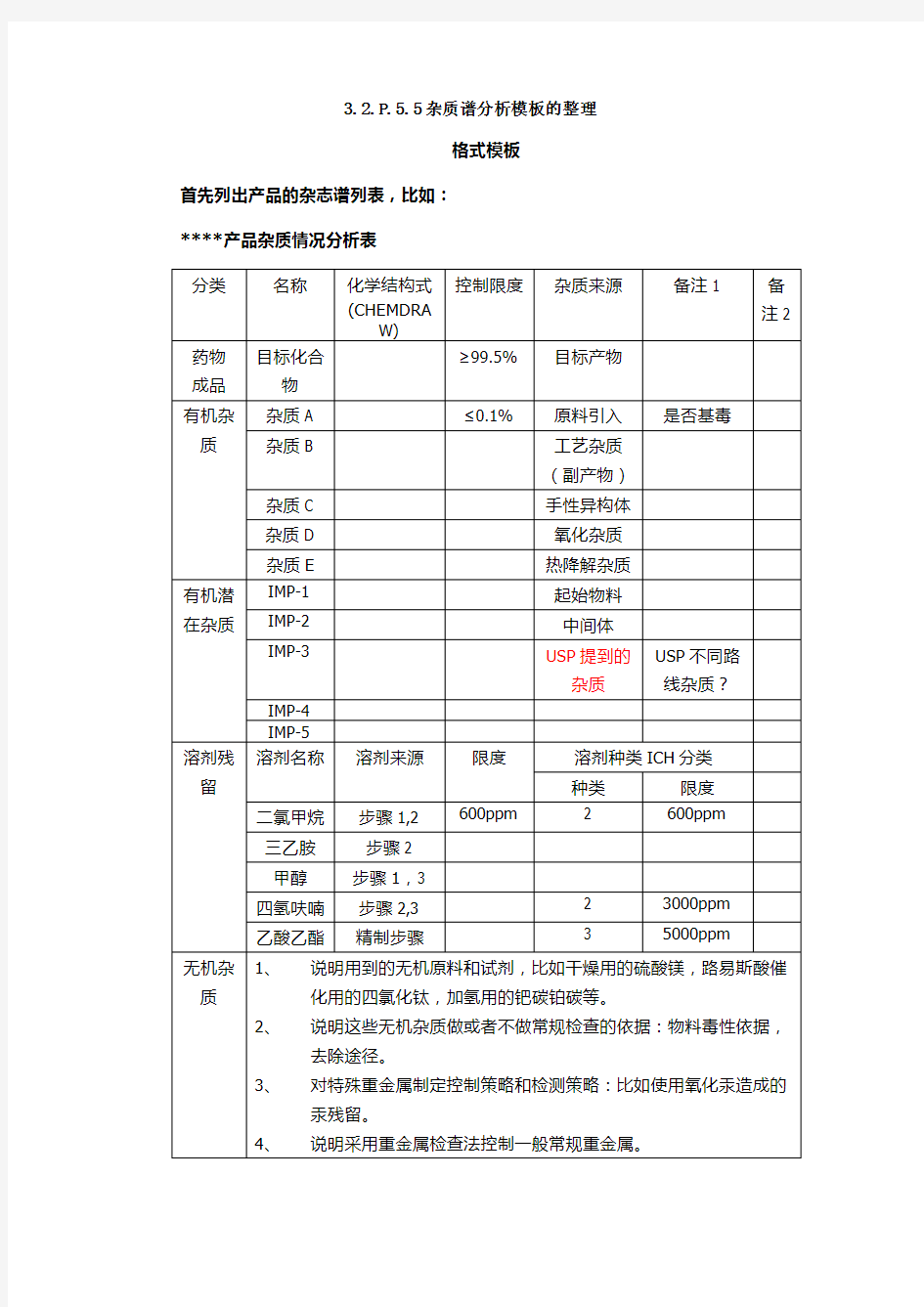
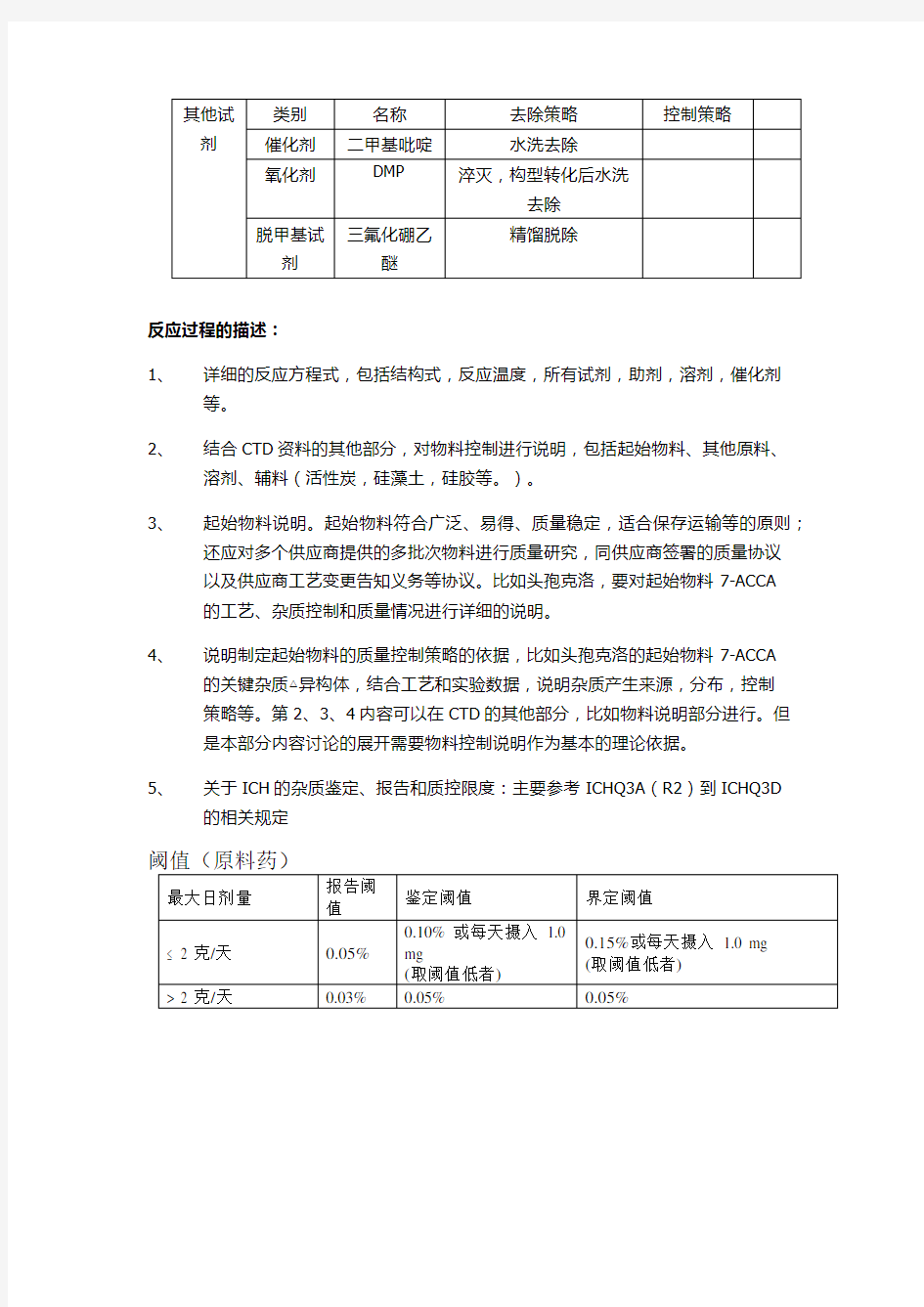
3.2.P.5.5杂质谱分析模板的整理
格式模板
首先列出产品的杂志谱列表,比如:
****产品杂质情况分析表
反应过程的描述: 1、
详细的反应方程式,包括结构式,反应温度,所有试剂,助剂,溶剂,催化剂等。
2、
结合CTD 资料的其他部分,对物料控制进行说明,包括起始物料、其他原料、溶剂、辅料(活性炭,硅藻土,硅胶等。)。
3、
起始物料说明。起始物料符合广泛、易得、质量稳定,适合保存运输等的原则;还应对多个供应商提供的多批次物料进行质量研究,同供应商签署的质量协议以及供应商工艺变更告知义务等协议。比如头孢克洛,要对起始物料7-ACCA 的工艺、杂质控制和质量情况进行详细的说明。
4、
说明制定起始物料的质量控制策略的依据,比如头孢克洛的起始物料
7-ACCA 的关键杂质△异构体,结合工艺和实验数据,说明杂质产生来源,分布,控制策略等。第2、3、4内容可以在CTD 的其他部分,比如物料说明部分进行。但是本部分内容讨论的展开需要物料控制说明作为基本的理论依据。
5、
关于ICH 的杂质鉴定、报告和质控限度:主要参考ICHQ3A (R2)到ICHQ3D 的相关规定
结合上述反应过程对杂质谱进行分析,主要分起始物料引入杂质,反应杂质,降解杂质等。
第一部分:起始物料引入的杂质分析(比如头孢克洛的起始物料7-ACCA 引入的杂质)
N S
OH O
O
NO 2
O
H N O
C 22H 19N 3O 7S MW: 469.47
O
C 16H 17KN 2O 4S MW: 372.48
Br
NO 2
+
O
2
MW: 469.51
C 7H 6BrNO 2MW: 216.0322
C 23H 23MW: 485.51
N S N H H
OH O
O
NO 2
O
C 22H 19N 3O 6S MW: 453.47
CH 2Cl 2/TEBAC TsCl
morpholine
N
S N H
H
N O
O
NO 2O
O
C
26H 26N 4O 6S MW: 522.57
1) Br 2-pyrindine
CH 2Cl 2
2
2C 14H 1235MW: 406.00
HCl 1) (PhO)3P/CH 2Cl 2
22) N,N-dimethyl aniline, PCl 5 i BuOH
Na 2S 2O 42H 2C 7H 7ClN 2O 3S MW: 234.66
1、 无机杂质:说明引入情况和消除渠道;以及相关的控制方法和标准以及依据。比如上述列表中的钯元素控制。
2、 普通有机杂质。
3、
对映异构体(根据品种的情况具体分析);考察不同的对应异构体对最终产品质量的影响情况。
4、 非对映异构体:比如ACCA 的△异构体;还包括非对应异构体自身的各种对映体。
第二部分:反应的每个步骤引入的杂质:需要结合实际反应监控(HPLC,LC-MS为主)过程对杂质消除过程以及对后续的影响进行实际说明。这个内容主要在工艺描述部分进行,本部分引用工艺描述内容。
1、步骤1引入的杂质。
2、步骤2引入的杂质。
3、步骤3引入的杂质。以及后续的步骤产生的杂质以及消除过程和简单的控制描
述……
4、精制过程引入的杂质。
第三部分:结合小试中试数据汇总列表、方法适用性、实际检测结果等内容说明各个有机杂质的分布情况。
第四部分:潜在杂质在成品中的检测结果
第五部分:自制原料与原研片杂质对比:
行对比性说明。
第六部分:制剂中的特定杂质:主要分析制剂与原料药储存过程中降解产生的杂质,以及因为辅料和原料药相容性而产生的特定杂质:比如纤维素类的曼德拉反
应产生的杂质等。
储存降解产生的杂质包括酸碱氧热光降解杂质,空气中水分造成的水解杂质,或
者跟空气中二氧化碳反应产生的杂质。
1.说明各杂质的限度:
****杂质阈值(举例)
2. 3.2.P.5.5.1潜在杂质的推测
由****公司提供**原料药简单工艺路线,分析整理本品的杂质见下表。
表3.2.P.** ,-------------潜在杂质推测
3.潜在杂质特征的鉴定
a)原料合成过程带入的相关杂质特征的鉴定
由厂家提供的原料药合成过程中所用的起始原料及各步中间产物的色谱行为和紫外光谱特征进行汇总分析,初步鉴定注射用头孢地嗪钠成品中原料合成过程中带入的杂质。起始原料及各步中间产物鉴定特征如下表:
表3.2.P.5-X ****原料合成过程相关杂质特征鉴定
b)注射用头孢地嗪钠强制降解试验中杂质的特征鉴定
试品与对照品强制降解试验
表3.2.P.** 热破坏试验主要降解产物色谱信息
表3.2.P.5-50 酸破坏试验主要降解产物色谱信息
表3.2.P.5-51 碱破坏试验主要降解产物色谱信息
表3.2.P.5-52 氧化破坏试验主要降解产物色谱信息
表3.2.P.5-53 光照破坏试验主要降解产物
4.制剂杂质列表
a)以列表的方式列明产品中可能含有的杂质,分析杂质的产生来源,结合相关
指导原则要求,对于已知杂质给出化学结构并提供结构确证资料,并提供控制限度。
杂质的结构和种类的鉴定在其他模块或者附件中体现。按照以前惯例,自制杂质的结构确认是通过工艺描述加氢谱确认即可,特殊的需要提供其他佐
证手段。外购杂质的结构确认一般是要提供完整的结构鉴定谱图。
b)说明各杂质在最终质量标准中是否进行控制以及控制的限度,并提供充分的
依据。
报告阈值是指所有高于此阈值的杂质及其含量均应在每批产品的检验报告中记录,并在申报资料中反映;鉴定阈值是指所有高于此阈值的杂质均应对其结构进行确证;合理限度是指只要质量标准中制订的杂质限度不高于此限度,就不需要提供该限度的制订依据,认为该限度是合理的。
c)提供详细的降解途径与降解产物研究资料与图谱。应在对原料药的降解途径
与降解产物有充分了解的基础上,进行系统的降解途径与降解产物研究,明
确说明本品的降解途径与降解产物
5.对比自制制剂片和原料药杂质谱以及纯度数据,
6.对比自制制剂片和参比制剂杂质谱以及纯度数据,
7.对比自制制剂片和参比制剂强制降解和长期稳定性考察的杂质谱变化、图
谱以及纯度数据。
长
期
6
个
月
************************************************************************************ ************************************************************************************ 附件1:
《CTD申报资料中杂质研究的几个问题》这篇需要我们认真研究一下:下面做了简单的抄录。(https://www.doczj.com/doc/088522480.html,/dzkw.do?method=largePage&id=312898)
杂志谱研究需要以杂质谱分析为主线,安全性为核心,按照风险控制的策略,将杂质研究与CMC各项研究,乃至药理毒理及临床安全性研究等环节关联思考、综合考虑,而不仅仅拘泥于提供准确分析数据的传统思维,不是一项孤立的分析工作。
1、杂质控制要做到源头控制、过程控制与终点控制相结合的杂质控制理念:
CTD格式中杂质控制的考虑要体现在CMC的各个环节,而不是仅仅局限在“质量控制”模块。比如原辅料首先要满足制剂质量要求;关键工艺步骤及参数的确立、工艺开发过程等要考虑以杂质是否得到有效控制为重点关注。制剂相关特性中要体现与原研产品杂质谱等的对比情况;包材、贮藏条件以及有效期的确立等也要以杂质是否处于安全合理的可控范围内为核心。
2、关注杂质分析与控制的系统性与整体性,不能割裂各项内容的必然联系和有机统一:
杂质分析与控制的相关内容会分布在分析方法(3.2.S.4.2)、方法学验证
(3.2.S.4.3)、杂质对比研究与杂质谱分析(3.2.S.4.5)、杂质情况分析总结
(3.2.S.3.2)、样品检测与数据积累(3.2.S.4.4)、控制限度(2.3.S.4.1)等各模块中,但杂质研究又是一项系统工程,具有统一的整体性。因此,不要因为申报资料格式的模块化而人为割裂各项研究内容的相互联系。
3、从杂质谱分析入手确立科学的杂质研究基本思路。杂质谱包括药物中所有杂质的种类、来源及特性等信息。有针对性地选择合适的杂质分析方法,以确保杂质的有效检出和确认;通过与原研产品杂质谱的对比研究,根据各相应杂质的一致性求证,或跟踪杂质
谱对安全性试验或临床试验结果产生的影响,评估各杂质的安全性风险和可接受水平;结合规模化生产时杂质谱的变化情况,确立安全合理的杂质控制水平。
4、分析方法的验证应具备针对性和全面性。
5、杂质限度的确定应符合相关技术指导原则的要求。在分析方法相同的情况下,各特定杂质应不超过同品种或同类品种法定标准的相应限度;非特定杂质应采用LC/UV、LC/MS、PDA等多种方法与原研产品或ICH成员国产品进行杂质比较,说明各杂质与原研产品的一致性,相同杂质限度可参考ChP、USP、BP/EP、JP等药典同品种最严格的单个杂质限度确定。
附件2:引用的5.30文件的要求:
化学仿制药电子通用技术文档申报指导原则(征求意见稿)
A.以列表的方式列明产品中可能含有的杂质,分析杂质的产生来源,结合相关指导原则要求,对于已
知杂质给出化学结构并提供结构确证资料,并提供控制限度
B.说明各杂质在最终质量标准中是否进行控制以及控制的限度,并提供充分的依据。
C.提供详细的降解途径与降解产物研究资料与图谱。应在对原料药的降解途径与降解产物有充分了解
的基础上,进行系统的降解途径与降解产物研究,明确说明本品的降解途径与降解产物
化学药品新注册分类申报资料要求(试行)-4类
A.以列表的方式列明产品中可能含有的杂质,分析杂质的产生来源,结合相关指导原则要求,对于已
知杂质给出化学结构并提供结构确证资料,并提供控制限度。可以表格形式整理
B.说明各杂质在最终质量标准中是否进行控制以及控制的限度,并提供充分的依据。
C.提供详细的降解途径与降解产物研究资料与图谱。应在对原料药的降解途径与降解产物有充分了解
的基础上,进行系统的降解途径与降解产物研究,明确说明本品的降解途径与降解产物
化学药品新注册分类申报资料要求(试行)-3类
A.以列表的方式列明产品中可能含有的杂质,分析杂质的产生来源,结合相关指导原则要求,对于已
知杂质给出化学结构并提供结构确证资料,并提供控制限度。
B.对于最终质量标准中是否进行控制以及控制的限度,应提供依据。
3.2.P.5.5杂质谱分析模板的整理 格式模板 首先列出产品的杂志谱列表,比如: ****产品杂质情况分析表
反应过程的描述: 1、 详细的反应方程式,包括结构式,反应温度,所有试剂,助剂,溶剂,催化剂等。 2、 结合CTD 资料的其他部分,对物料控制进行说明,包括起始物料、其他原料、溶剂、辅料(活性炭,硅藻土,硅胶等。)。 3、 起始物料说明。起始物料符合广泛、易得、质量稳定,适合保存运输等的原则;还应对多个供应商提供的多批次物料进行质量研究,同供应商签署的质量协议以及供应商工艺变更告知义务等协议。比如头孢克洛,要对起始物料7-ACCA 的工艺、杂质控制和质量情况进行详细的说明。 4、 说明制定起始物料的质量控制策略的依据,比如头孢克洛的起始物料 7-ACCA 的关键杂质△异构体,结合工艺和实验数据,说明杂质产生来源,分布,控制策略等。第2、3、4内容可以在CTD 的其他部分,比如物料说明部分进行。但是本部分内容讨论的展开需要物料控制说明作为基本的理论依据。 5、 关于ICH 的杂质鉴定、报告和质控限度:主要参考ICHQ3A (R2)到ICHQ3D 的相关规定
结合上述反应过程对杂质谱进行分析,主要分起始物料引入杂质,反应杂质,降解杂质等。 第一部分:起始物料引入的杂质分析(比如头孢克洛的起始物料7-ACCA 引入的杂质) N S OH O O NO 2 O H N O C 22H 19N 3O 7S MW: 469.47 O C 16H 17KN 2O 4S MW: 372.48 Br NO 2 + O 2 MW: 469.51 C 7H 6BrNO 2MW: 216.0322 C 23H 23MW: 485.51 N S N H H OH O O NO 2 O C 22H 19N 3O 6S MW: 453.47 CH 2Cl 2/TEBAC TsCl morpholine N S N H H N O O NO 2O O C 26H 26N 4O 6S MW: 522.57 1) Br 2-pyrindine CH 2Cl 2 2 2C 14H 1235MW: 406.00 HCl 1) (PhO)3P/CH 2Cl 2 22) N,N-dimethyl aniline, PCl 5 i BuOH Na 2S 2O 42H 2C 7H 7ClN 2O 3S MW: 234.66 1、 无机杂质:说明引入情况和消除渠道;以及相关的控制方法和标准以及依据。比如上述列表中的钯元素控制。 2、 普通有机杂质。 3、 对映异构体(根据品种的情况具体分析);考察不同的对应异构体对最终产品质量的影响情况。 4、 非对映异构体:比如ACCA 的△异构体;还包括非对应异构体自身的各种对映体。
3.2.P.5.5 杂质谱分析模板的整理 格式模板首先列出产品的杂志谱列表,比如: **** 产品杂质情况分析表
反应过程的描述: 1、详细的反应方程式,包括结构式,反应温度,所有试剂,助剂,溶剂,催化剂等。 2、结合CTD 资料的其他部分,对物料控制进行说明,包括起始物料、其他原料、溶剂、辅料(活性 炭,硅藻土,硅胶等。)。 3、起始物料说明。起始物料符合广泛、易得、质量稳定,适合保存运输等的原则;还应对多个供应商 提供的多批次物料进行质量研究,同供应商签署的质量协议以及供应商工艺变更告知义务等协议。 比如头孢克洛,要对起始物料7-ACCA 的工艺、杂质控制和质量情况进行详细的说明。 4、说明制定起始物料的质量控制策略的依据,比如头孢克洛的起始物料7-ACCA 的关键杂质△异构体,结合工艺和实验数据,说明杂质产生来源,分布,控制策略等。第2、3、4 内容可以在CTD 的其他部分,比如物料说明部分进行。但是本部分内容讨论的展开需要物料控制 说明作为基本的理论依据。 5、关于ICH 的杂质鉴定、报告和质控限度:主要参考ICHQ3A (R2)到ICHQ3D 的相关规定 阈值(原料药)
结合上述反应过程对杂质谱进行分析,主要分起始物料引入杂质,反应杂质,降解杂 质等。 第一部分:起始物料引入的杂质分析(比如头孢克洛的起始物料 7-ACCA 引入的杂质) 比如上述列表中的钯元素控制。 3 、 对映异构体(根据品种的情况具体分析);考察不同的对应异构体对最终产品 质量的影响情况。 4、 非对映异构体:比如 ACCA 的△异构体;还包括非对应异构体自身的各种对映体。 第二部分:反应的每个步骤引 入的杂质:需要结合实际反应监控( HPLC ,LC-MS 为 主)过程对杂质消除过程以及对后续的影响进行实际说明。这个内容主要在工艺描述 部分进行,本部分引用工艺描述内容。 1、 步骤 1 引入的杂质。 PAA O H N O H N S O OK C 16 H 17 KN 2 O 4 S MW: 372.48 O HN CH 2 Cl CH TsCl Cl /TEBAC n-methylmorpholine morpholine 1) (PhO) 3 P/CH Cl 2 /Pyridine 2) N,N-dimethyl aniline, PCl BuOH Br NO 2 C 7 H 6 BrNO MW: 216.03 H O O N S O O O O C 23 H 23 N 3 O 7 S MW: 485.51 2 Cl 2 DMF/CH 2 Cl 2 TMP toluene NO O N O N O S O 6S NO C 23 H 23 N 3O MW: 469.51 C 22 H 19 N 3 O 7 S MW: 469.47 HCl H 2 N Na 2 S 2 O 4 acetone/H 2O C 14 H 12 ClN 3O 5 S MW: 406.00 7H 7 ClN 2 O 3 S MW: 234.66 1、 无机杂质:说明引入情况和消除渠道; 以及相关的控制方法和标准以及依据。 2、 普通有机杂质。 NO 2 MW: 522.57 H N 2 Cl O
景德镇陶瓷学院 半导体课程设计报告 设计题目n型半导体材料的设计与性能分析专业班级 姓名 学号 指导教师 完成时间
一﹑杂质半导体的应用背景 半导体中的杂质对电离率的影响非常大,本征半导体经过掺杂就形成杂质半导体,半导体中掺杂微量杂质时,杂质原子的附近的周期势场的干扰并形成附加的束缚状态,在禁带只能够产生的杂质能级。能提供电子载流子的杂质称为施主杂质,相应能级称为施主能级,位于禁带上方靠近导带底附近。 一、N型半导体在本征半导提硅(或锗)中掺入微量的5价元素,例如磷,则磷原子就取代了硅晶体中少量的硅原子,占据晶格上的某些位置。 磷原子最外层有5个价电子,其中4个价电子分别与邻近4个硅原子形成共价键结构,多余的1个价电子在共价键之外,只受到磷原子对它微弱的束缚,因此在室温下,即可获得挣脱束缚所需要的能量而成为自由电子,游离于晶格之间。失去电子的磷原子则成为不能移动的正离子。磷原子由于可以释放1个电子而被称为施主原子,又称施主杂质。 在本征半导体中每掺入1个磷原子就可产生1个自由电子,而本征激发产生的空穴的数目不变。这样,在掺入磷的半导体中,自由电子的数目就远远超过了空穴数目,成为多数载流子(简称多子),空穴则为少数载流子(简称少子)。显然,参与导电的主要是电子,故这种半导体称为电子型半导体,简称N型半导体。 二、P型半导体在本征半导体硅(或锗)中,若掺入微量的3价元素,如硼,这时硼原子就取代了晶体中的少量硅原子,占 据晶格上的某些位置。硼原子的3个价电子分别与其邻近的3个硅原子中的3个价电子组成完整的共价键,而与其相邻的另1个硅原子的共价键中则缺少1个电子,出现了1个空穴。这个空穴被附近硅原子中的价电子来填充后,使3价的硼
P杂质谱分析的 集团文件版本号:(M928-T898-M248-WU2669-I2896-DQ586-M1988)
格式模板首先列出产品的杂志谱列表,比如:****产品杂质情况分析表
1、详细的反应方程式,包括结构式,反应温度,所有试剂,助剂,溶 剂,催化剂等。 2、结合CTD资料的其他部分,对物料控制进行说明,包括起始物料、 其他原料、溶剂、辅料(活性炭,硅藻土,硅胶等。)。 3、起始物料说明。起始物料符合广泛、易得、质量稳定,适合保存运 输等的原则;还应对多个供应商提供的多批次物料进行质量研究,同供应商签署的质量协议以及供应商工艺变更告知义务等协议。比如头孢克洛,要对起始物料7-ACCA的工艺、杂质控制和质量情况 进行详细的说明。
4、说明制定起始物料的质量控制策略的依据,比如头孢克洛的起始物 料7-ACCA的关键杂质△异构体,结合工艺和实验数据,说明杂质 产生来源,分布,控制策略等。第2、3、4内容可以在CTD的其他部分,比如物料说明部分进行。但是本部分内容讨论的展开需要物料控制说明作为基本的理论依据。 5、关于ICH的杂质鉴定、报告和质控限度:主要参考ICHQ3A(R2) 到ICHQ3D的相关规定 杂质,降解杂质等。 第一部分:起始物料引入的杂质分析(比如头孢克洛的起始物料7-ACCA 引入的杂质) 1、无机杂质:说明引入情况和消除渠道;以及相关的控制方法和标准 以及依据。比如上述列表中的钯元素控制。 2、普通有机杂质。 3、对映异构体(根据品种的情况具体分析);考察不同的对应异构体 对最终产品质量的影响情况。 4、非对映异构体:比如ACCA的△异构体;还包括非对应异构体自身 的各种对映体。 第二部分:反应的每个步骤引入的杂质:需要结合实际反应监控(HPLC,LC-MS为主)过程对杂质消除过程以及对后续的影响进行实际说明。这个内容主要在工艺描述部分进行,本部分引用工艺描述内容。 1、步骤1引入的杂质。 2、步骤2引入的杂质。 3、步骤3引入的杂质。以及后续的步骤产生的杂质以及消除过程和简 单的控制描述…… 4、精制过程引入的杂质。 第三部分:结合小试中试数据汇总列表、方法适用性、实际检测结果等内容说明各个有机杂质的分布情况。
杂质谱分析模板的整理 格式模板 首先列出产品的杂志谱列表,比如: ****产品杂质情况分析表
反应过程的描述: 1、详细的反应方程式,包括结构式,反应温度,所有试剂,助剂,溶剂,催化剂 等。 2、结合CTD资料的其他部分,对物料控制进行说明,包括起始物料、其他原料、 溶剂、辅料(活性炭,硅藻土,硅胶等。)。 3、起始物料说明。起始物料符合广泛、易得、质量稳定,适合保存运输等的原 则;还应对多个供应商提供的多批次物料进行质量研究,同供应商签署的质量协议以及供应商工艺变更告知义务等协议。比如头孢克洛,要对起始物料7- ACCA的工艺、杂质控制和质量情况进行详细的说明。 4、说明制定起始物料的质量控制策略的依据,比如头孢克洛的起始物料7-ACCA 的关键杂质△异构体,结合工艺和实验数据,说明杂质产生来源,分布,控制策略等。第2、3、4内容可以在CTD的其他部分,比如物料说明部分进行。但是本部分内容讨论的展开需要物料控制说明作为基本的理论依据。 5、关于ICH的杂质鉴定、报告和质控限度:主要参考ICHQ3A(R2)到ICHQ3D的 相关规定 阈值(原料药) 2 克/天
结合上述反应过程对杂质谱进行分析,主要分起始物料引入杂质,反应杂质,降解杂质等。 第一部分:起始物料引入的杂质分析(比如头孢克洛的起始物料7-ACCA 引入的杂质) N S OH O O NO 2 O H N O C 22H 19N 3O 7S MW: 469.47 N O N O H S O OK C 16H 17KN 2O 4S MW: 372.48 Br NO 2 + N O N O H S O O NO 2 C H N O S MW: 469.51 C 7H 6BrNO 2MW: 216.03PAA 22 N O H N O H S O O NO 2 O C 23H 23N O S MW: 485.51 TMP toluene N S N H H O O O 23H 21N 3O 5S N S N H H OH O O 2 O C 22H 19N 3O 6S MW: 453.47 CH 2Cl 2/CH 3OH O 3, TMP CH 2Cl 2/TEBAC TsCl morpholine N S N H H N O O 2O O C 26H 26N 4O 6S MW: 522.57 1) Br 2-pyrindine CH 2Cl 2N S Cl O O NO 2 O 2N C 14H 12ClN 3O 5S MW: 406.00 HCl HCl 1) (PhO)3P/CH 2Cl 2 2) N,N-dimethyl aniline, PCl 5 i BuOH Na 2S 2O 4N S OH O Cl O H 2N H C 7H 7ClN 2O 3S MW: 234.66 1、 无机杂质:说明引入情况和消除渠道;以及相关的控制方法和标准以及依据。比如上述列表中的钯元素控制。 2、 普通有机杂质。
药物杂质研究方法详解 近年来,随着药物研究的不断深入以及杂质研究要求不断提高,杂质的分析技术以及研究方法正发生着重要的改变。在对杂质建立分析方法时,清晰的杂质研究过程是方法建立的基础,而且选择合适的分析技术也至关重要。 杂质的来源分析 药物中的杂质可能来源于药物生产以及销售等各个环节(图1)。根据ICH 指导原则可将药物杂质分为有机杂质、无机杂质、残留溶剂以及其他杂质。本文主要针对有机杂质进行探讨。 对药物杂质研究时引入“质量源于设计( Quality byDesign,QbD)”的理念,可在药物生产之前根据具体工艺的合成机制、起始物料及各中间体的基本结构,初步勾画出产品的杂质谱。 杂质来源分析是制定药物杂质控制策略的基础,尤其是在对毒性杂质来源分析时,应分析所有合成和生产工艺中的试剂、中间体、副产物,推测可能产生的潜在杂质以及分析实际存在的杂质。 在原料药合成结束后,药物的活性化合物虽然经过毒性分析已不含有“警示结构”(alerting structure),但是在生产过程中使用到含有警示结构的化合物则还需考虑其遗传毒性。 杂质的研究方法 在药物研发过程中,药物杂质的分析是关键。因此,在杂质研究中清晰的杂质结构研思路(如图2)以及合适的杂质分析技术可极大地缩短杂质研究时间,推动着药物研究的快速发展。
1、杂质前处理技术 杂质的前处理是伴随着药物活性成分前处理而存在的,然而药物中杂质的含量低且其结构与主成分差异较大,因此常规药物活性成分的前处理和检测方法(如初始流动相溶解后直接进行HPLC-UV 分析)并不一定适用于药物杂质,应针对不同的样品选择不同的前处理技术。 (1)检测灵敏度低的样品 对检测灵敏度低的样品通常使用衍生化的前处理方式,比如引入生色团产生紫外响应,或增加易离子化基团增加离子化效率等。 虽然常规衍生化方式能够满足日常检测的需求,但是为了实现对低浓度的基因杂质进行快速筛选和定量,可对传统的衍生化试剂进行改变以增加其专属性和灵敏度,也可使用气-固衍生化来弥补液-固衍生化的不足。 (2)低浓度的杂质 低浓度杂质前处理方法的选择根据其杂质类型所决定,如降解产物利用强制降解等方法提高降解物的浓度等,但是常规的降解方法往往会引入其他杂质,因而会干扰特殊杂质的杂质谱研究,为了得到单一的杂质研究机制,Ueya-ma 等提出了一种新型的固体药物氧化降解平台,该平台排除了常见氧化方式(例如H2O2 主导)引起的水解、溶剂解或热效应等,可用于氧化降解机制的特异性研究。 (3)易污染仪器的样品 不同仪器有不同的使用条件,因此对复杂样品进行前处理工作能够延长仪器的使用寿命,例如质谱检测器不能使用含有非挥发性盐的流动相,因此在建立液质联用条件时可利用二维液相色谱技术在第一维将各峰进行分离并将样品保留至样品环中,第二维液相使用质谱可接受的流动相以及脱盐柱来洗脱样品环中的样品从而实现了被分析物“脱盐”来保护质谱。 2、杂质分离技术
杂质谱的分析 在药品研发及药品评价的过程中,杂质研究是一项非常重要的内容。因为药物在临床使用过程中所发生的不良反应除了与药品本身的药理活性有关外,有时还与药品中所含有的杂质有很大的关系。众所周知,从事药品研发及药品评价所要遵循的一个基本原则就是要保证上市药品的安全性和有效性,由于药品质量的稳定可控是保证药品安全有效的前提和基础,而杂质研究又是药品质量研究的一项重要内容,所以杂质研究及杂质控制是药品质量保证的关键要素,是确保药品安全有效性的重要体现。 2005年SFDA颁布的《化学药物杂质研究技术指导原则》中明确说明任何影响药物纯度的物质统称为杂质。具体的解释就是指药物中所含有的没有治疗作用、可能影响药物的稳定性和疗效,甚至是对人体健康有害的物质。杂质的来源有工艺杂质和降解产物等,工艺杂质指的是药品在制备工艺过程中引入的杂质,它包括没有反应完全的反应物、反应过程中所生成的中间体及副产物、反应过程中所使用的试剂及催化剂等。降解产物指的是药品在生产和贮藏过程中发生化学变化而产生的杂质,如发生水解、氧化、开环等反应,降解产物主要与药物的结构特征密切相关。 由于杂质研究与药品的质量及安全有效性直接相关,为了提高药品的质量,保障公众的用药安全,因此,在药品研发过程中需规范地进行杂质研究,并将其控制在安全、合理的限度范围内。在杂质研究总体原则的指导下,其中杂质谱的分析应是杂质研究的重要内容之一。 一、杂质研究的总体原则 杂质研究的总体原则就是要结合在研产品具体的工艺以及产品的特点开展研究。首先,要结合具体工艺及产品特点来分析产品中可能产生什么样的杂质,通过杂质谱的分析对产品中杂质的来源及结构情况有较为全面的了解;然后,在杂质谱分析的基础上,有针对性地选择合适的分析方法,以确保杂质的有效检出及控制;最后,需综合药学、药理毒理及临床研究结果确定合理的杂质限度,从而保证药品的质量及安全性。 二、杂质谱的分析 前已提及,对于杂质谱的分析需结合具体的工艺及产品特点展开,下面简要
第2章 区熔提纯
吉林大学电子科学与工程学院 半导体材料
区熔技术的提出
区熔-区域熔炼(zone melting)
加热环移动方向 再凝固 熔化区 固体
加热环 区域熔炼(示意图)
?半导体工业对原料纯度的要求达到 8 个 “9” (99.999999)以上
一般化学提纯方法无法满足此要求。 ?区熔提纯-1952年蒲凡(W.G.Pfann)提出的一种物理提纯方法
?是制备极高纯度物质的重要方法,可以制备8个“9”以上的半
导体材料(如硅和锗)。
吉林大学电子科学与工程学院 半导体材料
区熔技术的提出
区熔提纯(zone refining)
(1917-1982)
Zone refining was developed by William Gardner Pfann in Bell Labs as a method to prepare high purity materials for manufacturing transistors.
吉林大学电子科学与工程学院 半导体材料
区熔技术的提出
1957 年,中科院物理所半导体 室 ( 现中科院半导体所 ) 获得了纯 度为7个“9”的高纯锗。采用金属 锗铸锭进行区域熔炼提纯工艺制 得。 1957 年下半年,中科院物理所 半导体室在林兰英的指导下,以 区熔提纯制备高纯锗为原料研制 出籽晶,拉制出完整的锗单晶。
中国半导体 材料之母— 林兰英
吉林大学电子科学与工程学院 半导体材料
发布日期20070628 栏目化药药物评价>>化药质量控制 标题杂质谱的分析 作者于红 部门 正文内容 审评四部审评七室于红 在药品研发及药品评价的过程中,杂质研究是一项非常重要的内容。因为药物在临床使用过程中所发生的不良反应除了与药品本身的药理活性有关 外,有时还与药品中所含有的杂质有很大的关系。众所周知,从事药品研发 及药品评价所要遵循的一个基本原则就是要保证上市药品的安全性和有效 性,由于药品质量的稳定可控是保证药品安全有效的前提和基础,而杂质研 究又是药品质量研究的一项重要内容,所以杂质研究及杂质控制是药品质量 保证的关键要素,是确保药品安全有效性的重要体现。 2005年SFDA颁布的《化学药物杂质研究技术指导原则》中明确说明任何影响药物纯度的物质统称为杂质。具体的解释就是指药物中所含有的没有 治疗作用、可能影响药物的稳定性和疗效,甚至是对人体健康有害的物质。 杂质的来源有工艺杂质和降解产物等,工艺杂质指的是药品在制备工艺过程
中引入的杂质,它包括没有反应完全的反应物、反应过程中所生成的中间体及副产物、反应过程中所使用的试剂及催化剂等。降解产物指的是药品在生产和贮藏过程中发生化学变化而产生的杂质,如发生水解、氧化、开环等反应,降解产物主要与药物的结构特征密切相关。 由于杂质研究与药品的质量及安全有效性直接相关,为了提高药品的质量,保障公众的用药安全,因此,在药品研发过程中需规范地进行杂质研究,并将其控制在安全、合理的限度范围内。在杂质研究总体原则的指导下,其中杂质谱的分析应是杂质研究的重要内容之一。 一、杂质研究的总体原则 杂质研究的总体原则就是要结合在研产品具体的工艺以及产品的特点开 展研究。首先,要结合具体工艺及产品特点来分析产品中可能产生什么样的杂质,通过杂质谱的分析对产品中杂质的来源及结构情况有较为全面的了解;然后,在杂质谱分析的基础上,有针对性地选择合适的分析方法,以确保杂质的有效检出及控制;最后,需综合药学、药理毒理及临床研究结果确定合理的杂质限度,从而保证药品的质量及安全性。 二、杂质谱的分析 前已提及,对于杂质谱的分析需结合具体的工艺及产品特点展开,下面简要介绍关于杂质谱分析的若干途径。 1.对于原料药,需依据所采用的具体合成工艺来分析在研产品中可能产生的杂质。 例如:抗心绞痛药物盐酸曲美他嗪质量标准中哌嗪的检查,曲美他嗪的合
关于仿制药杂质分析方法的几点注意事项 :1、在仿制药杂质谱的对比研究中,需关注该产品是否在ICH成员国药典收载,收载的检测方法与申报方法有无明显差异,是否进行了方法比较研究。如果申报方法与ICH成员国药典方法之间存在较大差异,应进行包括检测能力和样品测定结果的方法对比研究,在此基础上优选专属性好、灵敏度高,能够充分检出相关杂质的检测方法。 需要注意的是:在杂质一致性的研究求证中,分析手段不能等同于日常检测,分离技术(如HPLC法)应与质谱分析(或二极管阵列检测)相结合或使用分析标识物(如杂质对照品),以便从色谱行为、UV特征、分子量及分子碎片特征等信息共同把握其物质一致性。 2、如采用HPLC法的相对保留时间识别某特定未知杂质,需要进行充分的方法耐用性的验证,并在质量标准中规定色谱柱的品牌、规格、粒径、流动相流速等分析条件,以保证检测方法具有足够的重现性,仅仅按照药典标准格式规定色谱填料的类型是不够的。 3、关于杂质分析的定量方式,通常有以下几种: (1)杂质对照品法,即外标法。用于对已知杂质的控制,如采用该法,则应注意对该对照品进行定性和定量研究,需对含量进行准确标定,并提供相关研究信息。 (2)加校正因子的主成分自身对照法,即以主成分作对照的内标法,校正因子可在检测时测定,但需提供杂质对照品,也可在建立方法时将测得的校正因子载入质量标准,供以后常规检验使用,无需长期提供杂质对照品,但也仅适于已知杂质的控制。 (3)不加校正因子的主成分自身对照法,实质上也是以主成分作对照的内标法,但其前提是假定杂质与主成分的响应因子相同,适用于具有与主成分相同或类似发色团的杂质,在有关物质与主成分具有相似的分子结构的情况下,此法不致发生太大误差。 需要关注的是稳定性考察中采用自身对照法考察有关物质变化的相关问题,由于主药本身含量也会降低,因此以主药作为杂质计算的参考标准会影响到
杂质研究与控制是把控药品质量风险的重要内容之一,基于杂质谱分析的杂质控制是“质量源于设计”基本理念在杂质研究与控制中的具体实践,需要与CMC各项研究乃至药理毒理及临床安全性研究等环节关联思考、综合考虑,而不仅仅拘泥于提供准确的分析数据。本文针对当前CTD申报资料中杂质研究方面存在的问题与不足,结合CTD过程控制和终点控制相结合、研究和验证相结合、全面系统的药品质量控制理念,探讨仿制药杂质研究与控制的基本逻辑思路,提出CTD申报资料中杂质研究与控制方面几个需要关注的问题。 关键词:杂质研究与控制杂质谱CTD格式 杂质研究与控制是一项系统工程,需要以杂质谱分析为主线,安全性为核心,按照风险控制的策略,将杂质研究与CMC各项研究,乃至药理毒理及临床安全性研究等环节关联思考、综合考虑,而不仅仅拘泥于提供准确分析数据的传统思维,不是一项孤立的分析工作。CTD(Common Technical Document)申报格式体现了过程控制和终点控制相结合、研究和验证相结合、全面系统的药品质量控制理念,更加符合杂质研究与控制的基本规律和逻辑思路。自2011年4月起,药审中心陆续发布了多项有关CTD格式及技术审评的相关要求及电子刊物,对于国内研发单位正确理解CTD格式内含的基本精神起到了一定的促进作用,但就目前阶段的申报情况看,有些申报资料在杂质研究方面仍存在一些不足,仅仅是形式上的CTD 格式,尚未实质性贯彻CTD的基本逻辑思路。以下是针对目前CTD申报资料中杂质研究相关问题的一些考虑。 1、CTD格式中杂质控制的考虑要体现在CMC的各个环节,而不是仅仅局限在“质量控制”模块。如制剂的原辅料控制中,原辅料的选择与控制要考虑以符合制剂质量要求(杂质等)为核心,必要时进行精制处理并制定内控标准;关键工艺步骤及参数的确立、工艺开发过程等要考虑以杂质是否得到有效控制为重点关注之一;制剂相关特性中要体现与原研产品杂质谱等的对比情况;包材、贮藏条件以及有效期的确立等也要以杂质是否处于安全合理的可控范围内为核心等等。实际上这正是源头控制、过程控制与终点控制相结合的杂质控制理念的体现,在研发工作及申报资料的整理中都需要针对性的贯彻实施。 问题与案例:有些申报资料在某种程度上未能充分体现杂质研究的整体性,对杂质控制措施仅强调了终点控制措施,尚未充分体现源头控制与过程控制的基本思路,具体表现在如下方面: (1)制剂杂质控制受制于原料药质控水平的约束,以目前国内批准的原料药杂质水平现状为由,未能根据该品当前杂质控制的水平与趋势,对原料药提出较为严格的针对性的杂质控制要求,并进行质量内控,因而难以确保制剂杂质控制水平与目前国际水平相适应; (2)在论述说明制剂相关特性时,未提供与原研产品杂质谱的对比分析情况; (3)关键工艺步骤及参数的确立、工艺开发过程相关内容中未详细说明杂质谱的变化情况,缺失关键质量数据的支持。 2、CTD格式的特点之一是研究内容模块化呈现,但需关注杂质分析与控制的系统性与整体性,不能割裂各项内容的必然联系和有机统一。比如对原料药而言,杂质分析与控制的相关内容会分布在分析方法(3.2.S.4.2)、方法学验证(3.2.S.4.3)、杂质对比研究与杂质谱分析(3.2.S.4.5)、杂质情况分析总结(3.2.S.3.2)、样品检测与数据积累(3.2.S.4.4)、控制限度(2.3.S.4.1)等各模块中,但杂质研究又是一项系统工程,具有统一的整体性,因此,不要因为申报资料格式的模块化而人为割裂各项研究内容的相互联系,甚至遗漏相关研究内容,要高度关注杂质分析与控制的系统性与整体性,将杂质研究与控制的全部内容和信息体现在相应模块中。如详细的杂质研究报告可以体现在3.2.S.4.5中;3.2.S.3.2要报告杂质研究的结果;杂质分析方法的筛选、研究与验证内容要在3.2.S.4.3中体现;对仿制药而言,杂质限度确定的论证与依据需要在与原研产品进行全面的杂质谱对比研究基础上进行论证说明,因此,与原研产品的对比研究及结论要在3.2.S.4.5中体现。
格式模板首先列出产品的杂志谱列表,比如:****产品杂质情况分析表
1、详细的反应方程式,包括结构式,反应温度,所有试剂,助剂,溶 剂,催化剂等。 2、结合CTD资料的其他部分,对物料控制进行说明,包括起始物料、 其他原料、溶剂、辅料(活性炭,硅藻土,硅胶等。)。 3、起始物料说明。起始物料符合广泛、易得、质量稳定,适合保存运 输等的原则;还应对多个供应商提供的多批次物料进行质量研究,同供应商签署的质量协议以及供应商工艺变更告知义务等协议。比如头孢克洛,要对起始物料7-ACCA的工艺、杂质控制和质量情况 进行详细的说明。 4、说明制定起始物料的质量控制策略的依据,比如头孢克洛的起始物 料7-ACCA的关键杂质△异构体,结合工艺和实验数据,说明杂质 产生来源,分布,控制策略等。第2、3、4内容可以在CTD的其他部分,比如物料说明部分进行。但是本部分内容讨论的展开需要物料控制说明作为基本的理论依据。 5、关于ICH的杂质鉴定、报告和质控限度:主要参考ICHQ3A(R2) 到ICHQ3D的相关规定 杂质,降解杂质等。
第一部分:起始物料引入的杂质分析(比如头孢克洛的起始物料7-ACCA 引入的杂质) 1、无机杂质:说明引入情况和消除渠道;以及相关的控制方法和标准 以及依据。比如上述列表中的钯元素控制。 2、普通有机杂质。 3、对映异构体(根据品种的情况具体分析);考察不同的对应异构体 对最终产品质量的影响情况。 4、非对映异构体:比如ACCA的△异构体;还包括非对应异构体自身 的各种对映体。 第二部分:反应的每个步骤引入的杂质:需要结合实际反应监控(HPLC,LC-MS为主)过程对杂质消除过程以及对后续的影响进行实际说明。这个内容主要在工艺描述部分进行,本部分引用工艺描述内容。 1、步骤1引入的杂质。 2、步骤2引入的杂质。 3、步骤3引入的杂质。以及后续的步骤产生的杂质以及消除过程和简 单的控制描述…… 4、精制过程引入的杂质。 第三部分:结合小试中试数据汇总列表、方法适用性、实际检测结果等 内容说明各个有机杂质的分布情况。 第四部分:潜在杂质在成品中的检测结果
CTD申报资料中杂质研究的几个问题 张哲峰成海平宁黎丽田洁 化药药学二部 摘要:杂质研究与控制是把控药品质量风险的重要内容之一,基于杂质谱分析的杂质控制是“质量源于设计”基本理念在杂质研究与控制中的具体实践,需要与CMC各项研究乃至药理毒理及临床安全性研究等环节关联思考、综合考虑,而不仅仅拘泥于提供准确的分析数据。本文针对当前CTD申报资料中杂质研究方面存在的问题与不足,结合CTD 过程控制和终点控制相结合、研究和验证相结合、全面系统的药品质量控制理念,探讨仿制药杂质研究与控制的基本逻辑思路,提出CTD申报资料中杂质研究与控制方面几个需要关注的问题。 关键词:杂质研究与控制杂质谱CTD格式 杂质研究与控制是一项系统工程,需要以杂质谱分析为主线,安全性为核心,按照风险控制的策略,将杂质研究与CMC各项研究,乃至药理毒理及临床安全性研究等环节关联思考、综合考虑,而不仅仅拘泥于提供准确分析数据的传统思维,不是一项孤立的分析工作。CTD(Common Technical Document)申报格式体现了过程控制和终点控制相结合、研究和验证相结合、全面系统的药品质量控制理念,更加符合杂质研究与控制的基本规律和逻辑思路。自2011年4月起,药审中心陆续发布了多项有关CTD格式及技术审评的相关要求及电子刊物,对于国内研发单位正确理解CTD格式内含的基本精神起到了一定的促进作用,但就目前阶段的申报情况看,有些申报资料在杂质研究方面仍存在一些不足,仅仅是形式上的CTD格式,尚未实质性贯彻CTD的基本逻辑思路。以下是针对目前CTD申报资料中杂质研究相关问题的一些考虑。 1、CTD格式中杂质控制的考虑要体现在CMC的各个环节,而不是仅仅局限在“质量控制”模块。如制剂的原辅料控制中,原辅料的选择与控制要考虑以符合制剂质量要求(杂质等)为核心,必要时进行精制处理并制定内控标准;关键工艺步骤及参数的确立、工艺开发过程等要考虑以杂质是否得到有效控制为重点关注之一;制剂相关特性中要体现与原研产品杂质谱等的对比情况;包材、贮藏条件以及有效期的确立等也要以杂质是否处于安全合理的可控范围内为核心等等。实际上这正是源头控制、过程控制与终点控制相结合的杂质控制理念的体现,在研发工作及申报资料的整理中都需要针对性的贯彻实施。
Eurasian Journal of Analytical Chemistry Volume 2, Number 1, 2007 Impurity profile: Significance in Active Pharmaceutical Ingredient Sanjay B. Bari, Bharati R. Kadam, Yogini S. Jaiswal, Atul A. Shirkhedkar Department of Pharmaceutical Chemistry, R. C. Patel College of Pharmacy, Shirpur, Dist: Dhule - 425 405 (MS), India Abstract Various regulatory authorities like ICH, USFDA, Canadian Drug and Health Agency are emphasizing on the purity requirements and the identification of impurities in Active Pharmaceutical Ingredient’s (API’s). Qualification of the impurities is the process of acquiring and evaluating data that establishes biological safety of an individual impurity; thus, revealing the need and scope of impurity profiling of drugs in pharmaceutical research. Identification of impurities is done by variety of Chromatographic and Spectroscopic techniques, either alone or in combination with other techniques. There are different methods for detecting and characterizing impurities with TLC, HPLC, HPTLC, AAS etc. Conventional Liquid Chromatography, particularly, HPLC has been exploited widely in field of impurity profiling; the wide range of detectors, and stationary phases along with its sensitivity and cost-effective separation have attributed to its varied applications. Among the various Planar Chromatographic Methods; TLC is the most commonly used separation technique, for isolation of impurities; due to its ease of operation and low cost compared to HPLC. An advancement of thin layer chromatography HPTLC, is a well-known technique for the impurity isolation. Headspace GC is one of the most preferred techniques for identification of residual solvents. The advent of hyphenated techniques has revolutionized impurity profiling, by not only separation but structural identification of impurities as well. Among all hyphenated techniques, the most exploited techniques, for impurity profiling of drugs are LC-MS-MS, LC-NMR, LC-NMR-MS, GC-MS, and LC-MS. Keywords:Impurity, Analytical method development, Spectrophotometry, Chromatography Copyright ? 2007 by MOMENT ISSN: 1306-3057
基于杂质谱的仿制药质量一致性评价流程研究 张明媛1,2, 张军东3,陆峰1* 1第二军医大学药学院药物分析学教研室,上海 200433; 2 福建医科大学附属南平第一医院药学部,福建 南平 353000; 3.上海信谊药厂有限公司药物研究所,上海201206 摘要 目的利用杂质谱理念,参考化学计量学中评价中药指纹图谱相似度的方法,研究一套用于仿制药质量一致性评价的流程与方法。方法采用相关系数法、夹角余弦法、主成分分析法(PCA)、系统聚类分层法(HCA)、加权相关系数法等化学计量学方法对7个厂家16个批次的硝苯地平缓释片样品中杂质谱信息进行计算分析。结果经过色谱方法的选择、样品检测与数据采集、批次内相似度评价、仿制药相似度评价、加权后仿制药评价等5个步骤,逐步筛选出了仿制水平较高的仿制厂家。结论拟定的评价流程与方法可逐步筛选出仿制相对最优的厂家。 关键词杂质谱,一致性评价,化学计量学,硝苯地平缓释片 Evaluation procedure for quality consistency of generic drugs based on the impurity profile Zhang Mingyuan1,2, Zhang Jundong3,Lu Feng1* 1 School of Pharmacy, Second Military Medical University, Shanghai 200433; 2 Department of Pharmacy, the First Hospital of Naping, Fujian Medical University, Nanping 353000; 3 Shanghai Sine Pharmaceutical Laboratories Co., Ltd, Shanghai 201206 Abstract [第一作者] 张明媛,研究生,从事药物分析研究。 [通讯作者] * 陆峰,教授,E‐mail:fenglufeng@https://www.doczj.com/doc/088522480.html,,从事药物质量信息学研究。 [基金课题] 上海市科委产学研医合作项目(12DZ1930504)
..P..杂质谱分析模板的整理
————————————————————————————————作者:————————————————————————————————日期: 2
3.2.P.5.5杂质谱分析模板的整理 格式模板 首先列出产品的杂志谱列表,比如: ****产品杂质情况分析表 分类名称化学结 构式 (CHEM DRAW ) 控制 限度 杂质 来源 备注1 备 注 2 药物成品目标 化合 物 ≥ 99.5 % 目标 产物 有机杂质杂质 A ≤ 0.1 % 原料 引入 是否基 毒 杂质 B 工艺 杂质 (副 产物) 杂质 C 手性 异构 体 杂质 D 氧化 杂质 杂质热降
E解杂 质 有机潜在杂质IMP- 1 起始 物料 IMP- 2 中间 体 IMP- 3 USP提 到的 杂质 USP不 同路线 杂质?IMP- 4 IMP- 5 溶剂残留溶剂 名称 溶剂来 源 限度溶剂种类ICH 分类 种类限度二氯 甲烷 步骤 1,2 600p pm 2 600pp m 三乙 胺 步骤2 甲醇步骤 1,3 四氢 呋喃 步骤 2,3 2 3000p pm 乙酸 乙酯 精制步 骤 3 5000p pm
无机杂质 1、说明用到的无机原料和试剂,比如干燥用的硫酸镁,路易斯酸催化用的四氯化钛,加氢用的钯碳铂碳等。 2、说明这些无机杂质做或者不做常规检查的依据:物料毒性依据,去除途径。 3、对特殊重金属制定控制策略和检测策略:比如使用氧化汞造成的汞残留。 4、说明采用重金属检查法控制一般常规重金属。 其他试剂类别名称去除策略控制策 略催化 剂 二甲基 吡啶 水洗去除 氧化 剂 DMP 淬灭,构型转 化后水洗去 除 脱甲 基试 剂 三氟化 硼乙醚 精馏脱除 反应过程的描述: 1、详细的反应方程式,包括结构式,反应温度,所有试剂,助剂,溶剂, 催化剂等。
半导体材料中的杂质 半导体材料中的杂质(impurity in semiconductor material) 半导体晶格中存在的与其基体不同的其他化学元素原子。杂质的存在使严格按周期性排列的原子所产生的周期性势场受到破坏,这对半导体材料的性质产生决定性的影响。杂质元素在半导体材料中的行为取决于它在半导体材料中的状态,同一种杂质处于间隙态或代位态,其性质也会不同。电活性杂质在半导体材料的禁带中占有一个或几个位置作为杂质能级。按照杂质在半导体材料中的行为可分为施主杂质、受主杂质和电中性杂质。按照杂质电离能的大小可分为浅能级杂质和深能级杂质。浅能级杂质对半导体材料导电性质影响大,而深能级杂质对少数载流子的复合影响更显著。氧、氮、碳在半导体材料中的行为比较复杂,所起的作用与金属杂质不同,以硅和砷化镓为例叙述杂质的行为。 硅中的杂质主要有金属杂质和氧、碳。 金属杂质分为浅能级杂质和深能级杂质。Ⅲ族元素硼、铝、镓、铟和V族元素磷、砷、锑,它们在硅中的能级,位于导带底或价带顶的附近,电离能级小,极易离化,因此称为浅能级杂质。它们是硅中主要的电活性杂质。Ⅲ族元素起受主作用,V族元素起施主作用,常用作硅的掺杂剂。这两种性质相反的杂质,在硅中首先相互补偿,补偿后的净杂质量提供多数载流子浓度。 其他金属杂质,尤其是过渡元素(重金属),如铜、银、金、铁、钴、镍、铬、锰、钼等,在硅中的能级位置一般远离导带底或价带顶,因此称为深能级杂质。它们在硅中扩散快,并起复合中心作用,严重影响少子寿命。它们本身可产生缺陷,并易与缺陷络合,恶化材料和器件的性能。除特殊用途外,重金属元素在硅中都是有害杂质。 镍、钴、铜、铁、锰、铬和银所造成的“雾”缺陷,按次序降低。铜和镍具有高的扩散系数和高的间隙溶解度,在“雾”缺陷形成中,它们会溶解、扩散并沉淀在硅中,而铁、铬、钴则在热处理中将留在硅的表面。 锂、钠、钾、镁、钙等碱金属和碱土金属离子,在电场作用下易在p—n结中淀积,使结退化,导致击穿蠕变,MOs阈电压漂移,沟道漏电,甚至反型。 锗是替位式杂质,电中性,能有效地消除氧化片滑移,增加硅的机械强度。 氧氧在硅中是间隙型杂质,分散在硅中的氧原半bar1子呈电中性。是硅中含量最多又极为重要的杂质。硅中氧主要来源于熔融硅与石英坩埚的反应。因此直拉硅单晶比区熔硅单晶的氧含量要高得多。前者一般在1~1.7×1018cm-3,后者则可<5×1015cm-3。氧在硅中的极限溶解度约为2×1018cm-3。 间隙氧原子与最邻近的硅原子键合成为si-0-Si键,键角约162°,称为准线性分子。si—O键振动在室温下有三个对应的吸收峰,分别为1205cm-1、1106cm-1和515cm-1。 氧在硅中的行为及其状态与热处理过程有密切关系。表面氧沉淀会造成漏电,甚至使器件失效。体内低密度的氧沉淀有吸除金属杂质的作用。但高密度的氧沉淀则能产生位错,使硅片翘曲。氧施主的存在使硅片电阻率变化,器件的阈值电压漂移。晶体内的氧可起钉扎位错的作用,使硅片机械强度增加,因此制作集成电路多用直拉硅单晶。