
官方ATS/ERS/JRS/ALAT临床实践指南:特发性肺纤维化的治疗背景:
这是美国胸科协会/欧洲呼吸协会/日本呼吸协会/拉丁美洲胸科协会联合发布的的特发性肺纤维化治疗指南的更新版本。
方法:
运用系统性回顾和meta分析的方法来汇总与我们遇到的问题相关的证据。这些证据都经过GRADE评估,多学科专家小组对其进行了讨论。
采用了预先确定的利益冲突管理策略,指南建议的制定、编写、分级都是由专门的小组进行的。
结果:
指南推荐中支持或反对某种特殊的治疗干预措施,都考虑到了效果评价的可信度、结局研究的重要性、治疗结果的满意度、成本、可行性、可接受性、生存质量的影响。
总结:
全体成员阐述了支持或者反对特发性肺纤维化的各项诊疗建议的证据。
内容:
治疗疑问建议
问题1:IPF患者应该接受抗凝治疗吗?
问题2:IPF患者应该接受酪氨酸激酶抑制剂伊马替尼的治疗吗?
问题3:IPF患者应该接受强的松、硫唑嘌呤和N-乙酰半胱氨酸联合治疗吗?
问题4:IPF患者应该接受选择性ER-A内皮素手提的拮抗剂,安倍生坦的治疗吗
问题5:IPF患者应该接受酪氨酸激酶抑制剂尼达尼布的治疗吗?
问题6:IPF患者应该接受吡非尼酮的治疗吗?
问题7:IPF患者应该接受抗酸要的治疗吗?
问题8:IPF患者应该接受磷酸二酯酶5的抑制剂西地那非的治疗吗?
问题9:IPF患者应该接受波生坦或马西替坦,双重ERAs(ER-A、ER-B)吗?
问题10:IPF患者应该接受N-乙酰半胱氨酸单一疗法吗?
问题11:IPF患者应该接受双肺移植还是单肺移植?
问题12:IPF患者应该治疗PH吗?
概述
指南的目的是评估从2011年至今发表的临床证据。指南旨在让临床医师能够针对不同IPF患者,参考这些意见并作出合适的临床决策。在应用到特定的临床情境或决策前,每条建议都经过细致的回顾总结和委员会成员的讨论,他们重视每一个特定的治疗问题,包括患者的价值观念和意愿。
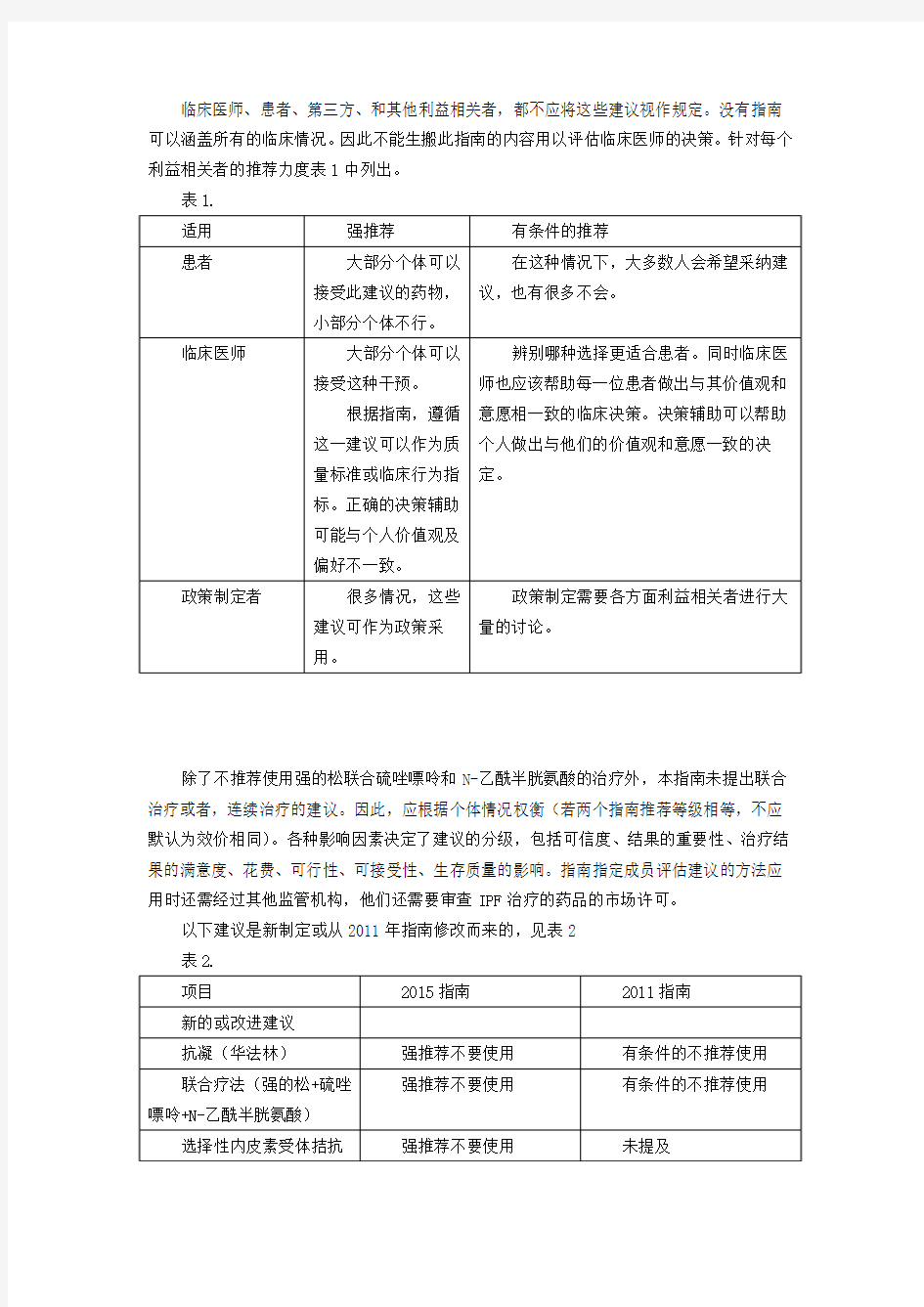
临床医师、患者、第三方、和其他利益相关者,都不应将这些建议视作规定。没有指南可以涵盖所有的临床情况。因此不能生搬此指南的内容用以评估临床医师的决策。针对每个利益相关者的推荐力度表1中列出。
表1.
除了不推荐使用强的松联合硫唑嘌呤和N-乙酰半胱氨酸的治疗外,本指南未提出联合治疗或者,连续治疗的建议。因此,应根据个体情况权衡(若两个指南推荐等级相等,不应默认为效价相同)。各种影响因素决定了建议的分级,包括可信度、结果的重要性、治疗结果的满意度、花费、可行性、可接受性、生存质量的影响。指南指定成员评估建议的方法应用时还需经过其他监管机构,他们还需要审查IPF治疗的药品的市场许可。
以下建议是新制定或从2011年指南修改而来的,见表2
表2.
1.强推荐不使用下列药物治疗IPF
a.抗凝药物(华法林)(+ + - -,作用的可信度低)
b.伊马替尼,选择性酪氨酸激酶抑制剂拮抗PDGF受体(+ + - -中可信度)
c.联合强的松、硫唑嘌呤和N-乙酰半胱氨酸(+ + - -可信度低)
d.选择性内皮素受体拮抗剂(安倍生坦)(+ + - -,可信度低)
2.有条件的推荐使用下列药物
a.尼达尼布,酪氨酸激酶抑制剂,以多酪氨酸激酶为靶位,包括内皮生长因子,成纤维细胞生长因子,和PDGF受体(+ + + -,中可信度)
b.吡非尼酮,(+ + + -,中可信度)
3.依据条件,不建议使用下列药物
a.5-磷酸酯酶抑制剂(西地那非)(+ + - -中可信度)
b.双重内皮素受体拮抗剂(马西替尼、波生坦)(+ + - -低可信度)
下列建议与2011指南相较没有变化(表2)
1.最新的与N-乙酰半胱氨酸单一疗法和抗酸治疗相关的证据经小组成员分析,两者与2011指南相比未做改动(有条件的不建议使用N-乙酰半胱氨酸单一疗法,低可信度,有条
件的不建议使用抗酸药,很低可信度)。
2.委员会了解了最新的与肺动脉高压治疗相关的证据,但最终的决定推迟到下次更新。
3.2011指南中提及的多种干预方法(糖皮质激素,吸氧,机械通气,肺疾病康复治疗以及肺移植治疗IPF急性加重)本指南未做更新。
一个关于单肺还是双肺移植的新问题被提出,但治疗建议还是推迟到下一次指南的修订,期间以获得更多必要的信息。由于资源有限,关于更新的治疗(如抗生素)的问题,被推迟到下次指南的修订。
简介
IPF是慢性进展性纤维化的间质性肺炎的一种特殊表现型,成人发生,病因不明。影像学和组织学的表现都和一般的间质性肺炎类似。
尽管2000年第一次发表的IPF指南是这一领域的国际专家共同制定的, 2011年指南的制定更是由ATS/ERS/JRS/ALAT共同制定。他们回顾了所有的文献证据,阐明了IPF的定义,给出了准确的诊断标准,描述了疾病发展的自然过程,提供了有依据的治疗建议。2011指南也表示会根据不断更新的证据进行更新。虽然2011指南清晰的给出了很多治疗建议,但很多新的很重要的IPF治疗依据在2011年以后不断出现。
这份文件重新评估了之前的治疗选择,并对新的药物选择做出建议。关于IPF的临床管理的依据不断更新进展,2011指南关于诊断、基因相关和其他问题都反复被提出来。本指南的最终目的是创立“活的文件”,通过不断更新,使新的证据不断融合进去,更及时指导临床管理工作。
构建临床问题
委员会使用2011指南文件的治疗部分作为起点,提出12个和目前的临床实践相关的问题,以更新IPF的治疗建议。这些问题大部分在2011文件中提及并给出相应的建议。关于IPF 患者的肺疾病康复、给氧、抗生素、姑息治疗、机械通气和其他2011指南强烈推荐或强烈否定的治疗,此次更新均未涉及,除非文献搜集发现新的相关证据。
以2011指南为指导,以及GRADE工作组的方法建议,委员会选择每个问题的有利结果。所有的结果都会明确优先级,委员会会明确他们的相对重要性(从IPF患者的角度)。这样可以帮助集中注意到与患者最相关的问题并有助于解决决策分歧。这些关键问题主要包括疾病的死亡率和进展情况。病情进展在2011指南中定义为加重的呼吸系统症状,恶化的肺功能、HRCT上显示进展的纤维化、进行的呼吸衰竭或者死亡。FVC随时间的变化或者肺CO弥散总量考虑为间接的病情进展的检测手段。所有结果的分级制定都是经委员会一致同意的。
临床证据审核和指南发展
麦克马斯特大学方法学团队遵循GRADE的方法使用GRADEpro指南发展工具在线软件针对每一个问题准备了证据摘要。委员会成员审查了证据摘要,并在适当的地方进行了修正。我们的证据建立在2011年的证据摘要的基础之上。如果有必要,这些摘要已经根据近期的RCTs 做了更新。委员会成员也查询了每个本次检索之外的研究。如果没有足够的RCTs的结果数据,观察性研究也用于支持指南。GM的两委位审核人员筛选文献的标题和摘要,审查评估全文,挑选出可能有关的文献(二者之一认为可能有关即可)。存在分歧时,MG组共同解决。数据提取一式两份,使用预先设计经过试用的数据提取方法。除了临床资料,存在偏倚的个人独立研究,两位审核人员要针对RCTs使用Cochrane偏倚分析工具,针对观察性研究使用渥太华——纽卡斯尔工具分别进行独立分析。
采取同种治疗药物的研究结果被合并进行研究,利用Cochrane Collaboration Review Manager, version 5.2进行的meta分析也一并纳入。MG独立进行研究数据的汇总分析与meta 分析。所有满足先验纳入标准的数据都包括在内,而这个指南中提出的汇总分析有时可能会与其他已发表的荟萃分析不同,这取决于纳入或排除标准。对于每个感兴趣的结果的效应估计信心(confidence in effect estimate),采用GRADE方法进行评估,评估时基于以下标准进行:偏倚,精度,一致性,证据的直接性,发表偏倚,剂量 - 效应关系,作用大小,以及合理的残余混杂或偏倚的影响评估。每个结果的效果估计信心在被分为以下四个级别:高,中,低或很低。
委员会制定的指南中的每一项建议都立足于GRADE证据框架。我们采用指南开发工具GRADE证据来指导决策框架,以便围绕每项建议展开讨论,并确保每个以下因素皆被考虑在内:证据的质量,每个管理方案相应的利弊后果的平衡,与决策相关的价值和优选的假设,资源利用和卫生公平,利益相关者介入的可接受性,以及实施的可行性(在线看补充)。指南推荐意见和详细说明通过协商一致决定,若果不能达成共识,投票表决。委员会就最终措辞和关于每项推荐意见的评论(例如,分组的考虑,论证,实施注意事项)取得一致意见。
按照GRADE方法,指南推荐要么是“强”要么是“有条件的”。有条件的推荐也就是薄弱的推荐。 2011年的指导方针已经使用术语“弱”,但是为了提高透明度(与指南实施有关的条件),便于指南翻译为其他语言,GRADE使用“有条件的”作为替代。影响的建议的强度的因素包括证据的强度,结果研究和与患者相关的重要性,治疗的期望和不期望的后果,治疗费用,卫生公平的实施,治疗的可行性,治疗重要的利益相关者的可接受性,和潜在的治疗监测和执行问题。
正如GRADE建议的那样,我们用“我们推荐”来表达强烈支持,用“我们建议”来表达有条件的推荐。表1向利益相关者提供了这些指南推荐的说明,包括病人,临床医生和卫生政策制定者。两个问题,委员会决定不提供建议,因为人们认识到,很多的证据,主要是间接和资源或者成本相关的证据,应告知委员会,我们将这种标记为“不推荐”。
指南推荐有两个重要方面需要考虑。首先,类似强度的推荐不应该被解释为“相当于推荐”。每个推荐的强度是考虑先前描述的多重因素的最终结果,因此两个推荐被认为具有相同的强度可能出与不同的原因(例如,一个推荐可能是有条件的,因为它是基于非常低的可信度的效果估计,而另一个推荐可以是有条件的,因为成本是如此之高,目前还不清楚对每个患者来说潜在益处是否超过所花费的成本)。第二,在指南中编写推荐意见支持或反对某种治疗应用的方法不是管理机构应用的方法(其目的是审查提交给他们的数据,并随后考虑批准患者使用新的治疗方法)。
具体治疗问题的建议
问题1:IPF患者应该接受抗凝治疗吗?
背景:研究表明高凝状态参与促进纤维化,通过细胞表面受体介导的通路,提供了一种生物学合理性的机制,提示血栓形成与肺纤维化之间可能存在联系。系统性抗凝在防止IPF 患者发生这类变化中起到的作用尚不明确。
临床证据总结:2011指南包括了一项研究,一项开放的随机试验比较了56位患者口服华法林加上强的松与单用强的松。华法林降低了IPF急性加重相关的死亡率。这项实验有存在的方法学问题,缺乏明确的描述如何随机分组,缺乏对如何处理中途退出的患者的处理,以及没有排除肺栓塞这一潜在恶化的原因。还有缺乏安慰剂对照组,考虑到这些原因,这一研究结果可能有很大的偏差,因此,将此实验从汇集的资料中排除。
2011年后一项随机对照试验,调查145名IPF患者口服华法林(INR2.0-3.0)对比安慰剂控制组。这项研究于平均随访28周后早早结束了。因为华法林没有明显益处,且对治疗存在潜在的损害。尽管相对的事件发生率较低,但期中分析,华法林组的死亡率升高(RR,4.73;95%可信区间1.42-15.77,低可信度),与出血并发症无关。两组相较,FVC变化没有明显差异(低可信度),研究期间FVC下降超过10%的患者比例也没有明显差异(低可信度)。华法林治疗的患者有更严重的不良反应的趋势。
建议:我们建议对于没有明确指征的IPF患者不要使用华法林抗凝治疗。(强推荐不要使用,其作用评估可信度低)。
依据及实行的注意事项
这一建议重视潜在的不良反应如死亡。委员会成员觉得死亡风险上升,因此强推荐不要使用口服华法林。然而,这条建议仅用于那些希望用华法林将INR控制在2.0-3.0的患者,不包括其他抗凝药物用于其他指征。患者存在其他抗凝治疗的指征如深静脉血栓性疾病或者房颤,应该遵循这类独立于IPF的情况的相关指南。考虑到这些情况,不能完全认为口服华法林没有益处。
未来的研究方向
委员会成员考虑,对IPF患者使用华法林的这类试验没有多大意义,因此很难有发展。
问题2:IPF患者应该接受伊马替尼,酪氨酸激酶抑制剂的治疗吗?
背景:伊马替尼是一种肺成纤维细胞-肌成纤维细胞分化增殖的有效抑制剂,同时也抑制细胞外基质的合成,通过抑制PDGF和转化生长因子-β信号通路。而关于另一种相对较低选择性的酪氨酸激酶抑制剂尼达尼布的使用见问题5,2011指南中均为提及这两种药。
临床证据总结:
伊马替尼治疗IPF患者已经通过一项119人的安慰剂控制的随机对照试验,随诊中位数日期为96周。两组的死亡率没有明显差异(RR,0.81;95%CI,0.35-1.92低可信度)。疾病分级,该研究最主要的结果就是超过10%的患者FVC下降或者96周时死亡,同时也显示伊马替尼治疗没有益处(HR,1.05;95%CI,0.56-1.96,中可信度)。伊马替尼组的患者不良反应发生率较对照组升高也具有统计学意义((RR, 1.54; 95% CI,1.25–1.90;高度可信);然而,大部分不良反应不影响药物的继续使用。严重不良反应发生率两组没有明显差异(低可信度)。
建议:我们建议临床医师不要对IPF患者使用伊马替尼治疗(强推荐,效果评估可信度中等)。
依据及实行的注意事项
伊马替尼是一种昂贵的药物,目前没有证据表明他可以延缓IPF患者的病情进展和降低死亡率。除了没有临床受益外,这一建议还考虑到不良反应事件和治疗成本的问题。
问题3:IPF患者应该接受强的松、硫唑嘌呤和N-乙酰半胱氨酸的联合治疗吗?
背景:在过去,免疫抑制剂被认为是IPF的重要治疗手段。还认为糖皮质激素加上硫唑嘌呤或者环磷酰胺可能优于单用糖皮质激素。早前还有很多研究支持N-乙酰半胱氨酸的治疗,临床医师和调查者评估了这三种药联合使用对IPF的治疗效果。
临床证据总结:2011指南包括了一项随机对照试验对比了在使用强的松和硫唑嘌呤的患者中再使用N-乙酰半胱氨酸和安慰剂对疗效的影响。在这项研究中,12个月后,尽管死亡率、呼吸困难指数和生活质量没有明显差异,但加用N-乙酰半胱氨酸组的肺总量及DLco 下降较少。但这项研究还有局限性,缺少其他有效药物的安慰剂对照组,更近一个随机对照试验,将患者随机分配使用联合治疗和其他药物的安慰剂治疗。这一多中心的研究提前结束了,因为其发现联合治疗与安慰剂对比,死亡率(HR, 9.26; 95% CI, 1.16–74.1极低可信度)和住院率(P<0.001)上升。两组FVC(中可信度)DLco(低可信度)和生活质量改
变(低可信度)改变没有明显的区别。
建议:我们建议临床医师不要使用N-乙酰半胱氨酸、硫唑嘌呤和强的松的联合治疗(强推荐,效果评价可信度低)
依据及实行的注意事项:这一建议主要基于单个实验结果。虽然因其损害作用,试验提前结束,但该实验明显的负面影响,也可视为一项重要结果。这一建议强调了治疗中的潜在不良反应。委员会认为,此建议只适用于试验中的IPF患者以及用于实验的药物剂量,可能无法推广到其他间质性肺疾病或者其他治疗药物的应用剂量。而对于长期接受联合治疗的IPF患者并有较好的耐受性,如何处理他们,委员会并未达成一致,并没有研究表明必须停止治疗。这种情况下,委员会建议有必要在患者和医师之间进行知情讨论,讨论联合治疗的潜在危险,并依据患者的意愿给出决策意见。对于个体患者判断受益情况有一定难度,而对那些似乎对联合治疗有一定反应的患者,则应该谨慎的重新考虑IPF这一诊断是否准确,并且重新考虑对这一治疗方案应答良好的其他疾病。
问题4:IPF患者应该接受选择性ER-A内皮素受体拮抗剂,安倍生坦的治疗吗?
背景:临床意义显著的内皮素受体主要分为两类。内皮素受体A(ET-A)可以诱导血管收缩,在血管平滑肌细胞表面表达。内皮素受体B(ET-B1)位于内皮细胞,可以刺激NO和环前列腺素的释放,从而产生血管舒张作用。ET-A受体还可以通过中介细胞因子促进内皮细胞向间叶细胞转化,导致一种纤维化的状态。ET-B2受体通过某种机制与ET-B1受体作用相反,可以导致血管收缩。临床上使用的内皮素受体拮抗剂(ERAs)包括选择性的ET-A拮抗剂(安倍生坦)和双重受体拮抗剂可以同时影响ET-A和ET-B受体(波生坦、马西替坦)。在IPF的肺中,ET-A和ET-B受体表达水平都升高,因此,无论是选择性还是双重受体拮抗剂都被研究其治疗IPF的作用。考虑到不同的作用机制,本指南分别着眼于这两种药物,并分别给予建议。2011指南没有给出选择性ERAs的建议。
临床证据总结
安倍生坦是唯一具有随机对照试验证实的选择性ERAs。该实验将492名IPF患者按照2:1分为两组,分别给予安倍生坦和安慰剂。试验还根据有无肺动脉高压进行了随机分层。由于欠缺疗效,且极有可能存在风险,试验最终提前终止。
随访中位数52周,使用安倍生坦的死亡风险比为2.08(95%CI 0.75-5.76 低可信度)。而且无论是否存在肺动脉高压,安倍生坦会导致疾病进展,DLco或FVC恶化(HR,1.74;95%CI,1.14-2.66;中可信度)。48周时,FVC、DLco、六分钟步行试验和生活质量指数两组没有明显差异。安倍生坦和安慰剂两组患者,不良反应(中可信度)和严重不良反应事件(低可信度)的发生率没有明显差异。
建议:我们建议临床医师针对IPF患者,不论患者是否存在肺动脉高压,都不要使用安倍生坦。(强推荐不要使用,作用估计可信度低)
依据及实行注意事项:
因为安倍生坦用于治疗除了合并IPF的肺动脉高压的患者,所以委员会建议不要使用安倍生坦治疗有肺动脉高压表现的IPF患者。考虑到欠缺疗效,潜在风险高等原因,正在接受安倍生坦治疗的患者应该停止给药。
委员会也建议未来不要再进行相关实验了。
问题5:IPF患者应该接受酪氨酸激酶抑制剂尼达尼布的治疗吗?
背景:尼达尼布(过去认为是BIBF1120)是一种胞内的多种酪氨酸激酶抑制剂,以多种生长因子为靶点,包括血管内皮生长因子,成纤维细胞生长因子和PDGF。
临床证据总结:两篇报道上提到了三组随机对照试验评估了尼达尼布对IPF患者的疗效。第一个是分2期,进行的安全性和有效性的试验,通过四个剂量的尼达尼布给药(50mg qd 、100 mg qd、150 mg qd、150 mg bid)与安慰剂组对比。各组之间的死亡率没有明显差异。12个月的随访期间,最高剂量的尼达尼布组,FVC下降超过10%的患者百分比较低(P=0.004),但其他剂量与安慰剂组相比没有显著差异。任一尼达尼布剂量组的患者与对照组相比,IPF的急性加重较少(HR,0.16;95%CI,0.04-0.70)。尼达尼布治疗的患者不良反应及严重不良反应事件较多,但都没有统计学意义。
INPULSIS I(高剂量BIBF1120用于特发性肺间质纤维化治疗的安全性和有效性)和INPULSIS II(高剂量BIBF1120用于特发性肺间质纤维化治疗的安全性和有效性 II)在第三阶段随机对照试验中被重复。该实验包括了1066名患者,以3:2的比例分别给予150mg尼达尼布一天两次和安慰剂。两组随访时间均为52周。尼达尼布对死亡率(RR, 0.70; 95% CI, 0.44–1.11)和急性加重的发生率(HR, 0.64; 95% CI, 0.39–1.05)没有显著影响。然而,少量患者经尼达尼布治疗后,研究期间FVC下降超过10% (RR, 1.16; 95% CI,1.06–1.27)。经过校正后的FVC年改变率,尼达尼布组与安慰剂组对比114.7ml对239.9ml(差值125.2 ml; 95% CI,77.7–172.8)。重要的是更多的患者经尼达尼布治疗后,出现不良反应(RR, 1.07; 95% CI, 1.03–1.11);但严重不良反应事件发生率没有明显上升。尼达尼布治疗的患者与安慰剂治疗的患者相比,恶心腹泻的发生率更高。
汇总分析这三组实验结果,死亡的相对风险0.70(95% CI,0.47–1.03; 中等可信度),急性加重风险比未0.47(95% CI,0.17–1.29; 低可信度)。尼达尼布可减少FVC下降超过10%的患者数(RR, 1.15; 95% CI,1.06–1.25; 中等可信度)。更多的患者经过尼达尼布治疗后出现不良反应(高可信度),但少有严重不良反应事件发生(高可信度)
建议:我们建议临床医师使用尼达尼布治疗IPF患者(有条件的推荐,作用估计可信度
中等)。
依据及实行注意事项:本建议强调患者的预后,如疾病的进展和死亡率,而不是不良反应,及治疗成本。与选择性更高的酪氨酸酶抑制剂相比,尼达尼布似乎疗效更好,但对总体死亡率影响不显著。治疗成本高可能也会限制他的可行性。针对这些情况,委员会也展开了讨论,将这些作为治疗建议的一部分,必须作为影响治疗选择的因素进行考虑。不良反应多件,尤其是腹泻,使用前应与患者说明。如前所述,尼达尼布不会增加严重不良反应事件的发生,相对较少的患者会因为不良反应而不能继续用药。一名委员会成员觉得这一建议需要强烈支持,其他的所有成员也支持有条件的用药。现有的证据都只显示在肺功能轻至中度受损的IPF患者会有效。而对于肺功能受损更严重或者有其他合并症的患者疗效是否会有差异还未可知。一些参与实验的患者,他们的HRCT有普通型间质性肺炎(UIP)征象(或被认为“UIP可能”)而不是明确的UIP(未经外科病理活检证实,仅在CT上表现出UIP征象)。而且没有证据可以给出合适的疗程,随着药物治疗进行,疗效的持续时间有多长也不明确。
未来的研究方向:
未来尼达尼布的研究方向应该着重于肺功能更差的IPF患者。还需要更多有关合适的疗程的信息。
问题6:IPF患者应该使用吡非尼酮吗?
背景:吡非尼酮是一种口服的多效性的抗纤维化药物,它已被证明在体外试验中可以调节纤维化和促进炎性细胞因子级联反应,同时减少在肺纤维化的动物模型中的成纤维细胞增殖和胶原合成。
临床证据总结:2011指南提到了两个相对小容量的RCTs比较了吡非尼酮和安慰剂对肺功能轻至中度损害的IPF患者的疗效。其中一项实验早期终止了,由于急性加重的频率高于安慰剂对照组。尽管数据不完整,但从6分钟步行试验氧饱和下降频率以及随着时间推移VC的下降都可以看出吡非尼酮的影响。第二个实验存在严重的方法问题,包括高度选择性的参与方法和最终目的在研究中途变更。但他也能反映吡非尼酮对降低VC的下降率的影响(290 mlvs. 2160 ml; P = 0.04)以及提高无进展生存期(P=0.03)。CAPACITY实验,是关于IPF的吡非尼酮的治疗,尚未发表,其囊括了两个大容量的RCTs结果(吡非尼酮治疗IPF患者的安全性和有效性、吡非尼酮的安全性和疗效在特发性肺纤维化的三组研究)。然而初步结果是可用的,且被认为是指南的最后一次迭代。
CAPACITY实验报道了两个独立的研究方案:研究004包括了435名患者随机分配到1-3组(高剂量吡非尼酮[2,403 mg/d],低剂量吡非尼酮[1,197 mg/d],安慰剂),而研究006包括344名患者随机分到2组(高剂量吡非尼酮[2,403 mg/d]、安慰剂)。低剂量吡非尼酮组的结果位于高剂量中间,为了避免治疗的异质性,我们着重观察两组试验高剂量吡非尼酮
组与安慰剂组的对比情况。研究004中,在72周的治疗期间,吡非尼酮可以减少FVC的降低。研究006并没有得出同样的结果。重要的是,两组实验的患者接受高剂量的吡非尼酮,恶心、消化不良、呕吐、食欲不振、光敏感和皮疹的发生率高于安慰剂组。ASCEND实验(一组随机双盲安慰剂对照的吡非尼酮治疗IPF的实验)随机将555名患者随机给予高剂量的吡非尼酮(2403mg/d)或安慰剂。与CAPACITY实验不同,ASCEND实验有严格的病人筛选条件,如FEV1/FVC低于0.8,因为这一条件,1562名患者被筛去1007位)。在52周的随访期间,对比安慰剂组,吡非尼酮可以降低FVC下降超过10%的患者比例。吡非尼酮还可以增加六分钟步行试验的距离,延长不进展生存期。死亡率或呼吸困难评分没有评估。与之前研究一致,吡非尼酮治疗相关的不良反应较多。
汇总这些实验结果,发现吡非尼酮会改善死亡率((RR, 0.70; 95% CI, 0.47–1.02;中可信度)。吡非尼酮可以减少FVC的下降量(标准差, 0.23; 95% CI, 0.06–0.41;高可信度)。汇总的信息不包括个别研究的阳性结果,因为异质性导致实验结果不准确。汇总分析表明,吡非尼酮治疗后光敏感(高可信度)、疲乏(中可信度)、腹部不适(中可信度)、厌食(高可信度)发生率上升。
建议:我们建议临床医师使用吡非尼酮治疗IPF患者(有条件的推荐,效果评估可信度中等。)
依据及实行注意事项:
指南修订得到新的证据支持条件性的使用这一治疗。一位委员强烈支持,其他成员也同意条件性的使用。本建议强调患者的预后,如疾病的进展,FVC的下降,而不是不良反应,及治疗成本。生活质量数据在实验过程中有零星的报道。不良反应较多,尽管FVC检查明确了药物的疗效,一些患者仍不愿意忍受这些不良反应。决策需要与患者共享,在开始治疗前,患者需要了解所有可能发生的不良反应。此外,吡非尼酮价格十分昂贵,这也是决策时需要考虑的因素,尤其在患者的经济压力比较重的时候。考虑到吡非尼酮实验患者筛选的条件比较严格,这些实验结果很难推广到肺功能更严重或有其他合并症的IPF患者。这些证据也没有提供合适的疗程,持续药物治疗疗效持续时间也不清楚。
未来的调查方向:
未来吡非尼酮的研究方向应该着重于合适的疗程,以及对肺功能更差及并存阻塞性通气功能障碍(FEV1/FVC<0.8)或者合并有肺气肿的IPF患者的治疗效果。
问题7:IPF患者应该进行抗酸治疗吗?
背景:胃食管反流包括无症状的胃酸增多(或者未经治疗的胃酸增多),可见于90%IPF 患者。GER是误吸的危险因素,并导致肺炎,这也是导致和加重IPF的一个重要机制。常规使用抗酸剂PPIs或H2RAs,可能可以降低误吸导致肺部损伤的风险。
临床证据总结:
观测研究尝试通过观察规律使用PPI和H2RAs在降低IPF患者病情进展方面的作用。一个
纵向的回顾性分析表明接受抗酸治疗的患者生存受益(HR,0.47; 95% CI, 0.24–0.93 ;调整分析),其中86人使用PPIs,12人使用H2RAs。另一个总量分析,检测了所有IPF患者包括安慰剂组还有其他3个不同药物的RCTs实验。124名患者接受了一种PPI或者H2受体阻断剂(91% PPI,9% H2RA)对比118名患者未使用抗酸治疗。这项分析表明,在研究期间,接受抗酸治疗的患者FVC的下降量更小(平均差 0.07 L; 95% CI,0–0.14; P = 0.05)。对比安慰剂,抗酸治疗不会增加急性加重。然而,研究也表明全因死亡率或住院率没有差异。
建议:我们建议临床医师对IPF患者规律使用抗酸治疗(有条件的推荐,疗效评估可信度很低)。
依据及实行注意事项:
这项建议重视改善肺功能及生存以及低治疗成本,而不是抗酸治疗引发肺炎的风险增高的可能性。观察研究表明,抗酸治疗的指征是基于医生的决定,其可以导致偏差。另外,不知道调查人员是如何控制这种联合干预的,同时这样的治疗效果也不明确。临床证据大部分着重于PPIs,很小一部分患者使用了H2RAs。其他的抗酸治疗可能也需要考虑。有必要注意的是,这项建议适用于所有的IPF患者,其以IPF为治疗指征而不是GER。抗酸治疗在有症状和无症状IPF患者之间是否有差异还不明确。然而,临床上GER/GERD的患者应该根据相应的GERD指南给予合理的治疗。此项建议还考虑到了PPI的安全性。最近一项观察研究的meta分析表明PPIs不会增加社区获得性肺炎的住院风险。PPIs与其他IPF药物的相互作用以及长期的治疗效果并不明确。
未来的研究方向:
需要更多的RCTs比较IPF患者使用抗酸治疗和安慰剂的效果。且需要更多的调查PPI 与其他IPF治疗药物的相互作用,以及长期使用对有或没有GER/GERD症状的患者的安全性,还有这一治疗对无酸反流以及异常GER和误吸在IPF发病,进展和或急性加重时所起的作用。还需要更多的研究来判断外科干预对降低IPF患者GER和误吸的风险的安全性和有效性。
问题8:IPF患者应该接受磷酸酯酶5抑制剂,西地那非治疗吗?
西地那非是一种口服的磷酸酯酶5的抑制剂,有两组RCTs研究过其对IPF患者的作用。2011指南涵盖了这一临床证据,会议至今仅完成了一项此类研究。因此无法提供IPF患者使用磷酸酯酶抑制剂的正式建议。
临床证据总结:
STEP-IPF(西地那非对IPF的运动性能影响的实验)是一个3期研究,他将180位IPF患者(DLCO,35%预计值)随机分配至西地那非(20mg 一天3次)和安慰剂组,进行12周的试验。初期结果,西地那非没有明显的作用,但在12周以后,超过20%的患者6分钟步行试验距离增加(10.1% vs. 6.6%; P = 0.39)。西地那非在改善气短,提高生活质量、改善DLco和提高氧饱和度方面有小的影响。两组之间严重不良反应的发生率没有显著差异。另一组分析了119名患者的超声心动图,以了解西地那非对有右心室肥厚或右室收缩功能障碍(RVSD)的IPF
患者的影响。对超声心动图显示有RVSD的患者,西地那非增加六分钟步行实验的距离(平均距离, 99.3 m; 95% CI, 22.3–176.2 m)。对没有RV功能障碍的患者,也有类似的结果。
第二个实验是一个小容量的29名轻至中度疾患的患者(平均DLco 42%预计值)的随机试验。接受6个月的治疗,西地那非(20mg 3次每天)和安慰剂。肺动脉高压或者RV功能障碍的患者被除外。这项研究发现,西地那非对六分钟步行试验、Borg呼吸困难评分、FVC、DLco 和氧饱和度没有影响。西地那非组不良反应发生率高,但并非严重不良反应。
汇总分析这两个实验,西地那非对死亡率(RR, 0.51;95% CI, 0.1–2.72;低可信度)或者急性加重(RR, 0.34; 95% CI, 0.04–3.22;低可信度)没有影响。经SGRQ评估西地那非可以显著该生生活质量(中可信度)。与个别研究相似,西地那非对FVC(中可信度)DLco (低可信度)Borg呼吸困难评分(中可信度)氧饱和度(低可信度)或六分钟步行试验(低可信度)没有影响。
建议:我们建议临床医师不要使用磷酸酯酶5抑制剂治疗IPF(有条件的不推荐使用,效果评估可信度中等)。
依据和实行注意事项:尽管对生活质量有所提高,但考虑到对死亡率、急性加重及呼吸困难评分都没有作用,认为只有净伤害。药物相关的不良反应和治疗成本也应考虑。本建议强调其对死亡率、急性加重、呼吸困难没有改善以及治疗成本高,而不是生活质量的改善。本建议需要委员会的投票表决:2名委员支持条件性的使用,5名主张有条件的不建议使用,还有两位弃权。这项建议不能用于那些用西地那非治疗其他疾病如PH或其他RV功能障碍的患者考虑到超声心动图不是诊断RV功能障碍或者PH的金标准,且仅有一组亚实验,对合并PH 的IPF患者,委员会并没有给出特殊建议。
未来的研究方向:
随机试验着眼于IPF合并肺动脉高压的患者。另外对西地那非对生活质量的影响需要进行更多的研究。
问题9:IPF患者应该接受双重内皮素受体拮抗剂(ER-A ER-B)波生坦、马西替坦治疗吗?
背景:2011指南记录了一个着眼于ERA(波生坦)疗效的试验,且发现没有益处,强推荐不要使用该治疗。有两组RCTs针对波生坦,只有一组关于马西替坦。BUILD-1(间质性肺病中波生坦的使用)将158位患者随机分配到波生坦和安慰剂组,随访12个月。对死亡率没有显著影响(RR, 1.14; 95% CI,0.24–5.54),但根据肺功能和临床状态分析死亡率和疾病进展综合结果得到改善(RR, 0.62;95% CI, 0.37–1.05)。波生坦治疗的不良反应或严重不良反应事件增多不具有临床意义。紧接着的BUILD-3研究,尝试阐明波生坦的潜在作用,包括了616名患者的大样本,为了提高特异性,只包括了活组织检查证实的间质性肺炎,病理诊断的IPF。尽管研究进行修改设计,但并没有表明对死亡率(RR, 1.25; 95% CI,
0.53–2.96))和疾病进展(RR, 0.86; 95% CI, 0.71–1.05)有确凿的影响。FVC、健康相关生活质量、呼吸困难评分、发生的不良反应或者严重不良反应都没有显著差异。
在一个178名经活检证实IPF患者的2期实验中,将马西替坦与安慰剂相对比。与波生坦类似,两组死亡率((RR, 0.74; 95% CI, 0.13–4.33)病情进展(RR 1.02;95% CI, 0.63–1.66)或者FVC改变(平均差, 0.00; 95% CI, 20.16 - 0.16)没有显著地差异。不良反应和严重不良反应的发生率也没有差异。
考虑到两种双重ERAs的类似机制和近似的结果,这三项研究汇总分析。双重ERAs对IPF 患者的总体死亡率没有影响(RR, 1.13;95% CI, 0.57–2.27;低可信度)。死亡和疾病进展的综合情况的得到改善,位于可信区间上段(RR, 0.85; 95% CI, 0.71–1.00; 低可信度)。FVC改变(中可信度)、不良反应(高可信度)或严重不良反应事件(高可信度)的发生,两组间没有显著差异。
建议:我们建议临床医师不要使用波生坦或马西替坦,双重ER-A和ER-B内皮素受体拮抗剂治疗IPF(有条件的推荐不使用,可信度低)。
依据及实行注意事项:
这一建议相较降低死亡风险和病情进展,更为重视患者的预后和药物高成本。考虑到实验结果关于死亡率和病情进展的不一致性,以及结果的不准确性,委员会建议不要使用这种治疗。同时他们也考虑到了双重受体ERAs的价格较高。他们还提到,波生坦和马西替坦没有效果,那其他双重ERAs是否会对IPF有效呢?委员会觉得IPF合并PH的患者可能比未合并的患者会更有益。然而没有证据支持这一建议。近期一项未被委员会考虑的研究报道,波生坦对IPF合并右心功能不全的患者的肺动脉高压没有作用。
未来的研究方向:
未来的研究包括评估治疗反应如死亡率和生存质量,用以评估双重ERAs治疗IPF合并PH 的患者的有效性。
问题10:IPF患者应该接受N-乙酰半胱氨酸的单一疗法嘛?
背景:
2011指南涵盖了唯一的RCT,它包含了30名患者随机分配接受雾化吸入的N-乙酰半胱氨酸和盐酸溴已新,持续12个月,记录下CT上磨玻璃影的变化范围,以及KL-6降低的水平。体格检查和步行距离没有差异。
临床证据总结:两组新的RCTs进行评估N-乙酰半胱氨酸单一疗法,并被收纳入此次更新。日本进行了一项多中心的前瞻性实验,随机分配患者接受352.4mg吸入N-乙酰半胱氨酸两次一天与对照组进行48周实验。FVC的变化与对照组相比没有显著差异。另一个RCT包括264名患者随机给予600mg口服N-乙酰半胱氨酸3次每天和安慰剂。这一实验的原目的是为了比较三组干预,其中包括一组联合治疗:强的松、硫唑嘌呤和N-乙酰半胱氨酸。因为考虑到安全性问题,联合治疗组研究中途被终止了,继续包括仅用N-乙酰半胱氨酸组和安慰剂组两组实
验。两组分析(包括研究设计更改前后)表明单用N-乙酰半胱氨酸FVC变化没有明显差异。死亡率和急性加重发生率也没有显著差异。汇总这三组RCTs,单用N-乙酰半胱氨酸对IPF患者死亡率没有影响(RR, 1.97; 95% CI, 0.50–7.71;低可信度)。FVC的改变(高可信度)、生活质量(中等可信度)和不良反应(低可信度)都没有明显差异。两项研究报道N-乙酰半胱氨酸单一治疗可以显著改善六分钟步行实验距离(平均差,44.33m;95%CI,2.92–85.75;低可信度)。
建议:我们建议临床医师不要使用N-乙酰半胱氨酸单一治疗IPF患者(有条件的推荐,效果评估可信度低)。
依据及实行注意事项:
本建议更为强调潜在风险,不便性和治疗的成本,而非患者某一方面的改善。制定这项建议,委员会成员们展开了激烈的讨论。临床证据只适用于肺功能轻到中度受损的IPF而推广到肺功能更严重的IPF患者时,需要谨慎对待。委员会没有找到足够的证据显示吸入和口服N-乙酰半胱氨酸的差异。因此,建议适用于两种给药方法。没有证据表明这一治疗方法有害,因此没有给出建议要求正在接受N-乙酰半胱氨酸单一疗法的患者停止治疗,但无论启用这项疗法或继续该疗法,似乎并没有影响。
未来的研究方向
对于氧化应激负担较重的IPF患者,N-乙酰半胱氨酸单一疗法的疗效并不明确。未来的实验应该证实某一亚群的患者更有可能受益于这一治疗。其中一种给药方式也有可能更优,可以考虑评估不同的给药方法的效用。
问题11:IPF患者应该接受双侧还是单侧肺移植?
背景:IPF会不断进展,无法治愈,因此中至重度患者常考虑肺移植治疗。对IPF患者,双侧肺移植是否优于单侧肺移植尚不明确。缺乏RCT证据指导建议,我们考虑观察接受双肺移植和单肺移植的IPF患者的死亡率,以评估两者差异。
临床证据总结:汇总三组观察研究的生存分析,表明两种肺移植之间没有差异((HR, 0.47; 95% CI,0.19–1.17)。此外有4组研究因为没有报道风险比所以没有被一起汇总,但与其他研究一致,双肺移植的存活率与单肺移植没有明显差异。委员会成立之后才有meta 分析发表,因此没有考虑进来,其结果与先前研究一致,因此对总体结论没有影响。
推荐:委员会未对IPF患者应接受单侧或双侧肺移植给出建议。。
依据:委员会认为应评估额外证据以指导临床决策。移植器官的短缺是一个普遍问题,而将双侧肺移植给同一患者还是将单肺移植给两位患者,医学的公平性也应该考虑。
未来的研究方向:
为了更好地解决这一问题需要RCTs。而未来这个问题的指南制定需要委员会加入肺移植方面的专家来共同解决这一临床问题。
问题12:IPF患者的肺动脉高压需要治疗吗?
背景:合并症PH在IPF患者中很常见,导致临床预后更差。
临床证据总结:2011指南仅凭借有限的资料建议不要治疗IPF患者的PH。初始的指南仅有一些短期血流动力学的研究,而没有长期患者的预后的资料,没有随机分配,没有合适的安慰剂、没有回顾性的分析或者存在各种各样方法学的问题。
之前提到的安倍生坦和西地那非治疗IPF的RCTs包括一个亚组分析了合并有PH的患者。ARTEMIS-IPF(随机安慰剂对照实验评估安倍生坦对IPF的安全性及有效性)实验是基于右心导管评估的PH状态,显示对平均肺动脉压高于25mmHg的患者,没有显著的影响。同时安倍生坦加速病情进展及提高患者住院率,因此强推荐这类患者不要使用安倍生坦。
在STEP-IPF实验中,调查者评估了西地那非对合并有右心室肥厚或RVSD的作用。如前所述,RVSD不伴有右心室肥厚的患者,西地那非可以明显改善六分钟步行实验的距离。没有其他方面有显著差异,由于缺乏PH诊断的金标准以及仅进行了分析考察,并未给IPF合并有PH 的患者使用磷酸酯酶抑制剂治疗的附加建议。
最后一组小容量非盲无对照的肺动脉血流动力学研究表明瑞司瓜特,一种可溶性的鸟苷酸环化酶刺激剂,在用于PH的患者及任何原因引起的间质性肺疾病治疗时,安全性可靠。在推广使用前,还需要进行3期4期的实验以评估对IPF患者的影响。
建议:关于IPF患者合并PH,委员会未作出任何建议。
依据:委员会认为还需要更多的临床证据来指导临床决策。
未来的研究方向:新型的PH的药物不断出现,未来的研究应该重视他们对IPF患者合并PH的作用。未来关于IPF表现出PH的患者的临床研究,应该考虑针对PH的药物尤其是在IPF 患者中已证实安全性可靠的药物(如双重ERAs、磷酸酯酶5抑制剂),而非有明确损害作用的药物(选择性ERA,安倍生坦)。临床研究应该考虑分析血管活性药物对明确PH的患者的影响。
总结
从2011指南修订开始,IPF的临床管理有了明显的改进。2011年制定的弱推荐的治疗方案有了新的临床证据,委员会仔细回顾了这些研究结果,并更新了治疗建议。尽管并不强推荐药物干预,新的药物如吡非尼酮和尼达尼布被有条件的推荐使用,抗酸治疗也被推荐用于IPF患者。
临床医师治应根据病人制定个体化的治疗方案,对有条件的推荐使用的药物,应仔细权衡利弊。各项研究中依据生理学和解剖学的变量而制定的标准,以及总体的可信度,是临床医师处理IPF患者需要考虑的重要因素。
指南中提及的药物,联合、连续和附加使用都未研究,因此未给出治疗意见。未来为解
决这些问题需要对各种治疗方案进行面对面的RCTs。新型药物的作用时间并不明确。需要进行更多的研究以明确合适的疗程。希望这些即将进行或正在进行的调查能够尽快明确这些问题。
一些治疗在IPF患者中有潜在的效益(如克霉唑),但并未在此次更新中提及。其他的治疗干预如急性加重期的治疗、肺疾病康复治疗、氧疗、机械通气、姑息治疗及其他新的相关临床证据会在另一更新中提及。
未来的方向
完全有必要进行更多长期的研究以明确各种治疗方法在不同程度肺功能损害的IPF患者中的安全和有效性。尤其是一些建议条件性使用的药物,包括吡非尼酮和尼达尼布。虽然已经明确,华法林用于没有指征的IPF患者是无益的,但研究其他抗凝药物,如新型的口服药物还是有价值的。强的松、N-乙酰半胱氨酸、和硫唑嘌呤三联治疗是有害的,但哪种成分或组合还是哪种单一成分的剂量引起损害尚不明确。进行另一个三联药物的实验的可行性尚有疑问,尤其是在明确强的松和硫唑嘌呤相关的不良反应的情况下,出现了新的抗纤维化药物。
N-乙酰半胱氨酸或其他抗氧化剂的治疗需要根据氧化应激负荷分级。
虽然用内皮素受体拮抗剂治疗不伴有PH的IPF患者时没有效果,但双重ERAs安全性可靠,且有明确的治疗PH的效果,尤其是马西替坦。未来的研究可以着眼于他们在IPF伴PH 的患者中的作用。由于大量的临床研究证实安倍生坦和选择性的ERA会削弱呼吸状态,所以不推荐用于治疗。未来的临床研究应该涉及其他潜在治疗方案来治疗IPF患者的PH。
GER与IPF关系密切,在IPF患者中异常胃酸增多GER很常见,而GER是否是IPF的原因或诱因并不明确。需要更多研究来确定抗酸治疗以降低误吸导致肺损伤时安全性和有效性以及外科手术降低GER的作用。
还需要进行关于联合治疗和IPF多靶位发病的研究。当联合给药时,新型药物可能导致累积效应甚至协同作用,在这一方面已有满意的结果(指南中已报道)。事实上,这类临床实验开始前应明确药物的相互作用、药代动力学和安全性。未来的研究应该包括所有IPF
患者并按肺功能损害和/或解剖学将疾病分级。
重要的是,绝大多IPF患者超过60岁,他们常表现出其他越来越多的合并症,包括PH,肺气肿,气流受限,冠脉疾病和肥胖。
对症姑息治疗如:气短、咳嗽和疲乏,以及临终关怀对IPF患者生命尾声十分重要。未来研究需要以这些作为起点,评估新治疗策略的反应。
一些IPF的患者有肺移植的指征,但单肺还是双肺移植长期预后更好尚不明确。众多混杂因素决定了单肺还是双肺移植的临床决策,需要进行多中心的研究来决定如何合理利用供者的器官。
最后,根据解剖、临床、外周循环或肺中(组织、肺泡灌洗液)的标志物的分级治疗、药物基因学还有药物经济学,针对这些的而研究在未来是很有价值的。这样可以帮助临床医师更好的理解为了改善IPF预后而不断增加的复杂性和不断提高的治疗成本。只要不断进行高水平和团结协作的临床和基础的科学研究、并具有奉献精神以及配备充足的资源和经费,终止疾病进展并治愈的愿望最终会实现的。
ATS/ERS/JRS/ALAT临床实践指南: 特发性肺纤维化的治疗(译文) 背景: 这是美国胸科协会/欧洲呼吸协会/日本呼吸协会/拉丁美洲胸科协会联合发 布的的特发性肺纤维化治疗指南的更新版本。 方法: 运用系统性回顾和meta分析的方法来汇总与我们遇到的问题相关的证据。这些证据都经过GRADE评估,多学科专家小组对其进行了讨论。 采用了预先确定的利益冲突管理策略,指南建议的制定、编写、分级都是由专门的小组进行的。 结果: 指南推荐中支持或反对某种特殊的治疗干预措施,都考虑到了效果评价的可信度、结局研究的重要性、治疗结果的满意度、成本、可行性、可接受性、生存质量的影响。 总结: 全体成员阐述了支持或者反对特发性肺纤维化的各项诊疗建议的证据。 内容: 治疗疑问建议 问题1:IPF患者应该接受抗凝治疗吗? 问题2:IPF患者应该接受酪氨酸激酶抑制剂伊马替尼的治疗吗? 问题3:IPF患者应该接受强的松、硫唑嘌呤和N-乙酰半胱氨酸联合治疗吗? 问题4:IPF患者应该接受选择性ER-A内皮素手提的拮抗剂,安倍生坦的治疗吗 问题5:IPF患者应该接受酪氨酸激酶抑制剂尼达尼布的治疗吗? 问题6:IPF患者应该接受吡非尼酮的治疗吗?
问题7:IPF患者应该接受抗酸要的治疗吗? 问题8:IPF患者应该接受磷酸二酯酶5的抑制剂西地那非的治疗吗? 问题9:IPF患者应该接受波生坦或马西替坦,双重ERAs(ER-A、ER-B)吗? 问题10:IPF患者应该接受N-乙酰半胱氨酸单一疗法吗? 问题11:IPF患者应该接受双肺移植还是单肺移植? 问题12:IPF患者应该治疗PH吗? 概述 指南的目的是评估从2011年至今发表的临床证据。指南旨在让临床医师能 够针对不同IPF患者,参考这些意见并作出合适的临床决策。在应用到特定的临 床情境或决策前,每条建议都经过细致的回顾总结和委员会成员的讨论,他们重视每一个特定的治疗问题,包括患者的价值观念和意愿。 临床医师、患者、第三方、和其他利益相关者,都不应将这些建议视作规定。没有指南可以涵盖所有的临床情况。因此不能生搬此指南的内容用以评估临床医师的决策。针对每个利益相关者的推荐力度表1中列出。 表1. 适用患者 强推荐有条件的推荐 大部分个体可以 接受此建议的药物, 小部分个体不行。 大部分个体可以 接受这种干预。 根据指南,遵循 这一建议可以作为质 量标准或临床行为指 标。正确的决策辅助 可能与个人价值观及 偏好不一致。 在这种情况下,大多数人会希望采纳建 议,也有很多不会。 临床医师辨别哪种选择更适合患者。同时临床医 师也应该帮助每一位患者做出与其价值观和 意愿相一致的临床决策。决策辅助可以帮助 个人做出与他们的价值观和意愿一致的决 定。 政策制定者很多情况,这些 建议可作为政策采 政策制定需要各方面利益相关者进行大量的讨论。
间质性肺病诊治指南 疾病简介: 间质性肺疾病(ILD)是以弥漫性肺实质、肺泡炎症和间质纤维化为病理基本病变,以活动性呼吸困难、X线胸片弥漫性浸润阴影、限制性通气障碍、弥散(DLCO)功能降低和低氧血症为临床表现的不同种类疾病群构成的临床-病理实体的总称。 ILD病谱的异质性(heterogeneity)具有多层含义,即病因学的多源性;发生或发病学的异质性;病种或表现型的多样性以及临床症状的异因同效的相似性。从异质角度的分类看,ILD病理组织学可呈肺泡炎、血管炎、肉芽肿、组织细胞或类淋巴细胞增殖。特发性肺纤维化(IPF)为肺泡炎,其病理异质性变化表现为普通型间质性肺炎(UIP)、脱屑型间质性肺炎(DIP)/呼吸性细支气管炎间质性肺病(RBILD)和非特异性间质性肺炎/纤维化 (NSIP/fibrosis)。此分类是由Katzenstein等(1998年)在Liebow(1970年)UIP、DIP、双侧性间质性肺炎 (BIP)、淋巴细胞间质性肺炎(LIP)和巨细胞间质性肺炎(GIP)分类基础上经修正后提出的新分类。Liebow原分类(1970年)中的BIP现已公认即为闭塞性细支气管炎并机化性肺炎(BOOP)。LIP与免疫缺陷有关,GIP与硬金属有关,已不属于IPF分类范畴。Katzenstein在新分类中指出DIP命名不当而应采用RBILD。UIP属IPF 的原型,多见于老年人,激素疗效不佳,而RBILD和NSIP患者年龄较低,对糖皮质激素有疗效反应,预后良好。 发病机制发病阶段 ILD确切的发病机制尚未完全阐明。假设ILD的演变过程可区分为三个阶段,即启动阶段、进展阶段和结局阶段。 启动阶段 启动ILD的致病因子通常是毒素和(或)抗原,已知的抗原吸入如无机粉尘与石棉肺、尘肺相关,有机粉尘与外源性过敏性肺泡炎相关等,而特发性肺纤维化(IPF)和结节病等的特异性抗原尚不清楚。 进展阶段 一旦暴露和接触了最初的致病因子,则产生一个复杂的炎症过程——肺泡炎,这是ILD发病的中心环节,肺泡炎的性质决定着肺损伤的类型、修复程度及纤维化形成等。炎性及免疫细胞的活化,不仅释放氧自由基等毒性物质,直接损伤I型肺泡上皮细胞和毛细血管内皮细胞,还释放蛋白酶等直接损伤间质、胶原组织和基底膜等。同时释放各种炎性介质,已发现的包括单核因子(monokines)、白介素-1(IL-1)、白介素-8(IL-8)、白介素-2(IL-2)、血小板衍化生长因子(platelet-derived growth factor,PDGF)、纤维连接蛋白(fibronectin,FN)、胰岛素样生长因子-1(insulin-like growth factor,IGF-1)、
·38·—一———一———一国外医学呼吸系统分册2003年第23卷革1期肺纤维化发病机制及治疗策略的新观念 沈阳军区总医院呼吸内科(沈阳1100153魏路清董彦‘综述陈萍审较 p}^与六摘要本文综述r肺纤维化发病机制的新观念,包括Tn,、|rnz型细胞斟子反应的平衡失嗣、TGF-p和CD40CLIO!。系统在成纤维细胞活化中的重要作用。并概述了由此产生的肺纤维化治疗的新策略,即TFN一7抑制2型细胞因子反应,抗TGF一口抗体及阻断CD40CD40L系统的治疗策略。 关键词肺纤维化;发病机制;治疗 众所周知.组织的损伤往往伴有炎症和修复过程。如损伤微小,修复后可恢复正常结构和功能。然而,当损伤较大或反复发生时,频繁的“修复”将导致纤维化或疤痕形成。纤维化是以大量的成纤维细胞聚集、细胞外基质沉积并伴有炎症和损伤所致组织结构破坏为特征”o。肺纤维化疾病包括特发性肺纤维化(IPF)、结节病、尘肺、过敏性肺炎、药物和放射线导致的纤维化,以及与胶原血管病有关的致纤维化肺泡炎等病因各异,范围广泛的疾病潜。其发病率和死亡率各不相同,严重性各异,但共同特点是缺少特异性治疗。目前对啼纤维化疾病的治疗仅限于非特异性抗炎、免疫抑制剂及糖皮质激素等,疗效尚不理想。因此,作者复习文献,就免疫和结构细胞产生的特异性细胞因子和CD40一CD40眄己体(CD40I.)系统在肺纤维化发病机制中的作用,并由此产生的新的治疗策略加以综述。 1关于肺纤维化的发病机制 1.1Tn。型、T。型细胞因子反应平衡失调2”。关于纤维化的发病机制有很多学说,其中之一认为在辅助性1型T细胞(T“-)和2型T细胞(Tnz)细胞因子反应之间存在着平衡失调,由此导致的对损伤的异常反应。这些反应最初是根据CD4T辅助淋巴细胞亚群所产生的细胞因子而命名[2]。然而现在更支持各种其它细胞类型包括成纤维细胞、巨噬细胞,特别是肥大细胞也能够产生这些细胞因子。因此称之为T。,和Tm型细胞因子反应似乎更恰当(简称1型反应、2型反应)。尽管这些细胞因子表型的表达在分类上Tw,与细胞免疫有关,T“z与体液免疫有关,但他们都参与r急性损伤以及由此激发的炎症反应的修复过程。1型反应可能促进正常组织结构的修复,2型反应可能有利于增生的成纤维细胞活化.最终细胞外基质蛋白沉积和纤维化。1型细胞因子包括IFN—T、1L一2、lL_12、IL-18、和TNF一 ·沈阳市卫生局8,后者也被称为淋巴毒素。而2型细胞因子包括IL一4、IL一5、11.一10、II.一13和MCP—l(单核细胞趋化蛋白一1)。1型和2型细胞因子反应一个重要的特点是各类细胞因子都能进一步激发和扩大各自的主要反应。例如,2型细胞园子IL_4表达增加将会导致2型细胞因子话性过度增加和l型细胞因子反应的抑制。反之,l型细胞因子IFN7则刺激1型细胞因子的产生,而抑制2型细胞因子反应。 体内、外研究资料表明,当细胞因子的乎衡以2型细胞因子反应占优势时,就会发生纤维化。已知原始类型的2趔细胞因子1L-4促进成纤维细胞增生、胶原基因表达以及胶原台成”。在体外¨.~13和MCP1也增加1型前胶原的合成“。MCP—l通过上调促纤维化的细胞因子TGFB而介导胶原的合成。而且,MCP一1增加IL4产生。1L_4抑制l型细胞因子,因而破坏了二者的平衡,有助于2型促纤维化反应。 实验动物研究使我们更进一步了解r肺纤维化发生过程中2型细胞因子反应的作用。在离子辐射、化疔药物博莱霉素及硅粉尘所致的肺纤维化动物模型中都有2型细胞因子表达增加。在小鼠的博莱霉素肺纤维化模型中,研究者报道了IL-5和MCP-lmRNA增加,使用针对II。5和MCPl的中和抗体在减少炎症细胞募集和抗肺纤维化方面取得成功”Ⅲ,这也提示2型细胞因子的表达,至少在部分程度上有助于纤维化发生。 Huaux等发现在硅导致的肺纤维化小鼠模型中2型细胞因子活性增加,同组研究还显示11.一10缺乏的鼠,硅诱导的肺纤维化明娩减少,提示2型细胞因子在肺纤维化发生过程中起着重要作用”J。尽管在纤维化模型中细胞因子之间的平衡是转向2型反应方向的,但2型反应不能独立发生。1型细胞因子在纤维化状态下亦可以见到,例如Davis等注 万方数据
间质性肺病诊治指南 疾病简介: 间质性肺疾病(ILD)是以弥漫性肺实质、肺泡炎症和间质纤维化为病理基本病变,以活动性呼吸困难、X线胸片弥漫性浸润阴影、限制性通气障碍、弥散(DLCO) 功能降低和低氧血症为临床表现的不同种类疾病群构成的临床-病理实体的总 称。 ILD病谱的异质性(heterogeneity)具有多层含义,即病因学的多源性;发生或发病学的异质性;病种或表现型的多样性以及临床症状的异因同效的相似 性。从异质角度的分类看,ILD病理组织学可呈肺泡炎、血管炎、肉芽肿、组织细胞或类淋巴细胞增殖。特发性肺纤维化(IPF)为肺泡炎,其病理异质性变化表现为普通型间质性肺炎(UIP)、脱屑型间质性肺炎(DIP)/呼吸性细支气管炎间质性肺病(RBILD)和非特异性间质性肺炎/纤维化(NSIP/fibrosis) 。此分类是由Katzenstein 等(1998年)在Liebow(1970年)UIP、DIP、双侧性间质性肺炎(BIP)、淋巴细胞间质性肺炎(LIP)和巨细胞间质性肺炎(GIP)分类基础上经修正后提出的新分类。Liebow原分类(1970年)中的BIP现已公认即为闭塞性细支气管炎并机化性肺炎(BOOP。LIP与免疫缺陷有关,GIP与硬金属有关,已不属于IPF分类范畴。Katzenstein在新分类中指出DIP 命名不当而应采用RBILBUIP属IPF 的原型,多见于老年人,激素疗效不佳,而R BILD和NSIP患者年龄较低,对糖 皮质激素有疗效反应,预后良好。 发病机制发病阶段 ILD确切的发病机制尚未完全阐明。假设ILD的演变过程可区分为三个阶段,即启动阶段、进展阶段和结局阶段。 启动阶段 启动ILD的致病因子通常是毒素和(或)抗原,已知的抗原吸入如无机粉尘与石棉肺、尘肺相关,有机粉尘与外源性过敏性肺泡炎相关等,而特发性肺纤维化(IPF)和结节病等的特异性抗原尚不清楚。 进展阶段 一旦暴露和接触了最初的致病因子,则产生一个复杂的炎症过程一一肺泡炎,这是ILD发病的中心环节,肺泡炎的性质决定着肺损伤的类型、修复程度及 纤维化形成等。炎性及免疫细胞的活化,不仅释放氧自由基等毒性物质,直接损伤I型肺泡上皮细胞和毛细血管内皮细胞,还释放蛋白酶等直接损伤间质、胶原组织和基底膜等。同时释放各种炎性介质,已发现的包括单核因子(monokines)、白介素-1(IL-1)、白介素-8(IL-8)、白介素-2(IL-2)、血小板衍化生长因子(platelet-derived growth factor,PDGF)、纤维连接蛋白(fibronectin,FN) 、胰岛素样生长因子-1(insulin-like growth factor ,IGF-1)、
特发性肺纤维化治疗 发表时间:2011-07-22T15:09:00.030Z 来源:《中外健康文摘》2011年第17期供稿作者:曲桂霞 [导读] 对激素治疗失败的病人,加用硫唑嘌呤(AZP)或环磷酰胺(CTX),并将激素减量,4~6周停用。 曲桂霞 (黑龙江省安达市医院 151400) 【中图分类号】R563【文献标识码】A【文章编号】1672-5085 (2011)17-0167-02 【摘要】特发性肺纤维化(idiopathic pulmonary fibrosis ,IPE)是一组原因不明的肺间质病,占DPLD尤其是IIP的大多数,它是一种不明原因的肺间质炎病性疾病,原因不明的肺泡纤维化和弥漫性肺间质纤维化均为其同义词。在欧洲,IPF常被隐原性致纤维化性肺泡炎(CFA)名称所替代。2000年ATS/ERS达成共识,将IPF的内涵局限于UIP,在大家再提到IPF时,指的是预后差的UIP,而不包括AIP、DIP、RBILD 或NSIP。从此,IPF局限为UIP的专用名词,而不包括其他间质性肺炎。由于特发性肺纤维化所占比例较大,现以IPF(UIP)为代表加以阐述。 【关键词】特发性肺纤维化肺间质炎病性疾病治疗 IPF的最佳治疗尚存在争议。治疗的目的主要是消除或抑制炎症成分。少数研究认为纤维化过程可逆转,但尚缺乏足够的证据。 1.肾上腺糖皮质激素激素用于IPF的治疗已30多年,但是仅10%~30%的病人对治疗有效,完全缓解很少见,大多数病人即使应用激素治疗,病情依然恶化。其剂量和方法多数学者的主张是:初始治疗时激素应用大剂量。泼尼松或泼尼松龙每天40~80mg,共2~4个月,然后逐渐减量。如果激素治疗有反应,一般在2~3个月显效。第4个月泼尼松减量至每天30mg;第6个月减量至每天15~20mg(或其他等效剂量的激素)。泼尼松用量及减量速度应由临床或生理学参数指导。 因为激素完全根除疾病是不可能的,所以,对治疗反应不一的病人均以最小剂量治疗1~2年是合理的。应用泼尼松每天15~20mg作为长期小剂量维持治疗已经足够。应用大剂量甲泼尼松龙(每次1~2g,每周1次或2次)静脉“冲击”治疗,但并不优于口服激素。 对激素治疗失败的病人,加用硫唑嘌呤(AZP)或环磷酰胺(CTX),并将激素减量,4~6周停用。当激素减量时,有些病人病情进展或恶化,对这些病人可隔天口服泼尼松(20~40mg),另外加用免疫抑制剂或细胞毒药物。 2.目前推荐的治疗方法在尚未证明哪些治疗方法最好的情况下,建议用激素加硫唑嘌呤或环磷酰胺,用于预期可能效果较好的病人。 (1)激素:起始的治疗方法,用泼尼松(或其他等效剂量的激素)每天0.5mg/kg(理想体重,LBM),口服4周;然后每天0.25mg/kg(LBM),口服8周;继之减量至每天 0.125mg/kg或0.25Img/kg,隔日1次。 (2)硫唑嘌呤:每天2~3mg/kg(LBM),口服,起始剂量为每天25~50mg,每7~14d增加25mg,直至最大剂量每天150mg;或环磷酰胺每天2mg/kg(LBM)口服,起始剂量为每天25~50mg,每7~14d增加25mg,直至最大剂量每天150mg。如没有副作用或并发症出现,治疗应持续至少6个月。在此期间应观察治疗反应,应注意监测药物副作用,尽可能以最小的剂量、最少的副作用,达到最好的疗效。 3.辅助治疗吸氧可减轻运动所致的低氧血症,提高运动能力。口服可待因和其他镇咳药对有些病人可能有用,也可用于咳嗽复发的病人。像所有慢性肺疾病的病人一样,可定期口服肺炎疫苗和流感疫苗。 4.肺移植单肺移植对某些内科治疗复发的终末期肺纤维化是一项重要的治疗选择。内科治疗失败的患者,其预后很差。肺功能严重受损(VC或TLC%60%预计值或 DLCO%40%预计值)和低氧血症的患者,2年死亡率超过50%。严重功能受损、低氧和病情恶化的患者,应考虑肺移植。除非特殊的禁忌证存在,如年龄大于60岁,一般情况不稳定,或有重要的肺外病变(肝、肾、心功能不全)。 5.治疗展望对肺纤维化的治疗,虽一致强调了现有治疗的好处,但存活方面的主要进展是等待发展新疗法。将来可能的治疗策略包括抑制细胞因子的药物、抑蛋白酶和(或)抗氧化剂、抗纤维化制剂等。 新制剂:如谷胱甘肽是一种氧自由基的有效清除剂,可抑制成纤维细胞对有丝分裂刺激引起的成纤维细胞增殖。牛磺酸烟酸可抑制动物模型中实验性肺纤维化发生、发展。N-乙酰半胱氨酸是谷胱甘肽的前体,IPF病人的治疗中有辅助的免疫抑制作用。 N-乙酰半胱氨酸(商品名富露施)每次600mg口服,每日3次。白细胞粘附分子抗体可以防止胶原沉积,抑制特异性成纤维细胞因子,可能有助于阻碍纤维化过程。血小板激活因子受体拮抗剂也有助于抗纤维化。其他抗纤维化制剂还有如甲苯吡啶酮(pir fenidone)一种脯氨酸消旋酶抑制剂修饰物等。 新疗法策略的产生,尚有赖于更好的理解这一疑难病的发病机制。对以上所提及的药物还需要多中心的临床上大量病例的前瞻性对照研究做出评价。 自然病史和预后 IPF的自然病史特征是经数月或数年肺功能不可逆的进行性损害,极少数病人呈暴发性,有时,疾病经过最初的消退期后可稳定下来,但自发性缓解相当罕见(<1%)。从出现症状到死亡,平均存活率3~5年。IPF的5年死亡率超过40%。呼吸衰竭是死亡的主要原因。支气管肺泡灌洗液(BAL)中淋巴细胞增多提示对糖皮质激素治疗有较高的反应率;若BAL淋巴细胞低则死亡率高。治疗的目的是最大限度地防止进行性纤维化和呼吸衰竭。IPF的其他死亡原因有缺血性心脏病、脑血管意外、肺栓塞、恶性肿瘤和感染。9%~1l%的IPF患者合并肺癌。 IPF在出现以下特征时,提示预后差:①病情呈进展性发展;②年龄>50岁;③性别为男性;④吸烟史;⑤中重度活动后呼吸困难;⑥查体发现爆裂音和杵状指;⑦中重度肺功能丧失;⑧就诊时的肺泡灌洗液细胞学为中性和嗜酸细胞增多为主;⑨HRCT表现为网状阴影或蜂窝肺;⑩病理上有更多的纤维化和成纤维细胞灶;⑩对皮质激素治疗反应差。以上特征更倾向于UIP。而年轻女性,肺功能损害轻,BALF细胞学以淋巴细胞为主,影像显示磨玻璃样改变,病理显示活跃炎症改变等因素提示可能有良好治疗反应。 参考文献 [1]曾林祥,颜春松,李羲.皮质激素联合大环内酯类治疗特发性肺纤维化的疗效[J];江西医学院学报;2004年04期. [2]刘卫敏,徐启勇,林宇辉,叶燕青,戴莉,刘瑛琳.浓当归注射液对博莱霉素致鼠肺纤维化治疗作用的研究[J];医学新知杂志;2001年01期. [3]潘金兵;王思勤;赵桂华;陈卓昌;冯可青;朱惠珍.特发性间质性肺炎肺功能的分析[J];实用诊断与治疗杂志;2006年03期.
对特发性肺纤维化患者的护理心得 发表时间:2012-07-16T10:19:14.593Z 来源:《中外健康文摘》2012年第9期供稿作者:王树玉陈志敏马富艳 [导读] 特发性肺纤维化(IPF)是一种慢性的、通常可致命性的间质性肺疾病。约50%IPF患者于诊断后3年内死亡。 王树玉陈志敏马富艳 (黑龙江省鸡西矿业集团总医院城子河中心医院 158100) 【中图分类号】R563【文献标识码】B【文章编号】1672-5085(2012)9-0338-02 【摘要】目的讨论对特发性肺纤维化患者的护理心得。方法配合治疗进行护理。结论监测安静和运动时的氧合情况。多数患者需要家庭氧疗。对出院计划、呼吸护理人员和家庭治疗设备提供方给予适当的参考意见,确保患者护理的连续。 【关键词】特发性肺纤维化护理 特发性肺纤维化(IPF)是一种慢性的、通常可致命性的间质性肺疾病。约50%IPF患者于诊断后3年内死亡。本病曾被认为是一种罕见疾病,而今检出率明显提高。IPF曾称为隐匿性肺纤维化肺泡炎、弥漫性肺间质纤维化、特发性间质性肺炎和Hamman-Rich综合征。 IPF是肺的炎症、免疫、纤维化过程一系列变化的结果。然而,尽管已有许多相关的研究和假说,其始动因素仍不清楚。虽然已证实慢性炎症起着重要作用,仍没有足够证据证明病毒感染或遗传因素所起的作用。损伤和纤维化导致的炎症,最终扭曲和损伤肺泡毛细血管气体交换面的结构和功能。 IPF患者中,男性稍多于女性,吸烟者明显多于不吸烟者。发病率随年龄增加显著上升。下面将特发性肺纤维化患者的护理心得报告如下。 1 病理生理学 间质性炎症包括淋巴细胞、浆细胞、组织细胞浸润肺泡间隔。纤维化区域由致密的非细胞胶原组成。蜂窝状区域由囊性纤维气腔构成,后者常被覆于细支气管上皮,并充满黏液。纤维化及蜂窝状组织可见平滑肌增生。 2 并发症 ·呼吸衰竭。 ·慢性低氧血症。 ·肺动脉高压。 ·肺源性心脏病。 ·红细胞增多症。 3 临床表现 3.1IPF常表现为劳力性呼吸困难,持续性干咳,通常为阵发性。 3.2大多数患者就诊时上述症状已存在数月到2年。 3.3早期可闻及呼气末啰音(即Velcro啰音),以肺底部较明显。 3.4在疾病晚期,出现呼吸道实变后,可闻及支气管呼吸音。 3.5在运动或静息时出现浅快呼吸,70%以上患者可有杵状指。 3.6疾病晚期常可出现发绀及肺动脉高压表现(第2心音增强和第3心音奔马律)。 3.7随着疾病的进展,严重的低氧血症和重度衰弱的呼吸困难为典型表现。 4 诊断性检查 诊断该疾病应首先通过病史除外引起间质性肺疾病的其他常见原因。 4.1动脉血气分析和脉搏氧饱和度仪提示低氧血症,在疾病早期,静状态下较轻,晚期较严重。运动时氧合水平常恶化,且较严重。 4.2X线胸片可表现为以下4种特征性表现之一:间质性改变、网状结节状阴影、毛玻璃样改变或蜂窝状改变。X线胸片虽然有助于证实现有的异常表现,但不能很好的与组织学改变或肺功能检查联系起来判断疾病的严重程度,亦不能鉴别炎症和纤维化。但是,动态观察X线胸片改变有助于观察疾病的进展。 4.3高分辨CT可更好地发现常规胸片中的4种改变,并常规地应用于确立IPF的诊断。表现为双侧毛玻璃样透过度减低的网状异常。CT 的这4种异常改变是否与治疗反应相关仍处于研究中。 4.4肺活体组织检查有助于IPF的诊断。过去,开胸的活体组织检查是惟一可行的方法,并被认为是完善诊断的优选方法,如今可通过胸腔镜或支气管镜获取活体组织检查标本。活组织检查的组织学改变是多样化的,这取决于疾病的分期和其他尚未完全明了的因素。单核细胞及多形核细胞等慢性炎细胞浸润导致肺泡壁肿胀。早期可见肺泡内炎性细胞,随着疾病的进展,过量的胶原和成纤维细胞填充于肺间质。进展期肺泡壁破坏,并被蜂窝状囊腔取代。 肺功能检查提示肺活量和肺总量减低,CO弥散量下降。动态观察肺功能(尤其是CO弥散量)和动脉血气分析有助于观察病程,评价患者对治疗的反应。 5 治疗 尽管治疗不能改变该疾病的病理学改变,氧疗在疾病进程的早期阶段可以预防呼吸困难和组织缺氧相关的问题。在疾病初期,患者在静息状态时可能只需少量或不需要额外供氧,但随着疾病进展和运动时,会需要更多的氧气。 目前尚无有效的治疗方法。大剂量糖皮质激素和细胞毒性药物可用于抑制炎症反应,但常常无效。最近,干扰素-γ-1B显示对该病治疗有一定希望。 肺移植对于年轻的或全身状况较好者可能有效。 6 护理措施 6.1向患者解释所有诊断性检查,他可能会对明确诊断需要做的许多检查感到焦虑。 6.2监测安静和运动时的氧合情况。医师可以针对患者安静和运动时各开出适宜氧流量,运动时的氧流量大些,维持充足的氧气供应。建议患者根据运动量调节适当的氧流量。随着IPF进展,患者需氧增加。他可能需要非再吸入型面罩提供高浓度氧。最终在最大氧流量时也
粉尘致肺纤维化机制的研究现状 07级卫生检验班蒙永胜指导教师:白钢 【摘要】鉴于尘肺病的高发病率、高危害性和不可逆转性,探讨其发病机制以达到早发现、早诊断、早治疗具有重要意义。本文对目前研究的较为深入的自由基学说和细胞因子网络学说进行综述。 【关键词】尘肺;肺纤维化;脂质过氧化作用;自由基;细胞因子网络尘肺是由于在职业活动中长期吸入生产性粉尘而引起的以肺组织弥漫性纤维化为主的全身性疾病,是我国发病率最高的职业病,已成为我国一个重大的公共卫生和社会问题。尘肺所造成的肺纤维化,目前认为是不可逆转的病理变化[1],一经传统的后前位胸大片确诊,肺部病变已经无法逆转。但目前尘肺的发病机理仍然不完全清楚,无早期诊断的特异性指标,也无特异性的治疗药物和方法。因此,进一步探讨粉尘致肺纤维化机制以寻找早期防治方法,具有重要意义。研究表明,尘肺发病涉及几个重要环节:肺泡巨噬细胞(AM)吞噬石英颗粒,AM 及有关细胞发生脂质过氧化,各种致纤维化活性因子的分泌,成纤维细胞增生与胶原合成肺泡Ⅰ、Ⅱ型细胞损伤与基底膜受损。这几个环节涉及许多分子机制,共同决定了尘肺病的发生发展。 1 自由基 自由基是指那些外层电子轨道上具有不配对电子的原子、分子或基团,性质不稳定,反应活性强。常见的自由基有超氧阴离子自由基(O2-·)、羟自由基(OH·)、氢过氧基、脂氧自由基、脂过氧自由基等。目前普遍认为SiO2可引发自由基反应,启动AM 细胞膜的过氧化,产生过氧化脂质,从而使AM 细胞膜结构破坏,表面电荷减少、膜通透性增强、细胞流动性降低,最终导致细胞破裂;而细胞破损后释放出的SiO2又被重新吞噬,如此再破裂、再吞噬,反复循环,引起AM 大量增殖和集聚,为矽结节的形成和发展提供了基础。 1.1 矽肺发生中自由基的来源[2] 1.1.1 石英自身可产生自由基石英粉尘经机械破碎后,再与水作用时,可产生OH·;粉尘表面存在的一些金属离子,特别是Fe、Gu 等过渡金属元素能催化氧化石英尘表面吸附的生物基质及化学吸附的氧,形成自由基;硅酸水化也能产生过氧化氢(H2O2)。 1.1.2 人体细胞内和质膜上存在着产生自由基的酶如质膜上的还原型辅(NADPH)作为电子供体,当SiO2作用于质膜时氧接受电子还原成为O2-·;细胞内的黄嘌呤氧化酶(XO)可作用于ATP 的代谢产物次黄嘌呤,使其转变为黄嘌呤、尿酸,同时产生O2-·和H2O2;SiO2被AM 吞噬时,与AM 中的溶酶体融合生成次级溶酶体,此时,溶酶体中的髓过氧化物酶(MPO)作用于被吞噬物,产生大量的O2-·;当AM 吞噬和游走时,由已糖氧化供给消耗能量,氧代谢会突然增长,并产生大量的O2-·(即呼吸爆发)。O2-·和H2O2通过Haber-Weiss 反应生成毒性很强的OH·;在体内有过渡性金属离子存在时,即使没有O2-·也可通过Fenton 氏反应产生OH·。OH·是一种活性极强的自由基,能与任何生物分子起反应,且由于半衰期极短,没有任何酶能够排除它。脂质被初始氧化后的中间产物,如有机自由基、烷氧自由基与O2-·等,也能持续产生毒性很强的H2O2。通过前述过程自由基可被放大和形成链式反应。 1.1.3 髓单核细胞和在肺中转化为AM 的细胞中都含有NO 合成酶NO 合成酶可催化精氨酸与分子O2生成NO·,当AM 被活化后可产生NO·,NO·与生物系统中产生 的O2-·相互作用可生成超氧亚硝酸阴离子(ONOO-),ONOO-一旦被质子化后可迅速分解成高活性的OH·和稳定的NO2·,产生细胞毒作用,导致细胞坏死和组织损伤。 1.2 自由基的损伤作用 1.2.1 对肺吞噬细胞的作用自由基对不饱和共价键有一种特殊的亲和力,因此,在体内自由基最易攻击生物膜磷脂的多不饱和脂肪酸,产生脂质自由基并自氧化,形成过氧化产物,
特发性肺纤维化(IPF)的护理常规 一定义: 间质性肺疾病是一组主要累及肺间质、肺泡和(或)细支气管的肺部弥漫性疾病,通常亦称作弥漫性实质性肺疾病。特发性肺纤维化(IPF)系指特发性间质性肺炎中病理表现为寻常型间质性肺炎的一种类型,且最常见。病变局限于肺部,引起弥漫性肺纤维化,导致肺功能损害和呼吸困难。 二临床表现: 通常为隐袭性起病,主要症状是干咳和劳力性气促,限制性通气功能障碍伴弥散功能降低、低氧血症和影象上的双肺弥漫性病变。病程多缓慢进展,逐渐丧失肺泡-毛细血管功能单位,发展为弥漫性肺纤维化和蜂窝肺,晚期出现肺心病,最终导致呼吸功能衰竭而死亡。 三辅助检查: X线胸片显示双肺弥漫性阴影 肺功能表现为限制性通气功能障碍和弥散量减少。 实验室检查为非特异性变化,可以有血沉加快,血乳酸脱氢酶增高和免疫球蛋白增高;有10%-26%的患者类风湿因子和抗核抗体阳性。 四治疗: IPF是一种持续发展的疾病,治疗原则主要在于积极控制肺泡炎症并使之逆转,进而防止发展为不可逆的肺纤维化,但迄今尚无特效疗法。 1.糖皮质激素仍为首选药; 2.其次为免疫抑制剂等。 3.对症治疗如出现继发感染时应根据细菌类型选择抗菌素;低氧血症可给予低流量吸氧。 六护理措施: 1.观察生命体征,呼吸形态。 2.痰的颜色,性状,粘稠度,气味及量的变化。 3.给予端坐位或半坐位,利于呼吸。 4.鼓励病人咳嗽,指导病人正确咳嗽,促进排痰。痰液较多不易咳出时,遵医嘱使用祛痰剂或超声雾化吸入,必要时吸痰。 5.合理用氧,采用低流量给氧,流量1-2L/min,吸入前湿化。 6.遵医嘱给予抗炎治疗,有效控制呼吸道感染。 7.多饮水,给予高热量、高维生素的流质、半流、软食,少量多餐,少吃产气食品,防止产气影响膈肌运动。 8.注意心理指导,护士应聆听病人的叙述,疏导其心理压力,必要时请心理医
特发性肺纤维化诊疗专家共识2020 2002年7月中华医学会呼吸病学分会制定了“特发性肺(间质)纤维化诊断和治疗指南(草案)”[1]。近十几年来对特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)的自然病程、病因学、发病机制和诊断方法都有了进一步的认识[2-3],特别是抗肺纤维化药物的研发取得了一定进展,使IPF的治疗进入了新阶段[4]。为规范IPF的诊断和治疗,中华医学会呼吸病学分会间质性肺疾病学组组织专家充分讨论后,结合国内外研究进展,参照国际最新指南,制定本专家共识。 一、概念 IPF是一种病因不明,慢性进行性纤维化性间质性肺炎,病变局限在肺脏,好发于中老年男性人群,主要表现为进行性加重的呼吸困难,伴限制性通气功能障碍和气体交换障碍,导致低氧血症、甚至呼吸衰竭,预后差,其肺组织学和胸部高分辨率CT(HRCT)表现为普通型间质性肺炎(UIP)[1-3]。在临床上,IPF的概念需要与以下疾病概念区分。 1.间质性肺疾病(interstitial lung disease,ILD):亦称作弥漫性实质性肺疾病(DPLD),是一组主要累及肺间质和肺泡腔,导致肺泡-毛细血管功能单位丧失的弥漫性肺疾病的总称。临床主要表现进行性加重的呼吸困难、通气功能障碍伴弥散功能降低、低氧血症和影像学上的双肺弥漫性病变。 2.特发性间质性肺炎(idiopathic interstitial pneumonias,IIPs):即目前病因不明的间质性肺炎,属于ILD疾病谱的一组疾病。2013年发表的有关IIPs的国际多学科分类,将IIPs分为主要的IIPs、罕见的IIPs和未分类的IIPs[3](表1)。主要的IIPs有6种类型,包括IPF、特发性非特异性间质性肺炎(iNSIP)、呼吸性细支气管炎伴间质性肺疾病(RB-ILD)、脱屑性间质性肺炎(DIP)、隐源性机化性肺炎(COP)、急性间质性肺炎(AIP)。罕见的IIPs有2
一、心理护理 多数患者由于受疾病的折磨,心情急躁,对预后担忧。学习和练习放松:焦虑和悲观情绪常见于慢性肺疾病患者,病逝病情加重;气短、活动力下降及悲观情绪可能会使患者脱离家人和朋友。学会放松有助于控制因气短而产生的恐惧;身体和精神放松可以避免因肌肉紧张而消耗过多的氧气。长期住院治疗,家庭经济困难,产生悲观、抑郁情绪。首先应多方面安慰,多与患者沟通,让患者增加治疗的信心,可以介绍效果好的患者治疗的过程、治疗前后的情况,消除患者的紧张情绪,建立信任的护患关系。尽量满足患者需求,护理操纵纯熟、正确、减轻其痛苦。 二、饮食护理 保持良好的营养和适当的体重:良好的营养对保持理想体重很有帮助,慢性肺疾病的患者,因为怕吃饭时气短,所以进食减少,导致营养不良,低营养使呼吸肌乏力,从而气短加重。另外,体重超重增加心肺供氧到全身的负担,也导致气短,超重也增加膈肌的压力而使呼吸不足。 ①重度肺纤维化病人可予软食或半流食:这样可以减轻呼吸急迫所引起的咀嚼和吞咽困难,既有利于消化吸收,又可防止食物反流。 ②少吃辛辣、煎炸等刺激性油腻食品:平时以清淡为宜,尤其对于肥胖患者,脂肪供应量宜低。吃肉以瘦肉为宜,以达到祛痰湿与适当控制体重为目的。辛辣、煎炸等食品,易生痰,导致热助邪盛,邪热郁内而不达,久之可酿成疾热上犯于肺,加重病情。 ③少量多餐。指导患者进食优质蛋白、高热量、高维生素的饮食。例如:蛋类、糙米、玉米面、荞麦面、水果和蔬菜等,可多给予木耳、紫菜、海带、蘑菇等。 ④多饮水:重度间质性肺炎/肺纤维化患者因张口呼吸,出汗多、饮食少,常使患者失水,并使痰液黏稠不易咯出,因此及时补充水分、增加液体摄进量,对于纠正或防止失水,具有非常重要的意义。要鼓励患者多饮水。如患者不能饮食时,可用静脉补液,这样有利于稀释痰液。伴有心衰的患者饮水要适量。 ⑤对某些已知会引起过敏、诱发哮喘的食物,应避免食用:所谓“忌口”就是忌“发物”。“发物”一般是指食后能引起宿病复发或新病加重的食物。“发物”包括的范围很广,对于不同的患者来说是因人而异的。例如:有些过敏体质者常因吃了鱼、虾、蟹等海腥类的食品而诱发咳、喘加重。所以肺纤维化患者应根据自己的实际情况,做好“忌口”,这样既可以避免由饮食不慎而导致咳、喘加重,又可以防止因过于讲究“忌口”而影响机体对多种营养物质的吸收。 ⑥忌烟酒、忌过咸食物:肺纤维化患者多数伴有气道高反应状态,烟、酒和过咸食物的刺激,轻易引发支气管的反应,加重咳嗽、气喘等症状。戒烟,停止刺激是阻止肺进一步损害的好方法,如果您仍吸烟,最重要的就是戒烟。戒烟困难者应该向医生寻求帮助。被动吸烟与您自己吸烟一样有害,劝告家人和朋友戒烟,至少不要在您周围吸烟。
肺纤维化诊治指南 特发性肺纤维化(idiopathic pulmonary fibrosis ,IPF)是一种不明原因的肺间质炎症性疾病,原因不明的肺泡纤维化和弥漫性肺间质纤维化均为其同义词。典型的IPF,主要表现为干咳、进行性呼吸困难,经数月或数年逐渐恶化,多在出现症状3~8年内进展至终末期呼吸衰竭或死亡。主要病理特点为肺间质和肺泡腔内纤维化和炎细胞浸润混合存在。尽管该病的发病机制还没有完全阐明,就其临床特征和病理足以说明这是一种特征性的疾病。IPF 的治疗尚缺乏客观的、决定性的预后因素或治疗反应,皮质激素(以下简称激素)或免疫抑制剂、细 胞毒药物仍是其主要的治疗药物,但不足30%的病人有治疗反应,且可表现毒副反应。病理 大多数间质性肺疾病都有共同的病理基础过程。初期损伤之后有肺泡炎,随着炎性-免疫反应的进展,肺纤维化泡壁、气道和血管最终都会发生不可逆 的肺部瘢痕(纤维化)。炎症和异常修复导致肺间质细胞增殖,产生大量的胶原和细胞外基质。肺组织的正常结构为囊性空腔所替代,这些囊性空腔有增厚的纤维组织所包绕,此为晚期的“蜂窝肺”。肺间质纤维化和“蜂窝肺”的形成,导致肺泡气体- 交换单元持久性的丧失。 肺纤维化发展过程中肺泡塌陷是失去上皮细胞的结果。暴露的基底膜可直接接触和形成纤维组织,大量肺泡塌陷即形成密集的瘢痕,形成蜂窝样改变。蜂窝样改变是瘢痕和结构重组的一种表现。 肺脏损伤后,修复的结果是纤维化还是恢复正常解剖结构,取决于肺泡内渗出物及碎屑能否有效清除。 如肺泡内渗出物未清除,成纤维细胞和其他细胞就会侵入并增殖,(免疫组织化学染色已经证实,在成纤维细胞灶里可发现蛋白聚糖、整合素、连结体等。这些特点表明纤维化是一种活动性进展,而不是一种“旧”纤维组织的后遗症。)从而使肺纤维化进行性进展。 特发性肺间质纤维化的西医病因病机:胶原蛋白是肺组织的主要ECM蛋白,约占肺脏五分之一。肺脏中何种类型的胶原蛋白与其他类型ECM成分等构成三维网状结构,作为肺组织结构的主要骨架,这些蛋白成分保持肺组织结构的完整性。并对维持肺上皮及内皮细胞分化状态起着十分重要的作用。肺纤维化是有早期组织损伤、症状变化发展而来的,虽然引起肺纤维化的病因或原发病有很大的差异,但归根结底是因为促进纤维化因子生成增多或抗纤维化因子产生相对不足,导致肺纤维化过程的ECM代谢异常。一般情况下,肺纤维化早期出现肺泡炎,肺泡内有浆液和细胞成分,肺间质内有大量单核细胞,部分淋巴细胞,浆细胞,肺泡巨噬细胞等炎性细胞浸润,肺泡结构完整。进入晚期,慢性炎症已减轻,肺泡结构为坚实的胶原代替,肺泡壁被破坏,形成扩张的蜂窝肺。胶原、细胞外基质、成纤维细胞分布在间质中,肺泡上皮化生为鳞状上皮。基于以上病理变化,临床上多表现为进行性呼吸困难或伴有刺激性干咳,胸部X 线显示两中下肺野网状阴影,肺功能为限制性通气功能障碍。病情呈持续性进展,最终因 呼吸衰竭而死亡。
!!作者单位">)####武汉$华中科技大学同济医学院附属协和医院呼吸内科血管生成在肺纤维化发病机制中的意义 何彦侠!综述!辛建保!审校 !!!摘!要"!肺纤维化是一组以肺间质弥漫性渗出,浸润和纤维化为主要病变的疾病$传统认为慢性炎症起着重要作用$ 但基于这种认识的非特异性抗炎$免疫抑制治疗不尽人意&近年有研究提出$肺部广泛损伤后修复初期$异常失控的血管生成是纤维化性修复的触发和推动因素$早期阻断异常血管生成有可能防止肺纤维化发生&但肺纤维化中的血管调节因素及其作用机制目前研究尚浅$有待于进一步探索& !关键词"!血管生成) 肺纤维化)血管生成因子!!机体的血管形成包括血管发生和血管生成两个过程&成血干细胞分化形成原始血管结构称为血管发生#W 9506834-+-5*5%$是胚胎发育中形成血管的主要形式&而由原始血管丛或已存在的血管出芽形成新血管的过程称为血管生成#9+4*34-+-5*5%$血管生成是成熟机体形成新生血管的唯一方式&生理条件下血管生成是一严格受控过程$只见于发育,再生和创伤修复等过程$而病理性的血管生成则是风湿性关节炎,角膜血管化,哮喘气道重构,慢性炎症,肺纤维化以及肿瘤等疾病的关键环节$它是一种持续,无控性过程$这种持续$无规则的新血管生成推动了疾病的发展& !!血管生成的过程 血管生成过程受到血管生成刺激因子和血管生成抑制因子的双重调节$即-血管形成的开关平衡假说.&正常组织中由于缺少血管生成刺激因子或者血管生成刺激因子被高水平血管生成抑制因子严格控制$血管生成的开关处于关闭状态)当生长因子浓度上升$或者血管生成抑制因子浓度下降$都会打破血管生成的调节平衡$使开关处于开放状态引起血 管生成过程$通常包括以下几个步骤*"+ ""内皮细胞 受各种刺激因子作用$激活分化)#血管局部细胞外基质,基底膜降解)$内皮细胞芽生$增殖和迁移直至管腔形成)/间叶细胞募集到管腔周形成内皮外膜细胞& %!肺纤维化中血管生成的诱因 多种刺激因素可以引起血管生成$如缺氧,广泛损伤,活化癌基因,细胞因子等&肺部广泛损伤后增生修复中伴有肺泡肉芽组织的形成&大量炎性渗出引起气体交换障碍)炎性细胞活化消耗大量氧引起相对氧不足&缺氧通过缺氧诱发因子#F S A % 刺激促血管生成因子释放$引起毛细血管代偿性增生以适 应组织代谢需要* $+ )同时炎性细胞释放大量促血管生成因子促进血管生成& )!肺纤维化中可能的血管生成调节因子 )+!!血管生成促进因子!多种因子可促进血管生成$主要有L Y %\O T O 趋化因子,血管内皮生长因子#K L ;A %,血管生成素#9+4*373*-,*+%,:&A <(,:;A <)等&但每种因子在肺纤维化血管异常生成中的地位如何目前尚无透彻研究&现分述如下")+!+!!L Y %\O T O 趋化因子家族!目前在肺纤维化血管生成机制中研究较多的是O T O 趋化因子家族&根据有无;86
特发性肺纤维化病因及发病机制 *导读:IPF病因不明,发病机制亦未完全阐明,但已有足 够证据表明与免疫炎症损伤有关。…… IPF病因不明,发病机制亦未完全阐明,但已有足够证据表明与免疫炎症损伤有关。不同标本所显示的免疫炎症反应特征不尽一致,周围血所反映出的是免疫异常比较突出,而支气管肺泡灌洗液显示炎症反应为主,而肺局部组织的异常又有所不同。因此在评估各种研究资料需要考虑到这种差异。 综合近年来的研究,关于IPF发病机制及其过程概括如下:①某种未知抗原激活B细胞,产生Ig并形成免疫复合物,进而刺 激和活化肺泡巨噬细胞。这一免疫反应在肺局部,如果肺泡壁B 淋巴细胞也产生抗体,则肺泡壁的某些成分可能会被错误地识别为异物。因此有人认为IPF可视为自身免疫性疾病。但IPF患者T细胞的变化及其作用不明确,仅B细胞参予不足以证明其为自身免疫性疾病。②活化的肺泡巨噬细胞释放多种介质,除蛋白水解酶、胶原酶、反应性氧代谢产物和某些细胞因子直接损伤肺细胞、细胞外基质、基底膜等结构外,尚有与纤维化形成密切相关的介质包括纤维连接蛋白(fibronectin,FN)、肺泡巨噬细胞原 性生长因子(alveolarmacrophagederivedgrowthfactor,AMDGF)、血小板衍生生长因子(plateletderivedgrowthfactor,PDGF)和 胰岛素样生长因子(insulinlikegrowthfactor,IGF)等,它们能
吸附成纤维细胞,并刺激其增殖,以及介导胶原基质收缩。③在肺泡巨噬细胞释放的IL-8、TNF等介导下中性粒细胞向着肺泡趋化、聚集和活化,形成以中性粒细胞比率增高(20%)为特征的肺泡炎,而中性粒细胞炎症反应又释放一系列介质,引起或加重肺损伤与纤维化。④成纤维细胞增生和产生胶原是本病的重要环节和结局。正常人成纤维细胞生长存在精确的调控,如前列腺素 E2、成纤维细胞移动抑制因子等均属于负调节因子。另在IPF发现一种编码PDGF的C-sis基因,与转移性病毒癌基因V-sis非常相似。因此IPF的发生是否代表了成纤维细胞的负调节失效、抑或成纤维细胞的"肿瘤性"增生,是十分饶有兴趣的问题。虽然有人发现IPF患者肺间质胶原的合成速度或总量并无增加,但Ⅰ型胶原增加、Ⅰ型胶原对Ⅱ型胶原的比率升高。因为Ⅰ型胶原是一种高张力强度、低顺应性、呈平行排列的交叉带状纤维,它的增加足以解释IPF的形态和生理学改变,而不论胶原总量增加与否。