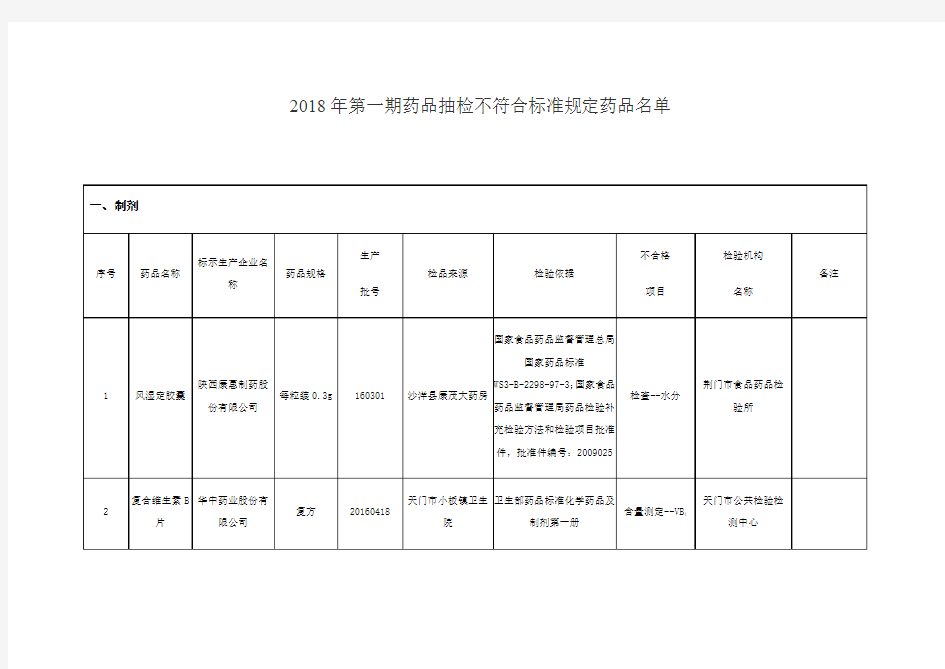
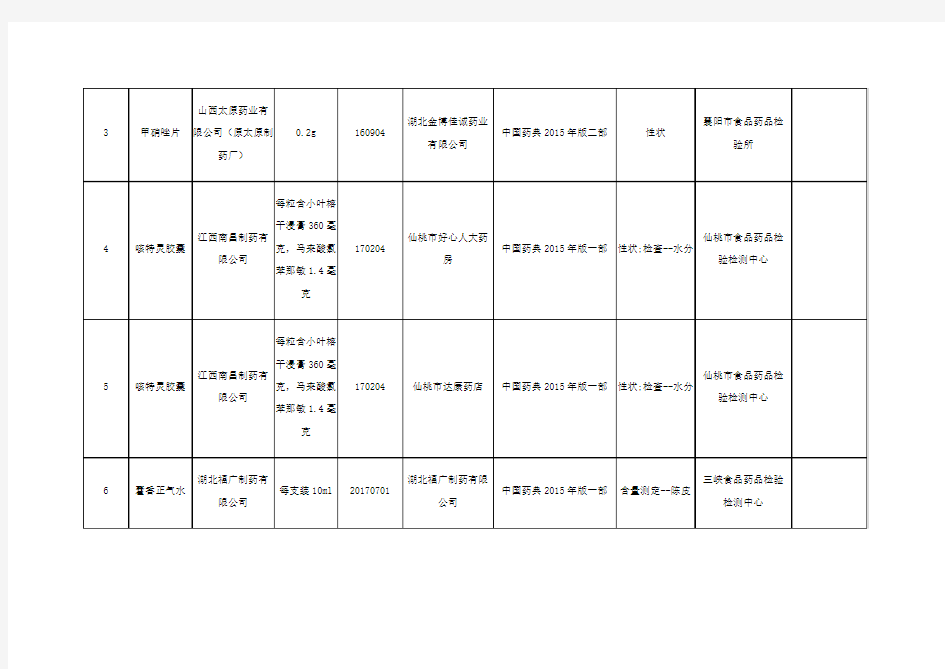
2018年第一期药品抽检不符合标准规定药品名单
备注:标“*”的药品为标示生产企业否认为该企业生产。
国家药品质量管理(GSP)制度范本 质量管理人员的任命 x x x x药店文件 经本企业研究决定,从发文之日起设立本企业质量管理人员,具体负责本企业的质量管理工作,在企业内部对药品质量进行裁决,对本企业经营药品的质量具有一票否决权,提出并监督实施采购合同的质量条款,规范本企业台帐、原始记录,接受企业内部关于质量技术问题的咨询。同时,任命XX为质量负责人。 质量负责人的职能是: 一、负责贯彻执行国家有关药品质量管理的法律、法规和行政规章。包括: 1、组织学习国家有关药品质量管理的法律、法规和行政规章。 2、宣传、贯彻、执行国家有关药品质量管理的法律、法规和行政规章。 3、熟练掌握药品法律、法规等基本知识。 4、指导本企业在药品的购进、验收、储存与养护中严格按药品的有关法律、法规办事。 二、负责起草、编制本企业药品质量管理制度,并指导、督促制度的执行。包括药品质量管理制度的起草和操作规程的制定。 三、负责首营企业的质量审核。 四、负责首营品种的质量审核。 五、负责建立企业所经营品种包含质量标准等内容的质量档案。
六、负责药品质量的查询和药品质量事故或质量投诉的调查、处理及报告。 七、负责药品的验收管理。 八、负责指导和监督药品保管、养护和运输中的质量工作。 九、负责质量不合格药品的审核,对不合格药品的处理过程实施监督。包括对不合格药品的确认、处理、报损和监督销毁。 十、负责收集和分析药品质量信息。包括本企业的外部信息和内部信息的收集、分析和报告。 十一、负责协助开展对本企业职工药品质量管理方面的教育或培训。 药品购进管理制度 XXXX药店文件 一、药品进货必须严格执行《药品管理法》、《产品质量法》、《合同法》及《药品经营质量管理规范》等有关法律法规,依法购进。 二、进货人员须经专业和有关药品法律法规培训,考试合格,持证上岗。 三、购进药品以质量为前提,从具有合法证照的供货单位进货。 四、购进药品要有合法票据,并依据原始票据建立购
对照药品(参比制剂)一次性进口公告解读 对照药品一次性进口公告有以下特点: 1.公告指出:受理审查及审批权力委托给各省局,极大程度简化了程序,节约时间; 2.公告规定了受理时限5个工作日,审查时限20个工作日。 3.进口药品批件有效期延长至12个月。 4.申报资料不再要求提供对照药品的质量标准和出厂检验报告书, 5.需向预进口口岸食品药品监督管理局进行备案。 进口备案要求 拟进口的对照药品应从《进口药品批件》载明的药品进口口岸进口。进口备案按照以下程序办理: (一)申请人向口岸食品药品监督管理局提出申请办理《进口通关单》,并同时提供以下资料: 1.所进口药品的《进口药品批件》; 2.申请人机构合法登记证明文件复印件(如营业执照、组织机构代码证等)。属于委托申请的,另须提供委托人的合法登记证明文件复印件及委托证明文件; 3.原产地证明复印件; 4.货物合同复印件; 5.装箱单、提运单和货运发票复印件; 6.药品说明书及包装、标签式样(原料药和制剂中间体除外); 7.经其他国家或者地区转口的进口药品,需要同时提交从原产地到各转口地的全部购货合同、装箱单、提运单和货运发票等。 上述各类复印件应当加盖申请人公章。 (二)口岸食品药品监督管理局应按照《药品进口管理办法》的相关规定办理对照药品的进口备案(进口单位取得《药品经营许可证》(生产企业应当取得《药品生产许可证》)和《企业法人营业执照》)。审查工作中,加强对原产地证明文件、购货合同以及发票等文件的审核,确认所进口对照药品的真实产地。 (三)口岸食品药品监督管理局审查全部资料无误后,准予进口备案,发出《进口药品通关单》。 6.明确说明:研究用对照药品的一次性进口申请,可不要求进行口岸检验的规定。 7.临床试验对照品检验规定放宽为申请人可自行检验,也可委托药品检验所进行委托检验。 8.明确规定临床试验对照药品的检验标准选择:(1)生产厂家的质量标准;(2)《中华人民共和国药典》2015年版中收载对的标准;(3)申请人自拟标准,且符合《中华人民共和国药典》2015年版的通用要求。 9.增加了麻醉药品、精神药品、临床试验用生物制品的对照药品不适用本公告的规定。 1
药品质量公告 (总第67期) 国家食品药品监督管理局发布 2006年10月 根据2006年全国药品抽验计划,国家食品药品监督管理局组织有关单位在全国范围内对相关品种进行了药品质量抽查检验,在抽验中发现18个批次的药品为假冒药品,涉及1家医疗机构、17家药品经营企业(见附表),现予以公告。 对公告中的假冒药品立即停止销售和使用,涉及经营、使用假冒药品的经营企业和医疗机构所在地的药品监督管理部门要认真追查假药的来源,依法从严查处。 附表:假冒药品名单
药品质量公告 (总第68期) 国家食品药品监督管理局发布 2007年3月
进行了药品监督抽验,本期公告内容为在流通和使用单位对15个品种的不同剂型进行抽查检验的结果。具体为: 阿司匹林(肠溶片、胶囊、泡腾片、维生素C分散片):41家生产企业的200批次为合格药品,5家生产企业的9个批次为不合格药品; 氨咖黄敏(胶囊、颗粒、片):44家生产企业的205批次为合格药品,1家生产企业的1个批次为不合格药品; 布洛芬缓释胶囊:34家生产企业的182批次为合格药品,1家生产企业的1个批次为不合格药品; 复方氨酚烷胺(片、分散片):149家生产企业的746批次为合格药品,5家生产企业的5个批次为不合格药品; 复方对乙酰氨基酚片:52家生产企业的165批次为合格药品,4家生产企业的5个批次为不合格药品; 交沙霉素片:13家生产企业的129批次为合格药品,1家生产企业的1个批次为不合格药品; 青霉素V钾(片、分散片):36家生产企业的394批次为合格药品,5家生产企业的27个批次为不合格药品; 维生素B12片:15家生产企业的314批次为合格药品,1家生产企业的1个批次为不合格药品; 维生素E烟酸酯(胶囊、胶丸):19家生产企业的53批次为合格药品,1家生产企业的1个批次为不合格药品; 盐酸利多卡因注射液、蚓激酶(胶囊、片)、甲硫氨酸片、胰激肽原酶肠溶片、左旋多巴注射液和对乙酰氨基酚颗粒6个品种抽验结果为符合规定。 药品质量公告 2007年度第一期 (总第69期) 国家食品药品监督管理局发布 2007年5月
国家食品药品监督管理局关于发布2003年第二季度国家药品 质量公告的通知 【法规类别】药品管理 【发文字号】国食药监市[2003]177号 【发布部门】国家食品药品监督管理局(原国家药品监督管理局)(已撤销) 【发布日期】2003.07.23 【实施日期】2003.07.23 【时效性】现行有效 【效力级别】XE0303 国家食品药品监督管理局关于发布2003年第二季度国家药品质量公告的通知 (国食药监市[2003]177号) 各省、自治区、直辖市药品监督管理局,解放军总后卫生部,武警总部卫生部,中国药品生物制品检定所: 根据2003年国家药品抽验计划,中国药品生物制品检定所组织各省级药品检验所在全国范围内对药品经营企业、医疗机构及中药材专业市场进行了抽查检验(结果详见附件1:药品质量公告,总第56期)。现将抽验结果予以公告并将有关事项通知如下: 一、2003年第二季度完成国家计划抽验概况 中国药品生物制品检定所组织省级药品检验所完成了对阿替洛尔片等20种化学药品在经营企业、医疗机构的统一抽验工作,共抽验2694个批次。其中,合格批次为2637批
次,抽验合格率(批次)为97.9%。 中国药品生物制品检定所组织省级药品检验所,完成了对17家中药材专业市场的监督检查抽验工作。共抽取样品2259件,涉及药材品种86种。其中,合格件次为1616件,不合格件次为643件。其中重点抽验了常用中药材丹参、金银花、山茱萸、防已、菟丝子、黄芩、黄柏、厚朴、黄连、黄芪、龙胆、南五味子、乌梅、栀子、枳壳等15个品种。共抽验了1598件样品,合格件次为1079件,不合格件次为519件。 从抽验结果看,流通领域化学药品的质量较为稳定。中药材专业市场的药材质量仍存在严重问题,市场管理仍存在混乱现象。这表明当前迫切需要对中药材专业市场进行整顿和规范。
《中国药典》2015年版实施公告 有关问题的解读(一) 1. 问:国家食品药品监督管理总局关于实施《中华人民共和国 药典》2015年版(以下简称“2015年版药典”)有关事宜的公告(以下简称“公告”)(2015年第105号)中规定,为符合2015年版药典而需进行补充申请的,应在2015年 12月1日前进行申报,2015年12月1日后是否仍可提交相应补充申请? 答:对2015年版药典发布前已上市药品,生产企业应在2015年12月1日前完成原标准与新版药典相关要求的研究和比对,并应按公告要求进行相应的备案或补充申报。2015年12月1日以后仍可以提交相应补充申请。 2. 问:企业的注册标准已经对2010年版药典相关品种进行评估 的,且2010年版与2015年版药典品种质量标准和检测方法无变化的,是否需要重新对产品进行评估? 答:虽然品种正文内容与2015年版药典品种规定无变化,但由于2015年版药典通用性要求,包括凡例、通则、制剂通则以及通用性检验方法等进行了全面的增修订,因此,生产企业仍需针对2015年版药典通用性要求方面对本产品进行相应的评估。 3. 问:关于药品执行标准的表述方式的问题
答:对于注册标准不低于《中国药典》项目的制品,执行注册标准,其执行标准表示方式为:“执行药品注册标准且符合《中国药典》2015年版要求”。 4. 问:对于进口药品生产企业,能否使用注册代理公司出具的 说明信来代替国外的声明信,进行备案或补充申请的申报?答:原则上注册代理公司应出具持证商的声明信。如使用说明信代替国外的声明信,应同时提供进口药品生产企业出具的委托注册代理公司办理该事项的委托书。 5. 问:国家食药总局2015年第67号公告中规定,2015年版药 典自2015年12月1日起实施”。如何界定产品的执行日期?答:按是历版药典执行惯例要求,自2015年12月1日起生产或进口的药品应符合2015年版药典的相关规定。 6. 问:按照实施公告要求提出备案或补充申请的品种,审评审 批期间是否仍可执行原标准,期间若有进口再注册申请的是否可按原注册标准核发新证。 答:申请人应按105号实施公告第五款规定执行。出现补充申请与再注册申请交叉情形者,建议补充申请与进口再注册合并审评,如2015年12月1日起前已提交补充申请,可在补充申请期间执行原标准的要求。 7. 问:制剂中间体是否也需要按照制剂的药典标准进行提高?
2015年执业药师考试真题《药事管理与法规》 一、最佳选择题(共 40 题,每题 1 分,每题的备选项中,只有一个最符合题意) 1.关于执业药师资格考试和注册管理的说法,正确的是 A.香港、澳门,台湾居民,按照规定的程序和报名条件,可以报名參加国家执业药师资格考试 B.不在中国就业的外国人,符合规定的学历条件,可以报名参加国家职业药师资格考试 C.执业药师执业单位包括医药院校、科研单位、药品检验机构 D.在香港、澳门注册的药剂师可以直接递交注册申请资料办理执业药师注册 【答案】A 【解析】执业药师注册管理 1.注册机构:各省级食药监部门为本辖区执业药师注册机构。 2.执业范围:药品生产、药品经营、药品使用。机关、院校、科研单位、药品检验机构不予注册。 2.下列内容属于执业药师职责范畴的是 A.指导公众合理使用处方药 B.指导公众合理使用非处方药 C.执行药品不良反应报告制度 D.为无处方患者提供用药处方 【答案】D 【解析】执业药师的职责 (1)执业药师必须遵守职业道德,忠于职守,以对药品质量负责、保证公众用药安全有效为基本准则。(2)执业药师必须严格执行《药品管理法》及国家有关药品研制、生产、经营、使用的各项法规及政策,对违反《药品管理法》及有关法规的行为或决定,有责任提出劝告、制止、拒绝执行并向上级报告。(3)执业药师在执业范围内负责对药品质量监督和管理,参于制定、实施药品质量监督和管理,参与制定、实施药品全面质量管理及对本单位违反规定的处理。 (4)执业药师负责处方的审核及监督调配,提供用药咨询与信息,指导合理用药,开展治疗药物的监测及药品疗效的评价等临床药学工作。 3、关于药品安全风险和药品安全风险管理措施的说法,错误的是 A.药品内在属性决定药品具有不可避免的药品安全风险 B.不合理用药,用药差错是导致药品安全风险的关键因素 C.药品生产企业应担负起药品整个生命周期的安全监测和风险管理工作 D.实施药品安全风险管理的有效措施是要从药品注册环节消除各种药品风险因素 【答案】D 【解析】药品安全风险可分为自然风险和人为风险。存在与药品研制、生产、经营、使用各个环节。 4、关于建立健全覆盖城乡居民的基本医疗卫生制度的基本内容的说法 A.建立健全公共卫生服务体系 B.加快建设多层次医疗保障体系 C.完善以县级公立医院为主的医疗服务体系 D.建立健全以国家基本药物制度为基础的药品供应保障关系
国家药品质量公告(2009年第4期,总第80号) 2010年01月14日发布 国家食品药品监督管理局 公告 2010年第2号 国家药品质量公告 (2009年第4期,总第80号) 为加强药品监管,保障公众用药安全,根据2009年国家药品评价抽验计划,国家食品药品监督管理局近期在全国范围内组织对布洛芬制剂、地高辛片、尼群地平片、盐酸溴己新制剂、格列本脲片、双黄连口服制剂、双黄连注射剂、消渴丸、参麦注射液、血塞通注射液、八珍益母制剂、格列吡嗪制剂、硝苯地平制剂、盐酸雷尼替丁胶囊、柴胡注射液、脉络宁注射液、硫酸沙丁胺醇制剂、奥美拉唑制剂18个国家基本药物品种进行了评价抽验。本次抽验的4868批次产品中,有4860批次产品符合标准规定,8批次产品不符合标准规定,结果显示,总体质量状况良好。现将抽验结果公告如下: 一、布洛芬制剂(片、胶囊、缓释片)
全国共有498个药品批准文号,368个生产企业。本次在流通领域抽样350批,涉及47个生产企业,经陕西省食品药品检验所检验,全部符合标准规定。 二、地高辛片 全国共有12个药品批准文号,3个生产企业。本次在流通领域抽样281批,涉及3个生产企业,经福建省药品检验所检验,全部符合标准规定。 三、尼群地平片 全国共有227个药品批准文号,191个生产企业。本次在流通领域抽样337批,涉及38个生产企业,经内蒙古自治区食品药品检验所检验,全部符合标准规定。 四、盐酸溴己新制剂(片、注射液) 全国共有50个药品批准文号,50个生产企业。本次在流通领域抽样198批,涉及10个生产企业,经北京市药品检验所检验,全部符合标准规定。 五、格列本脲片 全国共有85个药品批准文号,85个生产企业。本次在流通领域抽样298批,涉及16个生产企业,经贵州省食品药品检验所检验,全部符合标准规定。 六、双黄连口服制剂(片、颗粒、口服液) 全国共有27个药品批准文号,25个生产企业。本次在流通领域抽样325批,涉及21个生产企业,经黑龙江省药品检验所检验,全部符合标准规定。 七、双黄连注射剂(注射用双黄连〔冻干〕、双黄连注射液、粉针剂)
1.问:国家食品药品监督管理总局关于实施《中华人民共和国药典》2015年版(以下简称“2015年版药典”)有关事宜的公告(以下简称“公告”)(2015年第105号)中规定,为符合2015年版药典而需进行补充申请的,应在2015年12月1日前进行申报,2015年12月1日后是否仍可提交相应补充申请? 答:对2015年版药典发布前已上市药品,生产企业应在2015年12月1日前完成原标准与新版药典相关要求的研究和比对,并应按公告要求进行相应的备案或补充申报。2015年12月1日以后仍可以提交相应补充申请。 2.问:企业的注册标准已经对2010年版药典相关品种进行评估的,且2010年版与2015年版药典品种质量标准和检测方法无变化的,是否需要重新对产品进行评估? 答:虽然品种正文内容与2015年版药典品种规定无变化,但由于2015年版药典通用性要求,包括凡例、通则、制剂通则以及通用性检验方法等进行了全面的增修订,因此,生产企业仍需针对2015年版药典通用性要求方面对本产品进行相应的评估。 3.问:关于药品执行标准的表述方式的问题。 答:对于注册标准不低于《中国药典》项目的制品,执行注册标准,其执行标准表示方式为:“执行药品注册标准且符合《中国药典》2015年版要求”。 4.问:对于进口药品生产企业,能否使用注册代理公司出具的说明信来代替国外的声明信,进行备案或补充申请的申报?
答:原则上注册代理公司应出具持证商的声明信。如使用说明信代替国外的声明信,应同时提供进口药品生产企业出具的委托注册代理公司办理该事项的委托书。 5.问:国家食药总局2015年第67号公告中规定,2015年版药典自2015年12月1日起实施“。如何界定产品的执行日期? 答:按是历版药典执行惯例要求,自2015年12月1日起生产或进口的药品应符合2015年版药典的相关规定。 6.问:按照实施公告要求提出备案或补充申请的品种,审评审批期间是否仍可执行原标准,期间若有进口再注册申请的是否可按原注册标准核发新证。 答:申请人应按105号实施公告第五款规定执行。出现补充申请与再注册申请交叉情形者,建议补充申请与进口再注册合并审评,如2015年12月1日起前已提交补充申请,可在补充申请期间执行原标准的要求。 7.问:制剂中间体是否也需要按照制剂的药典标准进行提高? 答:生产企业根据需求,自行对制剂中间体的质量标准进行评估,确保其生产的制剂应符合2015版药典要求。按批准文号管理的制剂中间体必须执行105号公告的要求。
对照药品(参比制剂)一次性进口公告解读对照药品一次性进口公告有以下特点: 1.公告指出:受理审查及审批权力委托给各省局,极大程度简化了程序,节约时间; 2.公告规定了受理时限5个工作日,审查时限20个工作日。 3.进口药品批件有效期延长至12个月。 4.申报资料不再要求提供对照药品的质量标准和出厂检验报告书, 5.需向预进口口岸食品药品监督管理局进行备案。 进口备案要求 拟进口的对照药品应从《进口药品批件》载明的药品进口口岸进口。进口备案按照以下程序办理: (一)申请人向口岸食品药品监督管理局提出申请办理《进口通关单》,并同时提供以下资料: 1.所进口药品的《进口药品批件》; 2.申请人机构合法登记证明文件复印件(如营业执照、组织机构代码证等)。属于委托申请的,另须提供委托人的合法登记证明文件复印件及委托证明文件; 3.原产地证明复印件; 4.货物合同复印件; 5.装箱单、提运单和货运发票复印件; 6.药品说明书及包装、标签式样(原料药和制剂中间体除外); 7.经其他国家或者地区转口的进口药品,需要同时提交从原产地到各转口地的全部购货合同、装箱单、提运单和货运发票等。 上述各类复印件应当加盖申请人公章。
(二)口岸食品药品监督管理局应按照《药品进口管理办法》的相关规定办理对照药品的进口备案(进口单位取得《药品经营许可证》(生产企业应当取得《药品生产许可证》)和《企业法人营业执照》)。审查工作中,加强对原产地证明文件、购货合同以及发票等文件的审核,确认所进口对照药品的真实产地。 (三)口岸食品药品监督管理局审查全部资料无误后,准予进口备案,发出《进口药品通关单》。 6.明确说明:研究用对照药品的一次性进口申请,可不要求进行口岸检验的规定。 7.临床试验对照品检验规定放宽为申请人可自行检验,也可委托药品检验所进行委托检验。 8.明确规定临床试验对照药品的检验标准选择:(1)生产厂家的质量标准;(2)《中华人民共和国药典》2015年版中收载对的标准;(3)申请人自拟标准,且符合《中华人民共和国药典》2015年版的通用要求。 9.增加了麻醉药品、精神药品、临床试验用生物制品的对照药品不适用本公告的规定。
附件 关于药物研制过程中所需对照药品 一次性进口有关事项的公告 (征求意见稿) 根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)、《国务院办公厅关于开展仿制药质量和疗效一致性评价意见的通知》(国办发〔2016〕8号)、《药品进口管理办法》(国家食品药品监督管理局令2003年第4号)及《关于药品注册审评审批若干政策的公告》(国家食品药品监督管理总局2015年第230号)的规定,对符合条件的药物研制过程中所需对照药品,可予以一次性进口。现将有关事项公告如下: 一、适用范围 已在中国境外上市但尚未在中国境内批准上市的药品,用于以下用途的: (一)在中国境内药品注册相关研究中用于对照药品的药品; (二)仿制药质量和疗效一致性评价研究中用于对照药品的化学药品。 二、申报程序 (一)申请人按要求准备申请资料,向各省级食品药品监管
部门提出一次性进口申请。各省级食品药品监管部门对申请人提出的申请进行审核,并出具审核意见表(附件1)。 (二)申请人将资料和省级食品药品监管部门出具的审核意见表,一并向国家食品药品监督管理总局受理部门提交一次性进口申请。 (三)受理部门对申请人提交的相关材料进行形式审查,符合要求的,予以受理,并将申报资料送交国家食品药品监督管理总局审查。 (四)国家食品药品监督管理总局组织进行审查,符合要求的,发给《进口药品批件》。不符合要求的,发给《审批意见通知件》。 三、资料要求 申请人应当填写《进口药品批件申请表》(附件2),同时提交下列资料: (一)申请人登记证明文件(如营业执照等),《药品生产许可证》及变更记录页。申请人非进口药品使用者的,提供实际使用者的委托书。 (二)加盖申请人公章的书面申请报告。报告内容应包括:本申请所符合的情形、申请进口的原因和依据,申请进口的药品国外上市情况、申请进口药品的数量及使用方案。若申请用于开展药学研究、非临床研究的对照药品,申请人须承诺所进口药品
新标准未颁布前,仍执行原药典标准,但应符合新版药典的通用要求。 四、药品注册标准中收载检验项目多于(包括异于)药典规定或质量指标严于药典要求的,应在执行药典要求的基础上,同时执行原注册标准的相应项目和指标。因辅料及生产工艺等差异导致的检测项目差异,生产企业应基于科学、质量可控的原则开展研究,必要时申报药品补充申请。 药品注册标准收载检验项目少于药典规定或质量指标低于药典要求的,应执行药典规定。 五、《中国药典》2015年版发布之日(含当日)前已获批的药品应自2015年12月1日起执行新版药典相应要求。如涉及药品处方、原辅材料和生产工艺等变更的,应按照《药品注册管理办法》规定,在2015年12月1日前向国家食品药品监督管理总局提交补充申请,审评审批期间仍可执行原标准,审批通过者执行新标准,审批不通过者应立即停产。 仅涉及明确检测项目、指标限度调整但不涉及药品处方、原辅材料来源、生产和工艺变更的,应在实施之日前向省级食品药品监督管理部门(进口药品报国家食品药品监督管理总局)备案。 六、《中国药典》2015年版发布之日起(不含当日)新提交的药品注册申请,应按照新版药典相关要求开展研究并提交申报资料,技术审评部门应按照新版药典相关要求开展审评,不符合要求者不予批准。 《中国药典》2015年版发布之日(含当日)前已受理、技术审评部门尚未完成相关技术审评的注册申请,按照新版药典的相关要求开展审评;技术审评部门已完成相关技术审评的注册申请,药品批准上市后6个月内应符合新版药典的相关要求。 七、根据《中国药典》2015年版的增修订内容,药品生产企业应在上述规定期限前按《药品注册管理办法》规定及时提出变更药品说明书和标签的补充申请。规定期限后生产的药品必须使用变更
国家食品药品监督管理局 公告 2011年第6号 国家药品质量公告 (2010年第4期,总第84号) 为加强药品监管,保障公众用药安全,根据全国药品抽验工作计划,国家食品药品监督管理局在全国范围内组织对盐酸阿米替林片等20个国家基本药物品种进行了质量抽验。结果显示,本次抽验的2376批次样品,全部符合标准规定,被抽验产品总体质量良好。现将抽验结果公告如下: 本次抽取基本药物盐酸阿米替林片、叶酸片、更年安片、吲达帕胺制剂(片、缓释片)、金匮肾气丸、盐酸普萘洛尔片、吲哚美辛栓、伤科接骨片、酚酞片、附子理中制剂(片、丸、浓缩丸)、华佗再造丸、参芪扶正注射液、胃苏颗粒、熊去氧胆酸片、重酒石酸间羟胺注射液、蛤蚧定喘丸、甲睾酮片、氯解磷定注射液、乳癖消制剂(片、胶囊)、仙灵骨葆胶囊2 0个制剂品种,共计2376批次,全部符合标准规定。具体抽验结果如下:(一)盐酸阿米替林片 本次抽样58批次,涉及2家生产企业,经安徽省食品药品检验所检验,全部符合标准规定。
(二)叶酸片 本次抽样211批次,涉及16家生产企业,经安徽省食品药品检验所检验,全部符合标准规定。 (三)更年安片 本次抽样119批次,涉及9家生产企业,经辽宁省食品药品检验所检验,全部符合标准规定。 (四)吲达帕胺制剂(片、缓释片) 本次抽样219批次,涉及27家生产企业,经辽宁省食品药品检验所检验,全部符合标准规定。 (五)金匮肾气丸 本次抽样215批次,涉及34家生产企业,经北京市药品检验所检验,全部符合标准规定。 (六)盐酸普萘洛尔片 本次抽样193批次,涉及21家生产企业,经北京市药品检验所检验,全部符合标准规定。 (七)吲哚美辛栓 本次抽样147批次,涉及5家生产企业,经北京市药品检验所检验,全部符合标准规定。 (八)伤科接骨片 本次抽样30批次,涉及1家生产企业,经大连市药品检验所检验,全部符合标准规定。
卫生部等15部门关于印发《中国慢性病防治工作规划(2012-2015年)》的通知 卫疾控发〔2012〕34号 各省、自治区、直辖市及新疆生产建设兵团卫生厅(局)、发展改革委、教育厅(教委、局)、科技厅(科委、局)、工业和信息化主管部门、民政厅(局)、财政厅(局)、人力资源社会保障(人事、劳动保障)厅(局)、环境保护厅(局)、农业(农牧、农村经济)厅(委、局)、商务主管部门、广播电影电视局、新闻出版局、体育局、食品药品监督管理局(药品监督管理局): 为积极做好慢性病预防控制工作,遏制我国慢性病快速上升的势头,保护和增进人民群众身体健康,促进经济社会可持续发展,卫生部等15部门联合制定了《中国慢性病防治工作规划(2012-2015年)》(以下简称《规划》)。现印发给你们,请结合各地、各部门的工作实际认真组织实施,切实落实各项政策和保障措施,保证《规划》目标如期实现。 卫生部国家发展改革委教育部 科技部工业和信息化部民政部 财政部人力资源社会保障部环境保护部 农业部商务部广电总局 新闻出版总署体育总局国家食品药品监督管理局 二○一二年五月八日 中国慢性病防治工作规划 (2012-2015年) 为贯彻落实《中共中央国务院关于深化医药卫生体制改革的意见》,积极做好慢性病预防控制工作,遏制我国慢性病快速上升的势头,保护和增进人民群众身体健康,促进经济社会可持续发展,根据我国慢性病流行和防治情况,特制定《中国慢性病防治工作规划(2012-2015年)》。 一、背景
影响我国人民群众身体健康的常见慢性病主要有心脑血管疾病、糖尿病、恶性肿瘤、慢性呼吸系统疾病等。慢性病发生和流行与经济社会、生态环境、文化习俗和生活方式等因素密切相关。伴随工业化、城镇化、老龄化进程加快,我国慢性病发病人数快速上升,现有确诊患者2.6亿人,是重大的公共卫生问题。慢性病病程长、流行广、费用贵、致残致死率高。慢性病导致的死亡已经占到我国总死亡的85%,导致的疾病负担已占总疾病负担的70%,是群众因病致贫返贫的重要原因,若不及时有效控制,将带来严重的社会经济问题。 国内外经验表明,慢性病是可以有效预防和控制的疾病。30多年来,我国经济社会快速发展,人民生活不断改善,群众健康意识提高,为做好慢性病防治工作奠定了基础。多年来在我国局部地区和示范地区开展的工作已经积累了大量成功经验,并初步形成了具有中国特色的慢性病预防控制策略和工作网络。但是,慢性病防治工作仍面临着严峻挑战,全社会对慢性病严重危害普遍认识不足,政府主导、多部门合作、全社会参与的工作机制尚未建立,慢性病防治网络尚不健全,卫生资源配臵不合理,人才队伍建设亟待加强。“十二五”时期是加强慢性病防治的关键时期,要把加强慢性病防治工作作为改善民生、推进医改的重要内容,采取有力有效措施,尽快遏制慢性病高发态势。
农业部再次公布饲料、养殖中禁用药物和物质清单为保证动物源性食品安全,维护人民身体健康,根据《饲料和饲料添加剂管理条例》、《兽药管理条例》等有关规定,农业部2002年发布了176号和193号公告,2010年发布了1519号公告,向社会公布了禁止在饲料、动物饮用水和畜禽水产养殖过程中使用的药物和物质清单。清单主要包括克伦特罗、沙丁胺醇等兴奋剂类,己烯雌酚等激素类,呋喃唑酮、氯霉素等抗菌药物类,呋喃丹等杀虫剂类等四大类82种禁用药物和物质。现再次公布,各地畜禽、水产养殖企业、农民专业合作经济组织、基地以及养殖场(户)要严格遵照执行。 相关文件:农业部176号公告 农业部193号公告 农业部1519号公告 二〇一一年四月二十二日
中华人民共和国农业部公告 第176号 为加强饲料、兽药和人用药品管理,防止在饲料生产、经营、使用和动物饮用水中超范围、超剂量使用兽药和饲料添加剂,杜绝滥用违禁药品的行为,根据《饲料和饲料添加剂管理条例》、《兽药管理条例》、《药品管理法》的有关规定,现公布《禁止在饲料和动物饮用水中使用的药物品种目录》,并就有关事项公告如下: 一、凡生产、经营和使用的营养性饲料添加剂和一般饲料添加剂,均应属于《允许使用的饲料添加剂品种目录》(农业部第105号公告)中规定的品种及经审批公布的新饲料添加剂,生产饲料添加剂的企业需办理生产许可证和产品批准文号,新饲料添加剂需办理新饲料添加剂证书,经营企业必须按照《饲料和饲料添加剂管理条例》第十六条、第十七条、第十八条的规定从事经营活动,不得经营和使用未经批准生产的饲料添加剂。 二、凡生产含有药物饲料添加剂的饲料产品,必须严格执行《饲料药物添加剂使用规范》(农业部168号公告,以下简称《规范》)的规定,不得添加《规范》附录二中的饲料药物添加剂。凡生产含有《规范》附录一中的饲料药物添加剂的饲料产品,必须执行《饲料标签》标准的规定。 三、凡在饲养过程中使用药物饲料添加剂,需按照《规范》规定执行,不得超范围、超剂量使用药物饲料添加剂。使用药物饲料添加剂必须遵守休药期、配伍禁忌等有关规定。 四、人用药品的生产、销售必须遵守《药品管理法》及相关法规的规定。未办理兽药、饲料添加剂审批手续的人用药品,不得直接用于饲料生产和饲养过程。 五、生产、销售《禁止在饲料和动物饮用水中使用的药物品种目录》所列品种的医药企业或个人,违反《药品管理法》第四十八条规定,向饲料企业和养殖企业(或个人)销售的,由药品监督管理部门按照《药品管理法》第七十四条的规定给
附件 化学药生物等效性试验备案范围和程序 一、备案范围 (一)属于下列情形的化学药,应当进行BE试验备案: 1.仿制已上市的参比制剂,其活性成分、给药途径、剂型、规格应与参比制剂相一致。参比制剂应为原研药品。 2.已批准在境内上市,需通过BE试验开展相应变更研究的药品。 3.已在境内上市,需通过BE试验与参比制剂进行质量和疗效一致性评价的药品。参比制剂应为原研药或国际公认的仿制药。 (二)属于下列情形的化学药,如需开展BE试验,可按照《药品注册管理办法》的有关规定申报受理和审评审批。 1.放射性药品、麻醉药品、第一类精神药品、第二类精神药品和药品类易制毒化学品; 2.细胞毒类药品; 3.不适用BE试验方法验证与参比制剂质量和疗效一致的药品; 4.不以境内注册申请或仿制药质量和疗效一致性评价为目的进行BE试验药品; 5.注册申请人认为BE试验可能潜在安全性风险需要进行技术评价的药品。
二、备案程序 (一)注册申请人向具有资质的药物临床试验机构提出申请,获得该机构伦理委员会的批准,并签署BE试验合同。 (二)注册申请人开展生物等效性试验前30天,应当在国家食品药品监督管理总局指定的化学药BE试验备案信息平台进行化学药BE试验备案,按要求提交备案资料。 (三)备案资料主要包括注册申请人信息、产品基本信息、处方工艺、质量研究和质量标准、参比制剂基本信息、稳定性研究、原料药、试验方案设计、伦理委员会批准证明文件等。 (四)注册申请人BE试验的参比制剂及各参与方的基本信息等向社会公开。 (五)注册申请人在获得备案号后,应在第1例受试者入组前在国家食品药品监督管理总局药物临床试验登记与信息公示平台完成开展试验前的所有信息登记,并由国家食品药品监督管理总局向社会公示;1年内未提交受试者入组试验信息的,注册申请人须说明情况;2年内未提交受试者入组试验信息的,所获得备案号自行失效。 (六)注册申请人应严格执行《药物临床试验质量管理规范》(GCP),按照试验方案开展BE试验。BE试验过程中,参比制剂、原料药、制剂处方、工艺等发生变更,注册申请人应停止试验,通过备案平台提交试验中止的申请,国家食品药品监督管理总局将公示其中止试验。注册申请人根据变更情况,向国家食品药品监督管理总局提交备案变更资
国家药品质量公告(2009年第3期,总第79号) 2009年11月09日发布国家食品药品监督管理局 公告 2009年第68号 国家药品质量公告 (2009年第3期,总第79号) 为加强药品监管,保障公众用药安全,根据2009年国家药品评价抽验计划,国家食品药品监督管理局在全国范围内组织对克林霉素、洛伐他汀、抗病毒、消核片、妥布霉素、枸橼酸铋钾、西咪替丁等7个制剂品种进行了国家评价抽验。结果显示,总体质量状况良好。现将抽验结果公告如下: 一、克林霉素制剂(克林霉素磷酸酯注射液、盐酸克林霉素胶囊、盐酸克林霉素注射液、注射用克林霉素磷酸酯、注射用盐酸克林霉素) 全国共有292个药品批准文号、173家生产企业。本次流通领域抽样211批,涉及6家生产企业,经河北省药品检验所检验,全部合格。 二、洛伐他汀制剂(洛伐他汀胶囊、洛伐他汀片剂) 全国共有33个药品批准文号、28家生产企业。本次在流通领域抽样225批,涉及23家生产企业,经河北省药品检验所检验,221批次抽验结果为合格,4批次不符合标准规定,
分别为丽珠集团新北江制药股份有限公司生产的批号为0804005A的2批次,不合格项目为检查项(溶出度);河北华加药业有限公司生产的批号为20080601、20080101各1批次,不合格项目为检查项(溶出度)。 三、抗病毒制剂(口服液、颗粒) 全国共有25个药品批准文号,23家生产企业。本次在流通领域抽样317批,涉及20家生产企业,经辽宁省食品药品检验所检验,全部合格。 四、消核片 全国共有1个药品批准文号、1家生产企业。本次在流通领域抽样33批,涉及1家生产企业,经辽宁省食品药品检验所检验,全部合格。 五、妥布霉素制剂(复方妥布霉素滴眼液、硫酸妥布霉素注射液、妥布霉素滴眼液) 全国共有117个药品批准文号、102家生产企业。本次在流通领域抽样144批,涉及3 2家生产企业,经重庆市药品检验所检验,全部合格。 六、枸橼酸铋钾制剂(胶囊、颗粒、片剂) 全国共有80个药品批准文号、75家生产企业。本次在流通领域抽样304批,涉及29家生产企业,经重庆市药品检验所检验,全部合格。 七、西咪替丁制剂(胶囊、片剂) 全国共有674个药品批准文号、609家生产企业。本次在流通领域抽样365批,涉及7 8 家生产企业,经山西省药品检验所检验,全部合格。
为了对不合格药品实行严格管理,杜绝购进和销售不合格药品,特制定本制度。 2 适用范围 本制度适用于本公司各经营环节发现的所有不合格药品处理控制的全过程。 3 依据 《中华人民共和国药品管理法》及其实施条例。 《药品经营质量管理规范》(国家食品药品监督管理总局令第13号)。 《药品说明书和标签管理规定》等。 4 内容 4.1 职责 4.1.1 质量管理部负责对公司经营全过程发现的不合格药品实行有效控制管理。 4.1.2 仓储部部长或保管员负责落实不合格药品的有效控制和保管。 4.2 不合格药品的判断依据 4.2.1《中华人民共和国药品管理法》及其实施条例。 4.2.2《药品说明书和标签管理规定》。 4.2.3各级食品药品监督管理部门发文通知禁止销售的药品。 4.2.4各级食品药品监督管理部门公告抽检不合格的药品。 4.3不合格药品的定义:凡与法定的药品质量标准及以下有关规定不符的药品均属不合格药品,包括:4.3.1药品的内在质量不符合国家法定质量标准及有关规定的药品; 4.3.2药品的外观质量不符合国家法定质量标准及有关规定的药品; 4.3.3药品包装、标签及说明书不符合国家有关规定的药品。 4.4不合格药品的范围界定:包括内在质量不合格、外观质量不合格药品和包装不合格药品。 4.4.1内在质量不合格是指药品本身发生了物理变化或化学变化的药品,在本制度中是指质量管理部依据药品监督管理部门的质量公告及药品抽检结果而确认的不合格药品。 4.4.2 外观质量不合格是指药品出现破损、污染、变形、批号不清、标签破损或脱落等情况。 4.4.3 包装不合格是指包装不符合《药品说明书和标签管理规定》的药品。 4.5 不合格药品的认定:有以下情况之一的可确认为不合格药品: 4.5.1 在验收、陈列药品质量检查及销售过程中发现的外观质量及包装质量不符合法定质量标准的药品,如药品过期、失效、霉烂、变质、包装破损、污染、变形、批号不清、标签破损或脱落等情况; 4.5.2 各级食品药品监督管理部门公布的抽检检验不合格的药品; 4.5.3 各级食品药品监督管理部门发文通知禁止销售的药品; 4.5.4 质量管理部确认的不合格药品; 4.6不合格药品的处理按照《不合格药品确认和控制处理操作规程》进行,不合格药品一经确认,应
国家食品药品监督管理局关于发布2003年第一季度国家药品 质量公告的通知 【法规类别】药品管理 【发文字号】国食药监市[2003]176号 【发布部门】国家食品药品监督管理局(原国家药品监督管理局)(已撤销) 【发布日期】2003.07.23 【实施日期】2003.07.23 【时效性】现行有效 【效力级别】XE0303 国家食品药品监督管理局关于发布2003年第一季度国家药品质量公告的通知 (国食药监市[2003]176号) 各省、自治区、直辖市药品监督管理局,解放军总后卫生部,武警总部卫生部,中国药品生物制品检定所: 根据2003年度国家药品抽验计划,中国药品生物制品检定所组织各省级药检所在全国范围内对药品生产企业、经营企业和医疗机构的药品进行了抽查检验(结果详见附件1:药品质量公告)。 现将抽验结果予以公告并就相关事项通知如下: 一、本期药品质量公告所涉及药品抽验的基本情况 本期对药品生产企业、经营企业、医疗机构的4258批次药品进行了抽验,具体情况如
下: (一)中国药品生物制品检定所组织省级药品检验所对药品经营企业、医疗机构的大环内酯类抗生素、解热镇痛类化学药品、降糖类中成药及含人工牛黄类中成药共99个品种,4214个批次的药品进行了抽验。合格4132个批次,不合格82个批次,合格率(批次)为98.1%。其中:大环内酯类抗生素共9个品种,899个批次,合格890个批次,不合格9个批次,合格率(批次)为99.0%;解热镇痛类化学药品共20个品种,2562个批次,合格2506个批次,不合格56个批次,合格率(批次)为97.8%;降糖类中成药共26个品种,442个批次,合格427个批次,不合格15个批次,合格率(批次)为 96.6%;含人工牛黄类中成药共44个品种,311个批次,合格309个批次,不合格2个批次,合格率为99.4%。 (二)中国药品生物制品检定所对2001年度国家药品
《食品生产许可管理办法》(国家食品药品监督管理总局令第16号) 2015年08月31日 发布 国家食品药品监督管理总局令 第16号 《食品生产许可管理办法》已经国家食品药品监督管理总局局务会议审议通过,现予公布,自2015年10月1日起施行。 局长 毕井泉 2015年8月31日 食品生产许可管理办法 第一章 总 则 第一条 为规范食品、食品添加剂生产许可活动,加强食品生产监督管理,保障食品安全,根据《中华人民共和国食品安全法》《中华人民共和国行政许可法》等法律法规,制定本办法。 第二条 在中华人民共和国境内,从事食品生产活动,应当依法取得食品生产许可。 食品生产许可的申请、受理、审查、决定及其监督检查,适用本办法。 第三条 食品生产许可应当遵循依法、公开、公平、公正、便民、高效的原则。 第四条 食品生产许可实行一企一证原则,即同一个食品生产者从事食品生产活动,应当取得一个食品生产许可证。 第五条 食品药品监督管理部门按照食品的风险程度对食品生产实施分类许可。 第六条 国家食品药品监督管理总局负责监督指导全国食品生产许可管理工作。 县级以上地方食品药品监督管理部门负责本行政区域内的食品生产许可管理工作。 第七条 省、自治区、直辖市食品药品监督管理部门可以根据食品类别和食品安全风险状况,确定市、县级食品药
品监督管理部门的食品生产许可管理权限。 保健食品、特殊医学用途配方食品、婴幼儿配方食品的生产许可由省、自治区、直辖市食品药品监督管理部门负责。 第八条 国家食品药品监督管理总局负责制定食品生产许可审查通则和细则。 省、自治区、直辖市食品药品监督管理部门可以根据本行政区域食品生产许可审查工作的需要,对地方特色食品等食品制定食品生产许可审查细则,在本行政区域内实施,并报国家食品药品监督管理总局备案。国家食品药品监督管理总局制定公布相关食品生产许可审查细则后,地方特色食品等食品生产许可审查细则自行废止。 县级以上地方食品药品监督管理部门实施食品生产许可审查,应当遵守食品生产许可审查通则和细则。 第九条 县级以上食品药品监督管理部门应当加快信息化建设,在行政机关的网站上公布生产许可事项,方便申请人采取数据电文等方式提出生产许可申请,提高办事效率。 第二章 申请与受理 第十条 申请食品生产许可,应当先行取得营业执照等合法主体资格。 企业法人、合伙企业、个人独资企业、个体工商户等,以营业执照载明的主体作为申请人。 第十一条 申请食品生产许可,应当按照以下食品类别提出:粮食加工品,食用油、油脂及其制品,调味品,肉制品,乳制品,饮料,方便食品,饼干,罐头,冷冻饮品,速冻食品,薯类和膨化食品,糖果制品,茶叶及相关制品,酒类,蔬菜制品,水果制品,炒货食品及坚果制品,蛋制品,可可及焙烤咖啡产品,食糖,水产制品,淀粉及淀粉制品,糕点,豆制品,蜂产品,保健食品,特殊医学用途配方食品,婴幼儿配方食品,特殊膳食食品,其他食品等。 国家食品药品监督管理总局可以根据监督管理工作需要对食品类别进行调整。 第十二条 申请食品生产许可,应当符合下列条件: (一)具有与生产的食品品种、数量相适应的食品原料处理和食品加工、包装、贮存等场所,保持该场所环境整洁,并与有毒、有害场所以及其他污染源保持规定的距离。 (二)具有与生产的食品品种、数量相适应的生产设备或者设施,有相应的消毒、更衣、盥洗、采光、照明、通风、防腐、防尘、防蝇、防鼠、防虫、洗涤以及处理废水、存放垃圾和废弃物的设备或者设施;保健食品生产工艺有原料提取、纯化等前处理工序的,需要具备与生产的品种、数量相适应的原料前处理设备或者设施。 (三)有专职或者兼职的食品安全管理人员和保证食品安全的规章制度。 (四)具有合理的设备布局和工艺流程,防止待加工食品与直接入口食品、原料与成品交叉污染,避免食品接触有毒物、不洁物。 (五)法律、法规规定的其他条件。 第十三条 申请食品生产许可,应当向申请人所在地县级以上地方食品药品监督管理部门提交下列材料: (一)食品生产许可申请书; (二)营业执照复印件; (三)食品生产加工场所及其周围环境平面图、各功能区间布局平面图、工艺设备布局图和食品生产工艺流程图;