (Strr) endA1, nupG 6:HB101菌株 HB101菌株遗传性能稳定,使用方便,适用于各种基因重组实验 基因型:supE44,hsdS20(rB-mB-),recA13,ara-14,proA2,lacY1,galK2,rpsL20,xyl-5,mtl-1,leuB6,thi-1
为活细胞研究设计光学显微系统时,首要考虑的是检测器的敏感度(对信号乃至噪音的检测),图像获取的速度,以及在此基础上标本的可行性。对于固定细胞的成像,曝光时间及光强度相对来说都很高,这时可能会造成光漂白;然而对于活细胞成像,上述光的影响必须去除。几乎在所有情况下,活细胞显微镜都会在尽可能高的图像质量与尽可能好的细胞活性之间取得一个平衡。对于此类实验,时间及空间上的分辨率需要设定在能满足实验要求的水平上,而不是给予过度的光照或设定过多采样时间点。 基本上,一个理想的活细胞成像系统必需有足够的敏感度,来满足在弱荧光条件下仍能得到高图片质量;同时,系统也必需足够快,以记录整个动态过程。另外,这个系统还需要有足够高的分辨率以捕捉样品细节,并且能够准确的实时测量每个微小的光强变化。然而不幸的是,要改善上述的任意一条都需建立在牺牲其它性能的基础上。因此现在还不能够设计出一个可以满足所有要求的活细胞成像体系。研究人员现在只能在尽量减低不重要的信息的遗失的同时,尽可能的获得最优的重要参数。这样,显微镜的配置最终取决于成像的要求,对于样品在实验期间活性的要求,进行标记的难度水平,以及仪器的可用性等实际因素。 如图一(Figure 1)所示为一台倒置研究级显微镜,它配有四个相机接口,并可满足对培养的组织的研究。在四个接口上分别配有四个不同的相机,每一个都用来获取不同的图像。在大多数情况下,这种显微镜的分光设计是100%进入相机或以80:20的比例同时分配给相机和目镜。在弱光成像时,研究人员必需确保将最敏感的相机接在100%分光口上。在图一中,彩色CCD(Full color CCD)接在显微镜的底部(a),它从物镜接受的光信号不经过棱镜或反光镜的反射。这样的相机通常用来进行多色荧光或明场拍摄。显微镜右侧连接高效的电子倍增电荷偶联设备(Electron Multiplying Charge Coupled Device,EMCCD)(b),它通常用来检测极弱的荧光信号。接在显微镜左侧的相机(c)配有一个高量子效应的感应器,可以感应700-1000nm 波长范围内的光,所以这个相机可以用来进行微分干涉相称(differential interference contrast,DIC)观察方法下厚标本的红外线照明成像。最后,对于高分辨率的单色荧光成像,如全内反射荧光或其它荧光技术,图一中的显微镜在前部(d)
大肠杆菌电转化感受态细胞制备 第一天: 1. 将适合菌株(如XL1-Blue, DH5α)置于LB或其他营养丰富的培养基上,在37°C下过夜培养 2. 高温灭菌大的离心瓶(250-500ml)以备第二天摇瓶用 3. 准备几瓶灭菌水(总量约1.5升),保存于冷冻室中以备第二天重悬浮细胞用 第二天: 4. 转移0.2-1ml过夜培养物至装有500ml LB(或其他营养丰富的培养基)的1-2升挡板∑恐? 5. 37°C下剧烈振荡培养2-6小时 6. 定时监控O.D.600值(培养1小时后每半小时测定一次) 7. 当O.D.600值达到0.5-1.0时,从摇床中取出摇瓶,置于冰上冷却至少15分钟(需要的话这种方式可以存放培养液数小时) 8. 细胞在4°C 5000g下离心15分钟,弃上清液(如需要,沉淀可在4°C的10%甘油中保存一两天) 9. 用灭菌的冰水重悬浮细胞。先用涡旋仪或吸液管重悬浮细胞于少量体积中(几毫升),然后加水稀释至离心管的2/3体积。 10. 照上面步骤重复离心,小心弃去上清液 11. 照上面步骤用灭菌的冰水重悬浮细胞 12. 离心,弃上清液 13. 用20ml灭菌的、冰冷后的10%甘油重悬浮细胞 14. 照上面步骤离心,小心弃去上清液(沉淀可能会很松散) 15. 用10%甘油重悬浮细胞至最终体积为2-3ml 16. 将细胞按150μl等份装入微量离心管,于-80°C保存 转化方法: 1. 在冰上解冻电感受态细胞添加1-10μl DNA ,冰上培育约5分钟
2. 添加1-3μl DNA ,冰上培育约5分钟 3. 转移DNA/细胞混合物至冷却后的2mm电穿孔容器(无泡)中 4. 加载P1000,准备好300μl LB 或 2xYT 5. 对电穿孔容器进行脉冲(200 欧姆, 25μFd, 2.5 千伏)(检查时间常数,应该在3以上) 6. 立即添加300μl 的LB或2xYT至电穿孔容器中 7. 37°C下培养细胞40分钟至1小时以复原 大肠杆菌电转化感受态细胞制备及转化方法 大肠杆菌电转化感受态细胞制备 第一天: 1. 将适合菌株(如XL1-Blue, DH5α)置于LB或其他营养丰富的培养基上,在37°C下过夜培养 2. 高温灭菌大的离心瓶(250-500ml)以备第二天摇瓶用 3. 准备几瓶灭菌水(总量约1.5升),保存于冷冻室中以备第二天重悬浮细胞用 第二天: 4. 转移0.2-1ml过夜培养物至装有500ml LB(或其他营养丰富的培养基)的1-2升挡板∑恐? 5. 37°C下剧烈振荡培养2-6小时 6. 定时监控O.D.600值(培养1小时后每半小时测定一次) 7. 当O.D.600值达到0.5-1.0时,从摇床中取出摇瓶,置于冰上冷却至少15分钟(需要的话这种方式可以存放培养液数小时) 8. 细胞在4°C 5000g下离心15分钟,弃上清液(如需要,沉淀可在4°C的10%甘油中保存一两天) 9. 用灭菌的冰水重悬浮细胞。先用涡旋仪或吸液管重悬浮细胞于少量体积中(几毫升),然后加水稀释至离心管的2/3体积。 10. 照上面步骤重复离心,小心弃去上清液
转化医学着眼于将生物医学基础研究和解决临床问题结合起来,将基础研究的成果转化为疾病预防、诊断、治疗及预后评估的新手段,已经逐步成为了医学界关注的热点之一。细胞水平的研究,是转化医学研究的重要方向,而一些创新性技术手段在细胞研究领域的应用,正加速转化医学研究在细胞水平的进展。活细胞成像和超高分辨率成像技术,作为细胞水平研究的重要手段,也为转化医学的发展注入了新的活力。通过活细胞成像技术,对细胞内的蛋白的表达、细胞器的运动等动态过程进行长期动态观察,可为疾病诊断、新药开发提供更多的线索。以下就对几个典型的活细胞成像应用于转化医学中的实例进行介绍: 自噬在黑色素瘤治疗中的研究 细胞内的基本上所有的细胞器都通过自噬途径得到降解。一些重要的疾病如阿兹海默综合征、动脉粥样硬化都伴随着自噬途径的缺陷,因此一些自噬途径的重要调控因子已经成为最近医学研究和药物开发的热点。 化合物polyinosine-polycytidylic acid可以诱导黑色素瘤细
胞内的自噬途径激活。在加入polyinosine-polycytidylic acid后,细胞内很短时间内就可以看到自噬体标记蛋白LC3在细胞内出现,并伴随着细胞的凋亡。(Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell, 16, 103-114.) 特异性结核杆菌抗生素的研发 结合杆菌是造成肺结核的主要病原菌,开发低毒高特异性的抗生素是目前结核病防治的重要方向。 在对结核杆菌的繁殖进行连续观察检测的同时,在灌流培养基中中加入不同的化合物,从而监测不同化合物对结核杆菌的抑制作用,最终可以得到对结核杆菌有显著抑制作用的化合物并可进入下游的临床试验。(Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother, 54, 4150-4158.)
常用大肠杆菌感受态JM109,DH5a,BL21的区别 1:DH5a菌株 DH5a是一种常用于质粒克隆的菌株。E.coli DH5a在使用pUC系列质粒载体转化时,可与载体编码的β-半乳糖苷酶氨基端实现α-互补。可用于蓝白斑筛选鉴别重组菌株。 基因型:F-,φ80dlacZΔM15,Δ(lacZYA-argF)U169,deoR,recA1,endA1,hsdR17(rk-,mk+),phoA,supE44,λ-,thi-1,gyrA96,relA1 2:BL21(DE3) 菌株 该菌株用于高效表达克隆于含有噬菌体T7启动子的表达载体(如pET系列)的基因。T7噬菌体RNA聚合酶位于λ 噬菌体DE3区,该区整合于BL21的染色体上。该菌适合表达非毒性蛋白。 基因型:F-,ompT, hsdS(rBB-mB-),gal, dcm(DE3) 3:BL21(DE3) pLysS菌株 该菌株含有质粒pLysS,因此具有氯霉素抗性。PLysS含有表达T7溶菌酶的基因,能够降低目的基因的背景表达水平,但不干扰目的蛋白的表达。该菌适合表达毒性蛋白和非毒性蛋白。 基因型:F-,ompT hsdS(rBB-mB-),gal, dcm(DE3,pLysS ,Camr 4:JM109菌株 该菌株在使用pUC系列质粒载体进行DNA转化或用M13 phage载体进行转染时,由于载体DNA产生的LacZa多肽和JM09编码的LacZΔM15进行α-互补,从而显示β-半乳糖苷酶活性,由此很容易鉴别重组体菌株 基因型:recA1,endA1,gyrA96,thi-1,hsdR17,supE44,relA1,Δ(lac-proAB)/F’[traD36,proAB+,lacIq,lacZΔM15] 5:TOP10菌株 该菌株适用于高效的DNA克隆和质粒扩增,能保证高拷贝质粒的稳定遗传。基因型:F- ,mcrAΔ(mrr-hsd RMS-mcrBC),φ80 ,lacZΔM15,△lacⅩ74,recA1 ,araΔ139Δ(ara-leu)7697, galU ,galK ,rps, (Strr) endA1, nupG 6:HB101菌株 该菌株遗传性能稳定,使用方便,适用于各种基因重组实验 基因型:supE44,hsdS20(rB-mB-),recA13,ara-14,proA2,lacY1,galK2,rpsL20,xyl-5,mtl-1,leuB6,thi-1 7:M110或 SCS110 大多数大肠杆菌菌株中含有Dam甲基化酶和Dcm甲基化酶,前者可以在GATC序列中腺嘌呤N-6位上引入甲基,后者在CCA/TGC序列的第一个胞嘧啶 C-5位置上引入甲基。常用的菌株都会产生dam,dcm,从而受到甲基化的影
Science:活细胞代谢成像新方法 0 25细胞S-腺苷甲硫氨酸成像图,随着每个时间点蛋氨酸(右下)的增加,荧光强度也增高 通过基因工程技术使得细胞表达一种经修饰(改造)过的RNA,又称Spinach,研究人员能对活细胞中的小分子代谢物进行成像,并观察它们随时间变化是如何改变的。每个细胞新陈代谢都会产生代谢产物。假如能得知产物生成效率的话,就能辨识如癌症状态下细胞代谢的异常或确定药物能否将细胞的代谢状况恢复到正常状态。 康奈尔大学威尔医学院的研究人员说发表在3月9日的《科学》杂志上的相关论文详细描述了这种先进的技术方法,这一技术将有可能彻底颠覆以往对代谢组学的认识,提供数千种细胞内代谢产物的动态变化的化学指纹图谱。 威尔康乃尔医学院药理学副教授Samie R. Jaffrey博士说:“动态观察到代谢产物的变化将为我们提供新的和强大的武器,方便我们了解代谢在疾病状态下是如何改变的,并帮助我们找到可以将它们恢复到正常水平的方法”。Jaffrey博士领导威尔康乃尔的其他三名研究人员共同完成了这项研究。他说:“细胞的代谢水平调控着细胞诸多功能,也正因为如此,代谢水平的变化可以是细胞内在特定的时间内发生什么变
化的写照”。 例如生物学家都知道,肿瘤细胞存在代谢异常,这些细胞对葡萄糖能源的利用存在异常并产生独特的代谢产物如乳酸,从而有不一样的新陈代谢过程。Jaffrey博士说:“能够看到这些代谢异常的话,就可以了解癌症的发生发展。但是到现在为止,测量活细胞中代谢产物一直非常困难。 Jaffrey博士和他的团队展开的科学研究表明:可以用特定的RNA序列来检测细胞中代谢产物的水平。这些RNAs是基于能在细胞发出绿色光的Spinach RNA设计的。Jaffrey博士研究小组修改Spinach的RNAs,使得它们一旦遇到它们专属绑定的代谢物时就关闭,造成Spinach荧光开启。他们设计出了RNA序列以追踪细胞中五个不同代谢产物包括二磷酸腺苷、细胞能量分子ATP和参与调节基因活性的甲基化过程的SAM(S-腺苷蛋氨酸)水平的变化。他说:“在此之前,一直没有人能够实时观察到细胞中代谢产物水平是如何变化的”。 Jaffrey博士说:“在活细胞中运用RNA传感器,研究人员能够测量单个细胞中的目标代谢产物水平随着时间的变化而 发生的改变,你可以看到这些代谢物如何响应信号刺激或遗传变化进而发生动态变化的。你可以筛选出能使得这些基因异常发生正常化的药物,我们的一个主要目标是确定药物是否能使细胞的新陈代谢正常化。
大肠杆菌感受态细胞的制备实验具体步骤及详细说明 带有外源DNA 的重组质粒,在体外构建后,导入宿主细胞,随着细胞的大量复制、繁殖,才能够有机会获得纯的重组质粒DNA,该过程称之为转化过程。受体细胞经过一些特殊方法(如:CaCl2,RuCl 等化学试剂)的处理后,细胞膜的通透性发生变化,能容许外源DNA 的载体分子通过。 带有外源DNA 的重组质粒,在体外构建后,导入宿主细胞,随着细胞的大量复制、繁殖,才能够有机会获得纯的重组质粒DNA,该过程称之为转化过程。受体细胞经过一些特殊方法(如:CaCl2,RuCl 等化学试剂)的处理后,细胞膜的通透性发生变化,能容许外源DNA 的载体分子通过。 具体步骤 1、从37℃培养16-20 h的平板中挑取一个单菌落(直径2-3 mm),转到一个含有100 ml LB或SOB培养基的1L烧瓶中。于37℃剧烈振摇培养3 h。一般经验,1 OD600约含有大肠杆菌DH5α 109个/mL。 2、将细菌转移到一个无菌、一次性使用的、用冰预冷的50 ml聚丙烯管中,在冰上放置10 min,使培养物冷却至0℃。 3、于4℃用Sorvall GS3特头(或与之相当的转头)以4 100 r/min离心10 min,以回收细胞。 4、倒出培养液,将管倒置1 min以使最后的痕量培养液流尽。
5、每50 ml初始培养液用30 ml预冷的0.1 mol/LCaCl2-MgCl2溶液(80 mmol/L MgCl2,20 mmol/L CaCl2)重悬每份细胞沉淀。 6、于4℃用Sorvall GS3转头(或与之相当的转头)以41 00 r/min离心10 min,以回收细胞。 7、倒出培养液,将管倒置1 min以使最后的痕量培养液流尽。 8、每50 ml初始培养物用2 ml用冰预冷的0.1 mol/L CaCl2(或TFB)重悬每份细胞沉淀。 9、此时,可以用新鲜制备的感受态细胞直接做转化实验,也可以将细胞冻存于- 70℃。 注意事项 1. 为达到高效转化,活细胞数务必少于108个细胞/mL,对于大多散大脑杆菌来说,这相当于OD值为0.4左右。为保证细菌培养物的生长密度不致过高,可每隔15-20 min测定OD600值来监测,用监测的时间及OD值列一个图表,以便预测培养物的OD600值到0.4的时间,当OD600值达到0.35时,收获细菌培养物。 2. 在菌株与菌株之间,OD值与每毫升中活细胞散间的关系变化很大,因此有必要通过漶量特定脑杆菌的生长培养物在生长周期的不同时相的OD600值,并
CaCl2感受态细胞的制备实验步骤 ?1、从新活化的E.coli DH5α平板上挑取一单菌落,接种于3~5ml LB液 体培养中,37℃振荡培养至对数生长期(12h左右); ? 2、将该菌悬液以1:100~1:50转接于100ml LB液体培养基中,37℃振 荡扩大培养,当培养液开始出现混浊后,每隔20~30min测一次OD600,至OD600为0.3~0.5时停止培养,并转装到1.5 mL离心管中; ? 3、培养物于冰上放置20min; ? 4、 0~4℃,4000g离心10min,弃去上清液,加入1 mL冰冷的 0.1mo1/L CaCl2溶液,小心悬浮细胞, 冰浴20分钟; ?5、0~4℃, 4000g离心10min,倒净上清培养液,再用1.0 mL冰冷的 0.1mo1/L CaCl2溶液轻轻悬浮细胞,冰浴20min; ? 6、 0~4℃, 4000g离心10min,弃去上清液,加入100 μL冰冷的 0.1mo1/L CaCl2溶液,小心悬浮细胞,冰上放置片刻后,即制成了感 受态细胞悬液; ? 7、制备好的感受态细胞悬液可直接用于转化实验,如果在4℃放置 12~24h,其转化效率可以增高4~6倍;也可加入占总体积15%左右 高压灭菌过的甘油,混匀后分装于1.5ml离心管中,置于-70℃条件下, 可保存半年至一年。
CaCl2感受态细胞的制备实验步骤 1.前夜接种受体菌(DH5α或DH10B),挑取单菌落于LB培养基中 37℃摇床培养过夜(约16小时); 2 .取1ml过夜培养物转接于100ml LB培养基中,在37℃摇床上剧烈振 荡培养约2.5-3小时(250-300rpm); 3 .将0.1M CaCl2溶液置于冰上预冷;以下步骤需在超净工作台和冰上 操作; 4 .吸取1.5ml培养好的菌液至1.5ml离心管中,在冰上冷却10分钟; 5 .4℃下3000 g冷冻离心5分钟; 6 .弃去上清,加入100μl预冷0.1M CaCl2溶液,用移液枪轻轻上下吸动 打匀,使细胞重新悬浮,在冰放置20分钟; 7 . 4℃下3000 g冷冻离心5分钟; 8 .弃去上清,加入100μl预冷0.1M CaCl2溶液,用移液枪轻轻上下吸动 打匀,使细胞重新悬浮; 9 .细胞悬浮液可立即用于转化实验或添加冷冻保护剂(15% - 20%甘油)后超 低温冷冻贮存备用(-70℃)。 注意事项 1.细菌的生长状态:实验中应密切注视细菌的生长状态和密度,尽量使 用对数生长期的细胞(一般通过检测OD600来控制。DH5α菌株OD600为0.5时细胞密度是5×107/ml); 2 .所有操作均应在无菌条件和冰上进行; 3 .经CaCl2处理的细胞,在低温条件下,一定的时间内转化率随时间的 推移而增加,24小时达到最高,之后转化率再下降(这是由于总的 活菌数随时间延长而减少造成的); 4 .化合物及无机离子的影响:在Ca2+的基础上联合其他二价金属离子 (如Mn 2+或Co 2+)、DMSO或还原剂等物质处理细菌,可使转化效率大大提高(100-1000倍); 5 .所使用的器皿必须干净。迹量的去污剂或其它化学物质的存在可能 大大降低细菌的转化效率; 6 .质粒的大小及构型的影响:用于转化的应主要是超螺旋的DNA; 7 .一定范围内,转化效率与外源DNA的浓度呈正比;
全国免费电话:400-818-1148 TOP10 感受态细胞 Cat. No. JH0102-3 保存:-80℃ 组分说明 产品简介 本产品是大肠杆菌 TOP10 菌株经特殊工艺处理得到的感受态细胞,可用于 DNA 的热击转化。TOP10 是一种常用于质粒克隆的菌株,其 φ80lacZΔM15 基因产物可与载体编码的β-半乳糖苷酶氨基端实现α互补,可用于蓝白斑筛选。使用 pUC19质粒检测,转化效率可达 108,适用于高效的质粒 DNA 克隆并能保证高拷贝质粒的稳定复制。 注意事项 1.转化所有步骤均在无菌条件下操作。 2.感受态细胞应在-80℃下保存,不可多次冻融和放置时间过长,以免降低感受态细胞的转化效率。 3.包装中有0.1 ng/μl 的pUC19DNA ,供对照试验使用。 操作步骤 1.取感受态细胞置于冰浴中。一次转化感受态细胞的建议用量为50-100 μl ,可以根据实际情况分装使用。 以下实验以100 μl 感受态细胞为例。 2.待感受态细胞融化后,向感受态细胞悬液中加入目的DNA (根据实际情况加入适量的DNA ,通常100 μl 感受态细胞能够被1 ng 超螺旋质粒DNA 所饱和),用移液器轻轻吹打混匀,冰浴30分钟。 3.42℃热击90秒,迅速将离心管转移到冰浴中,冰上静置2-3分钟。 4.向每个离心管中加入900 μl 无菌的SOC 或LB 培养基(不含抗生素),混匀后置于37℃摇床,150 rpm 振荡培养45分钟使菌体复苏。 5.取100 μl 已转化的感受态细胞,加到含相应抗生素的SOC 或LB 固体琼脂培养基上,用无菌的涂布棒将细胞均匀涂开,直至干燥,倒置平板,37℃培养12-16小时。 注意:1) 涂布用量可根据具体实验调整。若转化的DNA 总量较多,可取少量转化产物涂布平板;若转化的DNA 总量较少,可取200-300 μl 转化产物涂布平板。若预计的克隆数较少,可通过离心(4,000 rpm ,2分钟)后吸除部分培养液,悬浮菌体后将其涂布于平板中。 2) 新制备的固体培养基不易涂干,可将平板正置于37℃直至液体被吸收后再倒置培养。 3) 涂布剩余的菌液可置于4℃保存,如果次日的转化菌落数过少,可以将剩下的菌液再涂布新培养基进行培养。 Cat. No. JH0102-3 Kit Size 10×100 μl TOP10 感受态细胞 10×100 μl Control DNA pUC19,0.1 ng/μl 10 μl
荧光共振能量转移(FRET)影像系统
Olympus(北京)销售服务有限公司上海分公司
PDF created with pdfFactory Pro trial version https://www.doczj.com/doc/2b15756042.html,
荧光共振能量转移(FRET)影像系统
一、研究目的
随着生命科学研究的不断深入, 光学显微镜使我们理解了细胞结构和有关功能。 但是分子 生物学研究已经显示了分子事件,例如信号传导和基因翻译,需要蛋白质的装配成特殊的大 分子复合体等。对各种生命现象发生的机制,特别是对细胞内蛋白质间相互作用的研究变得尤 为重要。 传统的生物物理或生物化学方法例如亲和色谱法或免疫沉淀反应法和近来的酵母双杂 交、磷酸化抗体、免疫荧光、放射性标记等方法等,都需要破碎细胞或对细胞造成损伤,无 法做到在活细胞生理条件下实时地对细胞内蛋白质-蛋白质间相互作用进行动态研究。 而基于强度的影像技术FRET方法,使得研究活细胞内的这些相互作用变得容易了,荧光 共振能量转移( FRET)是用于对生物大分子之间相互作用定性、定量检测的一种有效方法。根 据所基于的荧光显微镜配置不同而有不同的应用侧重,可在多细胞,单细胞,细胞膜,细胞 器等不同层次对生物大分子间的相互作用距离,动力学特性等进行研究。
二、FRET的原理和实现方法
FRET的原理和发生的基本条件:
1. 2. 3. 4. 发色团之间的距离在10A到100A 。 供体D的荧光光谱和受体A的吸收光谱足够多的重叠。 供体D的量子产率和受体A的吸收系数足够大。 D和A的跃迁偶极矩有最佳的相对取向,或者两者之一有一定的快速旋转的自由度。
FRET的实现方法:
1) 稳态方法(基于供体、受体的三通道计算校准) 供体荧光的减弱-主要的方法 受体荧光的增强 激发光谱和吸收光谱的比较 2) 3) 光漂白方法 (Pb-FRET) 时间分辨方法(TR-FRET) 供体荧光的衰减 受体荧光的增长
PDF created with pdfFactory Pro trial version https://www.doczj.com/doc/2b15756042.html,
感受态细胞的制备步骤和原理 实验目的 为了获得大量的质粒作为克隆载体,我们需要将购买来的质粒通过转化的方法导入细菌的细胞内。制备出感受态的细胞,我们就可以方便的将需克隆的质粒导入其中,使其繁殖,就能获得大量质粒。本次实验的目的就是增加质粒的数量。同时,我们能掌握大肠杆菌感受态细胞的制备及转化的基本原理和实验技术方法。 实验原理及过程 实验步骤 1挑取一个JM109单菌落于3ml LB(无抗生素)培养基中,37℃,180rpm培养过夜。 [让菌复苏,进入对数期的早中期。此时更容易制得感受态细胞。] 2取0.4mL过夜培养物加入到40mL LB(无抗生素)培养基中,37℃250rpm大约2.5小时。 [减少达到所需亮的均所需繁殖的世代数,以减少菌的变 GAGGAGAGGAFFFFAFAF
异。] 3取培养物置于40mL的灭菌离心管中,冰浴10min,4000rpm 4℃离心10min,弃上清。(将所有上清倒光,可以将离心管倒过来) [使细菌的生长停止,代谢减慢。因为这时已经没有培养基了,如果细菌仍有很高的代谢效率,则有大量细菌死亡。] 4菌体重悬于10mL 100mmol/L冰冷CaCl2中,轻吹,4℃30min,4000rpm离心5min弃上清。 [ 细菌处于0度,CaCl2低渗溶液中,菌细胞膨胀成球形,外源DNA形成抗DNA酶的羟基-钙磷酸盐混合物黏附于细胞表面(羟基来源于细胞壁上的糖,磷酸来源于DNA),经短时间420C热激处理,促进细胞吸收DNA复合物。(一定要轻吹,这时细胞壁打开,十分脆弱)] 5菌体重悬于1mL 100mmol/L冰冷的CaCl2中,轻吹,用于转化。 [除去所有的LB以及细胞破损后释放出来的细胞内含物。防止细胞分裂,以致大量细胞死亡。(一定要轻吹,这时细 胞壁打开,十分脆弱)] GAGGAGAGGAFFFFAFAF
感受态细胞的制备步骤和原理 实验目的为了获得大量的质粒作为克隆载体,我们需要将购买来的质粒通过转化的方法导入细菌的细 胞内。制备出感受态的细胞,我们就可以方便的将需克隆的质粒导入其中,使其繁殖,就能 获得大量质粒。本次实验的目的就是增加质粒的数量。同时,我们能掌握大肠杆菌感受态细 胞的制备及转化的基本原理和实验技术方法。实验原理及过程实验步骤 1挑取一个JM109单菌落于3ml LB(无抗生素)培养基中,37C, 180rpm培养过夜。[让菌复苏, 进入对数期的早中期。此时更容易制得感受态细胞。] 2取0.4mL过夜培养物加入到40mL LB(无抗生素)培养基中,37C 250rpm大约2.5小时。 [减少达到所需亮的均所需繁殖的世代数,以减少菌的变异。] 3取培养物置于40m L的灭菌离心管中,冰浴10min , 4000rpm 4C离心10min,弃上清。(将所 有上清倒光,可以将离心管倒过来) [使细菌的生长停止,代谢减慢。因为这时已经没有培养基了,如果细菌仍有很高的代谢效率,则有大量细菌死亡。] 4菌体重悬于10mL 100mmol/L 冰冷CaCl2中,轻吹,4 C 30min , 4000rpm离心5min弃上 [细菌处于0度,CaCl2低渗溶液中,菌细胞膨胀成球形,外源DNA形成抗DNA酶的羟基 -钙磷 酸盐混合物黏附于细胞表面(羟基来源于细胞壁上的糖,磷酸来源于DNA ),经短时 间420C热激处理,促进细胞吸收DNA复合物。(一定要轻吹,这时细胞壁打开,十分脆弱)] 5 菌体重悬于1mL 100mmol/L冰冷的CaCl2中,轻吹,用于转化。 [除去所有的LB以及细胞破损后释放出来的细胞内含物。防止细胞分裂,以致大量细胞死亡。 (一定要轻吹,这时细胞壁打开,十分脆弱)] 6取感受态细胞100uL,加入2uLpET-21a质粒,冰浴30 min受体菌:感受态细胞100uL, 加入2 uL 无菌双蒸水阴性对照:0.1mol/LCaCl2溶液100uL,加入2 uL阴性对照 [第一个阴性对照是为了验证LB中的抗生素有活性;第二个阴性对照是为了验证CaCl2溶 液和pET-21a质粒未被污染。][冰浴是为了降低细胞代谢率,防止细胞大量死亡。] 7热激42度,90s 【在低温处理后,热激,可以使细胞壁热胀冷缩,再低温,使细胞壁上黏附的质粒进入细胞体内。】8冰浴2 min 9加入800 uL无抗生素的LB, 37 C复苏1h 【用LB复苏细胞,使细胞恢复活力,从而使细胞中的质粒表达抗抗生素的基因,只有这样才能使 转化成功的细菌在有抗生素的LB平板上生长。】 10取100ul细胞直接在含50ug/mL Carbenicillin(短节西林)的LB培养基上铺平板,将平皿放置 37 oC温箱30min至液体被吸收。倒置平皿37 oC培养过夜,出现菌落。 【这些菌落为转化成功的菌落,而对照组应该没有菌落。在目的菌落的旁边有可能出现卫星菌落,这是因为亩的菌落的抗性基因似的菌落周围的抗生素失效,长出了转化为成功的菌 落。用短节西林因其对酸稳定、不易被细菌降解,所以可以降低卫星菌落的数量。 因为抗生素高温易失活,所以应等到LB600C时(热而不烫),加入抗生素。】
激光全息细胞成像及分析系统应用 细胞活力 激光全息细胞成像及分析系统可以实时监测细胞死亡过程,以及通过图像进行记录。全息技术再不需要荧光标记的情况下可以得到细胞形态学数据。Khmaladze A. et al(2012和Pavillion N. et al(2012使用DHM 研究细胞死亡过程,观察到死亡过程中细胞体积显著减小。Kuhn et al(2013使用DHM 研究活/死细胞特点时得到实验结果和和基于荧光标记方法结果相一致。他们使用PI 和Hoechst 标记细胞。染料法鉴定细胞死活是目前常见的鉴定方法,其中台盼蓝染色方法最常见。台盼蓝可穿透变性的细胞膜,与解体的DNA 结合,使其着色,而活细胞能阻止染料进入细胞内,故可以鉴别死细胞与活细胞。鼠成纤维细胞L929接种在μ-slide(Ibidi,Germany 上,肿瘤药物依托泊苷etoposide(100μM处理细胞,使用激光全息细胞分析系统(M3 分析细胞的死亡,并与台盼蓝染色法进行比较。图1中左图为台盼蓝染色结果,右图为全息结果,细胞越白,细胞越厚。死细胞是圆的,薄的。两种方法得到的结果是 一样的。
图1
图2细胞厚度VS 细胞体积,死亡细胞集中在绿色区域 细胞凋亡 细胞死亡起码有两种方式,即细胞坏死(necrosis)与细胞凋亡(apoptosis。细胞坏死是细胞受到强烈理化或生物因素作用引起细胞无序变化的死亡过程。表现为细胞胀大,胞膜破裂,细胞内容物外溢,核 变化较慢,DNA 降解不充分,引起局部严重的炎症反应。细胞凋亡是指为维持内环境稳定,由基因控制的细胞自主的有序的死亡。在这两种过程中,细胞体积都会减少,形态学都会发生变化。 前列腺癌细胞DU145和小鼠成纤维细胞L929分别接种在IBIDI-micro slides (IBIDI)上,接种24h 后,50μM依托泊苷(etoposide 处理细胞,HoloMonitor M3分析细胞死亡过程。
细胞迁移/侵袭实验分析 ——LumaScope活细胞成像系统 细胞迁移实验是普遍应用于评价损伤修复、贴壁肿瘤细胞转移或血管再生等的典型实验。传统方法是应用无菌枪头(Tip)在细胞培养容器上划痕来实现。但是此种方法无法实现在不同孔中划出同样大小的划痕。Oris TM迁移/侵袭试剂盒能够提供更加精确的方法,在培养容器中生成一个圆形区域。这种方法同样适用于观察不同方向的细胞迁移。LumaScope活细胞成像系统具备传统显微镜的功能,可应用于细胞或组织培养实验室的日常细胞观察。如细胞状态实时检测、远程传送和监控、细胞计数、形态观察、染色观察等。LumaScope可放置于培养箱中实现细胞的长时间的连续成像和定点监测。这种应用大大提高了实验过程检测的便捷性和结果的准确性。 本文将描述如何结合LumaScope与Oris TM迁移/侵袭试剂盒来实现细胞迁移/侵袭实验。实验结果可通过Image J软件来进行分析,最终得到细胞迁移的数量和速度数据。 细胞迁移分析: Day 1:am 9:00插入Stoppers,接种细胞; pm 4:00(根据细胞贴附状况),拔除stoppers,PBS洗一遍后加入新鲜培养液。Day 2:根据要观察的时间点设置LumaScope成像参数进行成像,并进行量化分析。 细胞侵袭分析: Day 1:am 9:00包被薄层BME(用无血清培养液制备),插入stoppers,接种细胞; pm 4:00 (根据细胞状况),拔除stoppers,包被厚层BME(含血清生长因子),再在第二层gel上面加上一层无血清培养液 Day 2:根据要观察的时间点设置LumaScope成像参数进行成像,并进行量化。
一、目的 1.了解感受态细胞生理特性及制备条件,掌握大肠杆菌感受态细胞制备方法。 2.掌握质粒DNA 转化大肠杆菌的方法,了解转化的条件和利用半乳糖苷酶基因插入失活选择重组质粒DNA 的原理。 二、原理 (一)大肠杆菌感受态细胞制备的原理 所谓感受态,是指细菌生长过程中的某一阶段的培养物,只有某一生长阶段中的细菌才能作为转化的受体,能接受外源DNA而不将其降解的生理状态。感受态形成后,细胞生理状态会发生改变,出现各种蛋白质和酶,负责供体DNA 的结合和加工等。细胞表面正电荷增加,通透性增加,形成能接受外来的DNA 分子的受体位点等。本实验为了把外源DNA(重组质粒)引入大肠杆菌,就必须先制备能吸收外来DNA分子 的感受态细胞。在细菌中,能发生感受态细胞是占极少数。而且,细菌的感受态是在短暂时间内发生。 目前对感受态细胞能接受外来DNA 分子的本质看法不一。主要有两种假说: 1、局部原生质体化假说――细胞表面的细胞壁结构发生变化,即局部失去细胞壁或局部溶解细胞壁,使DNA 分子能通过质膜进细胞。证据有: (1)发芽的芽孢杆菌容易转化; (2)大肠杆菌的原生质体不能被噬菌体感染,却能受噬菌体DNA 转化; (3)适量的溶菌酶能提高转化率。 2、酶受体假说――感受态细胞的表面形成一种能接受DNA 的酶位点,使DNA分子能进入细胞。证据是:(1)蛋白质合成的抑制剂如氯霉素,可以抑制转化作用;
(2)细胞分裂过程中,一直有局部原生质化,但感受态只在生长对数期的中早期出现;(3)分离到感受态因子,能使非感受态细胞转变为感受细胞。 目前对感受态细胞的转化理论尚未有统一结论,但是许多实验室一直进行探索,试图从实验中获得明确回答。有人根据pBR322 质粒DNA对E?coli K――12X1776菌株的转化结果,认为: 近来,在许多研究室都发现CaCl2对受体菌处理,可提高转化效率几十倍,通常把细胞悬浮在pH6.0 的100mmol/L CaCl2中,在冰浴条件下,放置过夜,转化率转高,但一过24小时,转化率测恢复为原来的水平。 (二)重组DNA 的转化原理 我们已经制备好大肠杆菌感受态细胞,接下的实验是把重组的DNA 引入受体细胞,使受体菌具有新的遗传特性,并从中选出转化子。作为受体的大肠杆菌C600 或DH5α,必须不同外来DNA分子发生遗传重组,通常是rec基因缺陷型的突变体,同时它们必须是限制系统缺陷或限制与修饰系统均缺陷的菌株。这样外来的DNA分子不会受其限制酶的降解。保持外来DNA 分子在受体细胞中的稳定性。制备的大肠杆菌细胞就具有这三种缺陷(rk- mk- rec-)同时此受体细胞还是氨苄青霉素敏感(Ap)。 在体外构建好的重组分子上具有分解氨苄青霉素(Ap)基因存在,当它导入受体细胞后,就赋于这些受体细胞新的特性,即Ap 抗性。同时载体质粒上具有乳糖操纵的β一半乳糖苷酶基因(lacZ,我们可以利用外源基因插入载体β一半乳糖苷酶基因(lacZ,使其失去β一半乳糖苷酶活性的原理来选择新构建的重组子。