
过渡金属催化的C-S的合成
摘要:过渡金属催化的C-S交叉偶联反应在有机合成方法学的研究中一直起着不可或缺的作用。这些经过交叉偶联反应所形成的一系列含碳-硫键结构的化合物,在染料、医药、农药、化工以及聚合物的制备中都有广泛的应用。不同过渡金属催化合成硫化物成为当前研究的一个热点。本文简单综述了不同过渡金属催化反应合成含C-S的化合物。
关键词:过渡金属;硫醇;催化;偶联反应;碳一硫键构建
Transition Metal Catalyzed Synthesis of C-S bond Abstract: transition metal catalyzed C-S cross coupling reaction plays an important role in organic synthetic methodology. The compounds synthesized through cross coupling reaction have very good biological activity and wide application in colorant, pharmaceutical, pesticide, and chemical industry , and the preparation of polymer.So transition metal catalytic synthesis of C-S bond becomes a hot issue. In this paper,transition metal-catalyzed reaction was briefly summarized.
Key words: transition-metal; thiols; catalyze; coupling reaction;C-S bond formation
许多含硫化合物具有生物活性,包括磺酰胺类抗生素和哮喘药物顺尔宁抗生素等[1-2]。多种含硫化合物的各类构建方法需要深入地研究,碳一硫键的构建和以及进一步的官能团化已经引起科学界的相当关注。硫化物,硫醇及它们的氧化衍生物在有机合成方面有广泛的应用[3-4]。与碳一氧键和碳一氮键的构建方法相比,有机金属试剂催化的碳一硫键的构建方依然是不足的。尽管人们始终认为硫能够毒化金属催化剂,但是金属催化的碳一硫键的构建方法研究有逐渐增强的趋势。
过渡金属催化通过偶联反应构建碳一硫键的各种方法有很多报道,我们接来将介绍不同的过渡金属催化合成碳硫键的这类反应最近进展。
1铜催化
乌尔曼偶联反应一般是亲核试剂和芳基卤代物在化学计量的铜催化剂下,高温反应合成二芳基化合物。此方面取得的进展基本上是扩展这种方法应用于芳基碳一杂键的形成,即乌尔曼缩合反应[5]。
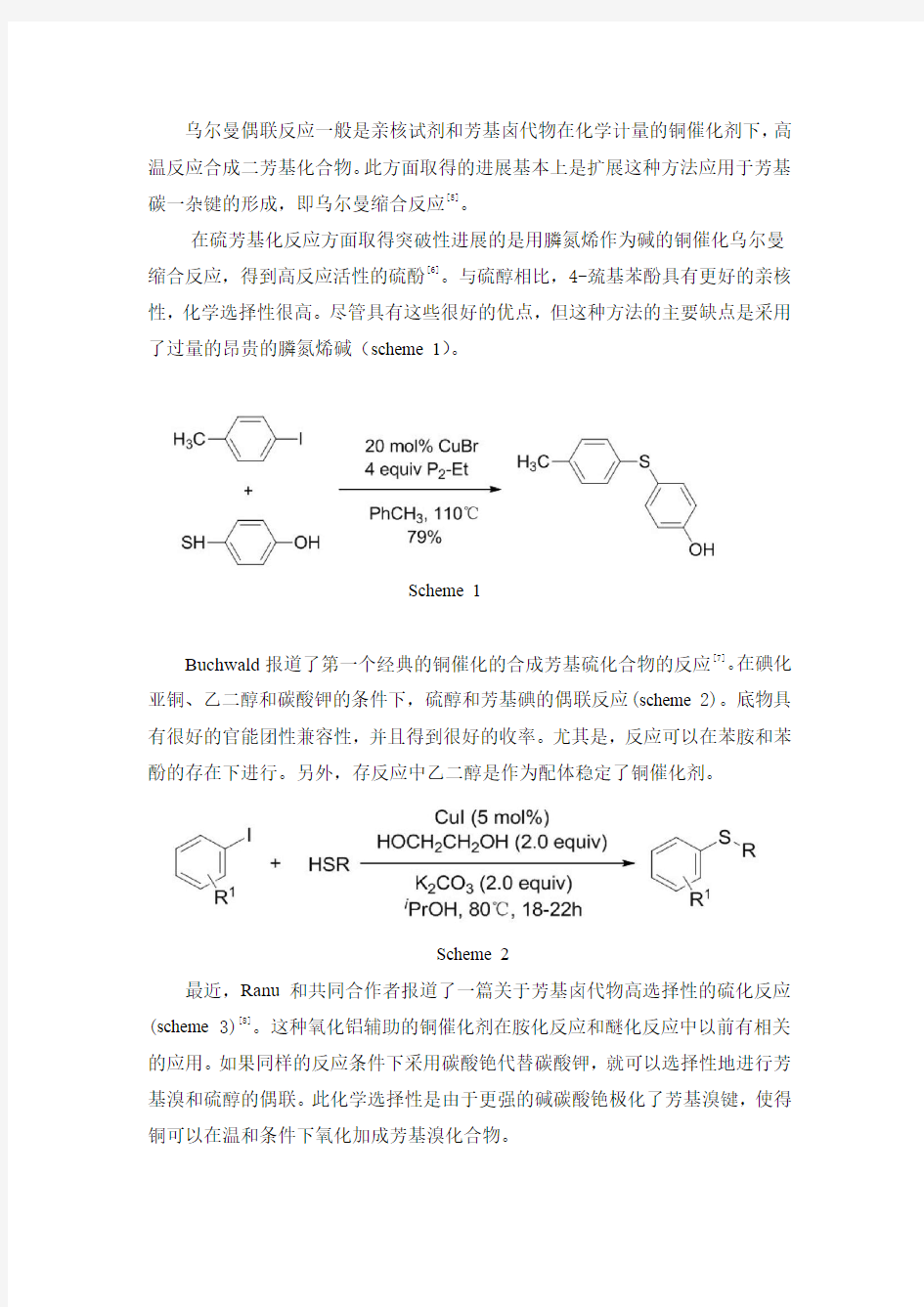
在硫芳基化反应方面取得突破性进展的是用膦氮烯作为碱的铜催化乌尔曼缩合反应,得到高反应活性的硫酚[6]。与硫醇相比,4-巯基苯酚具有更好的亲核性,化学选择性很高。尽管具有这些很好的优点,但这种方法的主要缺点是采用了过量的昂贵的膦氮烯碱(scheme 1)。
Scheme 1
Buchwald报道了第一个经典的铜催化的合成芳基硫化合物的反应[7]。在碘化亚铜、乙二醇和碳酸钾的条件下,硫醇和芳基碘的偶联反应(scheme 2)。底物具有很好的官能团性兼容性,并且得到很好的收率。尤其是,反应可以在苯胺和苯酚的存在下进行。另外,存反应中乙二醇是作为配体稳定了铜催化剂。
Scheme 2
最近,Ranu和共同合作者报道了一篇关于芳基卤代物高选择性的硫化反应(scheme 3)[8]。这种氧化铝辅助的铜催化剂在胺化反应和醚化反应中以前有相关的应用。如果同样的反应条件下采用碳酸铯代替碳酸钾,就可以选择性地进行芳基溴和硫醇的偶联。此化学选择性是由于更强的碱碳酸铯极化了芳基溴键,使得铜可以在温和条件下氧化加成芳基溴化合物。
Scheme 3
Wu等人报道了邻碘苯胺和异硫氰酸脂在碘化亚铜的催化下通过分子c-s 键的串联反应合成2-氨基苯并噻唑(scheme 4),反应条件比较温和,只有50 摄氏度,操作比较简单,官能团的兼容性良好。
Scheme 4
Lee等人报道了乙烯基卤化物和硫醇在氧化亚铜的催化下生成乙烯基硫的反应(scheme 5),芳基碘化物和硫醇只需要0.5 mol%的氧化亚铜而且不需要配体的参与就能进行偶联反应。在5 mol%氧化亚铜和10 mol%的1,10-啡罗琳的参与下,乙烯基溴化物和硫醇能够进行偶联反应,且产率较高。
Scheme 5
2镍催化
Migita报道了钯催化的硫醇的芳基化反应之后,紧接着Cris tau和合作者报道了一篇镍催化的合成二芳基硫化物的反应(scheme 6)[9]。采用二价镍和二配位的膦配体络合物作为催化剂,反应所需的催化剂用量很少。在此反应体系下需要较高温度和较长的反应时问,不过反应产率较高。
Scheme 6
最近,强配位能力的NHC-配体被研究发现可以很好地参与到镍催化的这类反应中。Zhang Y .G.等人就报道了关于这类的硫醇和芳基碘化物的偶联反应(scheme 7)[10]。
Scheme 7
镍催化剂同样被研究发现可以应用于插入双硫键来构建碳-硫键化合物方面。Taniguchi报道了在镍催化剂条件下二硫化物的芳基化反应(scheme 8)[11]。很多取代的碘化物和0.5个当量的二硫化物都能反应转化成相应的芳基硫化物。
Scheme 8
Taka gi,k.等报道了在碱性的碳酸钾和原位生成镍(0)催化剂dppf [dpp= 1,1'-双(二苯膦基)二茂铁]两者存在下,苯硫酚与等当量的碘苯反应,在25℃或在60℃溴苯中可以转化为有很好的得率的苯基硫化物(scheme9)。例如,苯硫酚与碘苯在25℃反应10小时,以97%的产率得到苯基硫化物。不对称硫化物也能由同一反应过程来制备。
Scheme 9
Taka gi等报道了在镍(0)催化剂催化下,芳基碘与硫脲的亲核取代得到芳基异硫脲碘化物。随后S-芳基异硫脲碘化物可以发生碱性水解得到相应的芳香硫醇,有几乎定量收率。用此方法可以很容易地由1,2一胺基碘苯和硫醇来制备2-苯硫脲胺碘。
由镍(II)催化的烯基卤化物与钠的苄基硫醇盐,经过类似的反应过程,可以用来制备二烯基硫化物和烯酮缩硫醛。在非质子溶剂中,如1,2-二甲氧乙烷和
甲苯,都获得了期望的烯基硫化物(scheme 10),并都有较好的收率。
Scheme10
对于二取代卤化物,无论是1,2-二溴乙烯或1,1-二滨乙烯化合物,都可以采用类似的反应条件(Scheme 11)。对于1,1-二溴乙烯,由镍(II)催化烯基的二取代溴原子能选择性的被硫醇负离子取代,以78%良好的收率得到相应的烯酮缩硫醛。
Scheme 11
3钯催化
钯催化偶联反应在碳碳键和碳卤键的构建方面有很广泛的应用。然而,在碳一氧键和碳一氮键的构建方面还没有广泛的研究时,钯催化的构建碳一硫键方面已经取得了成功。据悉Migita在1978年报道了首例钯催化的芳基卤代物的硫化反应[13,14]。这种方法能得到很好产率的二芳基硫化物,但是反应局限于芳基溴并且需要较高的反应温度和较长的反应时间(scheme 12)。Migita报道之后,型的钯催化方法是采用含有二配位的膦配体[15-17]。
Scheme 12
较先前的方法中有很重要进步的是Buchwald在2004年报道的芳基氯、溴和硫醇在醋酸钯催化下的高效偶联反应(scheme 13)[16]。在这篇报道中测试了许多的单配位和双配位的麟配体。采用富电了的芳基氯反应合成二芳基硫化物需要更弱的碱、较高反应温度和长的反应时间。值得注意是所有的单配位膦配在此体系下都不能反应。
Scheme 13
另一个非常成功的钯催化碳硫键构建方面的例了是2006年Hart wig运和共同合作者报道的芳基氯和硫醇的反应(scheme14)[18,19]。采用很好的Josiphos配体在钯催化体系中可以产生稳定性很高和催化活性强的催化剂。同时官能团的兼容性非常好,反应体系中存在的酚羟基、羰基、苯胺和酰胺都不需要用保护基团保护。
Scheme 14
Doi等人用硫代苯甲酞胺在把的催化下通过C-H键的活化和C-S键的形成合成了2一取代的苯并曝哩(scheme 15),反应体系用了10 mol%的氯化把和50 % mol 的碘化亚铜,生成了含有多种取代基的苯并曝哩,并且有良好的产率.
Scheme 15
Alper等人报道了把催化的分子内C-S键偶联反应及淡基化反应一锅法一步合成2-基苯并曝吩类化合物的反应(scheme 16),该反应具有很好的选择性和官能团兼容性,亲核试剂和硫酚类化合物都能够顺利地参与反应,并且得到相应的目标产物。
Scheme 16
4钴催化
虽然几种钯、镍和铜类催化剂已被证明在这种偶合过程中是非常有效的,但是通常反应温度高,催化剂的用量大或需要特别设计的膦配体,因此有必要寻找一个更好的催化剂。最近已证明钴的配合物能很好地催化碳一碳键的形成,因而将该试剂使用于碳一硫键的形成引起了科学家的兴趣。
钴催化的C-S偶联报道较少。Cheng等报道了利用CoI2/dppe体系在C-S偶联中的应用(scheme 17)。他们通过向反应体系中添加锌粉,将Co(II)还原为低价态的Co(I),随后低价态的钴发生氧化加成、配体交换和还原消除得到产物硫醚。这个催化体系对溴苯和含杂芳环的底物也有效,而且适用于脂肪族硫醇。这个体系具有良好的选择性,游离的羟基和氨基都不会参与反应;由于使用吡啶作碱,反应条件相对温和,能够耐受底物中多种宫能团的存在。
Scheme 17
Wong Y -C等在2006年就报道了一篇关于钴催化的芳基碘化物和硫醇的偶联反应(scheme 18[21]),反应中催化剂用量较少,并且反应有很广的底物范围,烷烃的硫醇类都可以很好地反应。
Scheme 18
5铁催化
Cornea等最近报道了用费用低廉、环境友好的铁盐,在配体的协助下实现了芳基硫醇的芳基化反应(scheme 19)。这个方法避免了使用昂贵的或对空气敏感的配体,并在大多数情况下,可以高产率的得到芳基硫化物。在目前阶段,成功的事例只是限于使用芳基碘化物作为亲电子试剂,但是,铁催化的芳基硫醇的芳
基化过程因其操作简单、环境和经济的优势而具用潜在的工业意义[22]。
Scheme 19
Bolm报道了三氯化铁催化的硫芳基化反应(scheme20)[23]。各类的硫醇,包括官能团化的硫醇都很好地和芳基碘化物反应。但是烷基硫醇类在此条件下不能发生反应。
Scheme 20
6镧催化
Shibasaki和研究组报道了镧三(联萘氧化物)催化硫醇和α,β-不饱和羰基化合物的不对称Michael加成反应[LSB=LaNa3三(联萘氧化物)[24](scheme 21)。
Scheme 21
7钌催化
钌催化的巯基与烯丙基碳酸酯的S-烯丙基化,可用于一般烯丙基硫化物的合成[25](scheme22)。与此类似的把催化反应,脂肪硫醇以及芳香和杂环硫醇都
能够顺利的发生烯丙基化反应,反应条件温和,得到的烯丙基烷基和烯丙基芳基硫化物都有很高的收率[26]。
Scheme 22
双核钌配合物Cp*Ru(μ-SR)2-Ru Cp*[Cp*=五乙基环戊二烯;R=Et, i-Pr, t-Bu]及相关的Cp*Ru(η1-C6F5)(μ-S)(μ-SC6F5)Ru Cp*作为硫醇和极化的炔加成的活性催化剂,如丙炔酸甲酯和炔二酸甲酯,在室温下就可完成加成反应。许多具有催化活性的钌配合物可以被分离,并且可循环使用和保持催化活性。
8铑催化
在硫醇和炔烃的区域和立体选择性加成中,RhCI-(PPh3)3也显示了良好的催化活性,能够生成专一的反马氏加成的产物。
Cao C.等发现,Tp*Rh(PPh3)2图示能催化芳基或烷基硫醇和一系列炔烃的硫氢化加成反应。与烷基硫醇进行反应有良好的区域选择性,容易制备有支链基取代的乙烯基硫化物.芳基硫醇和炔烃的硫氢化反应,分离产率有83-90% ,但选择性较低,在6:1到1.4:1之间,得到的是支链取代和直链的乙烯基硫化物混合的产物[27]。
9锰催化
硫醇和炔烃的加成是其中一个最简单的方法,可以得到乙烯基硫化物,这是重要的有机合成中间体。锰(III)的醋酸盐也可以催化1,l一二芳基乙烷与α-
巯基苯乙酮的反应(scheme 23)。乙烯和α-含巯基的苯乙酮混合后,在醋酸中用醋酸锰( III)催化发生环加成反应,以中等的产率得到环加成产物,同时还得到取代的产物。乙硫醇或苯硫酚和炔烃的加成,由锰(m)氧化引发容易形成自由基反应,生成更稳定的(E)-乙烯基硫化物,而且得到的几乎是定量的产率[28]。
Scheme 23
10小结
前面简单综述了各种过渡金属催化偶联方法构建碳一硫键化合物,经过人们的努力,碳一硫键的合成方法已经越来越成熟,操作条件越来越简单,这为碳一硫键的工业化应用特别是药物合成方面打下了很好的基础。但是还有很多方面需要研究和改进,如报道的很多是芳基卤代物和硫醇类化合物的偶联反应来构建碳一硫化合物,虽然有其他芳基供体如芳硼酸等底物进行这类反应的报道,但是相比较芳基卤代物在此方面的应用,芳硼酸等在此类反应的应用研究不是很充分,在反应中发现,硫醇类物质在反应中容易发生氧化等副反应,也不利于这类反应。因此在这类反应方面,人们以后的研究主要可能是进一步的优化和改进,比如采用便宜的金属、其它更好的芳基供体和优化反应的条件等。
参考文献
[1] Perry C.W.,Bader G.J.,Liebman A.A. Selective lithiation/carbonation Of polyhalobenzenes: animproved synthesis of furosem ide-7-14C. [J]. J. Org. Chem. 1978,43:4391-4391.
[2]Tsai W.-J.,Shiao Y,-J.,Lin S.-J,et al.selective COX-2 inhibitors. Part 1: Synthesis and biological evaluation of phenylazobenzene sulfonam ides [J]. Bioorg. Med.
Chem.Lett.2006,16:4440-4443.
[3]Trost B.M.,Bridges A. J. New. Synthetic reagents. 2-M ethoxy -3-phenylthiobuta-1,3-diene. A novel annelating agent [J].J Am Chem So 1976,98:5017-5019.
[4]Chou S.S.P.,Wey S.S.J. Intramolecular Diels-Alder reaction of sulfur-substituted dienes via 3-sulfolenes [J]. J Org Chem 1990,55:1207-1274.
[5] Prim D.,Campagne J. M., Joseph D, et al. Palladium-catalysed reactions of aryl halides with soft, non-organometallic nucleophiles[J]. Tetrahedron 2002, 58: 2041-2075.
[6] Palomo C., Oiarbide M., Lopez R. Phosphazene bases for the Preparation of biaryl thioethers from aryl iodides and arenethiols [J]. Tetrahedron Lett. 2000, 41: 1283-1286 .
[7] Kwong,F.Y.,Buchwald,S.L. A general, efficient, and inexpensive catalyst system for the coupling of aryl iodides and thiols [J]. Org. Lett. 2002, 4: 3517-3520.
[8] Bhadra,S.,Sreedhar, B.,Ranu, B.C.Recyclable Heterogeneous Supported Copper-Catalyzed Coupling of Thiols with Aryl Halides: Base-Controlled Differential Arylthiolation of Bromoiodobenzenes [J]. Adv. Synth. Catal. 2009, 351: 2369-2378.
[9] Cristau, H.J., Chabaud, B., Chene, A., Christol, H. Synthesis of Diaryl Sulfides by Nickel(Ii)-Catalyzed Arylation of Arenethiolates [J]. Synthesis-Stuttgart 1981, 13: 892-894.
[10]Percec,V.,Bae,J.Y.,Hill, D.H. Aryl Mesylates in Metal-Catalyzed Homo-Coupling and Cross-Coupling Reactions .4. Scope and Limitations of Aryl Mesylates in Nickel-Catalyzed Cross-Coupling Reactions [J]. J. Org. Chem. 1995, 60: 6895-6903.
[11]Zhang, Y.G., Ngeow, K.C., Ying, J.Y. The first n-heterocyclic carbene-based nickel catalyst for C-S coupling [J]. Org. Lett. 2007, 9: 3495-3498.
[12]Gomez-Benitez, V., Baldovino-Pantaleon, O., Herrera-Alvarez, C., et al. High yield thiolation of iodobenzene catalyzed by the phosphinite nickel PCP pincer complex: [NiCl{C6H3-2,6-(OPPh2)(2)}] [J].Tetrahedron Lett. 2006, 47: 5059-5062.
[13]Kosugi, M.,Shimizu, T., Migita, T.Reactions of Aryl Halides withThiolate Anions
in Presence of Catalytic Amounts of Tetrakis (Triphenylphosphine)Palladium Preparation of Aryl Sulfides [J]. Chem. Lett. 1978, 51:13-14.
[14]Migita, T., Shimizu, T., Asami, Y.,et al. The Palladium Catalyzed Nucleophilic-Substitution of Aryl Halides by Thiolate Anions. [J]. Bull. Chem. Soc. Jpn. 1980, 53: 1385-1389.
[15]Murahashi, S.I., Yamamura, M., Yanagisawa, K., et al. Stereoselective Synthesis of Alkenes and Alkenyl Sulfides from Alkenyl Halides Using Palladium and Ruthenium Catalysts [J]. J. Org. Chem. 1979, 44: 2408-2417.
[16]Murata, M.,Buchwald, S.L. A general and efficient method for the palladium-catalyzed crosscoupling of thiols and secondary phosphines. [J]. Tetrahedron 2004, 60: 7397-7403.
[17]Bryan,C.S., Braunger, J.A., Lautens, M. Efficient Synthesis of Benzothiophenes by an Unusual Palladium-Catalyzed Vinylic C-S Coupling. [J]. Angew. Chem. Int. Ed. 2009, 48: 7064-7068.
[18]Evano G.,Blanchard N.,Toumi M.Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis [J]. Chem. Rev. 2008, 108: 3054-3131.
[19]Buran aprasertsukP.,ChangJ.W.W.,Chavasir W.,et al. Copper-catalyzed Ullmann coupling under ligand- and additive-free conditions.Part 2: S-Arylation of thiols with aryl iodides [J]. Tetrahedron Lett. 2008, 49: 2023-2025.
[20] Luo P. S., Wang F., Li J. H., et al. Copper-Catalyzed Selective S-Arylation of 1,2-Bis(o-amino-1H-pyrazolyl) Disulfides with Arylboronic Acids [J]. Synthesis 2009, 6: 921-928.
[21] Wong Y.-C., Jayanth T. T., Cheng C.-H. Cobalt-Catalyzed Aryl?Sulfur Bond Formation [J]. Org. Lett. 2006, 8: 5613-5616.
[22] Correa A., Carril M., Bolm C. Iron-Catalyzed S-Arylation of Thiols with Aryl Iodides [J]. Angew. Chem. Int. Ed. 2008, 47: 2880-2883.
[23]Stymiest,J.L.;Mitchell,B.F.;WongS.;Vederas,https://www.doczj.com/doc/0318626330.html,.Lett.2003,5,47
[24] Ranganathan,D.;Lakshmi,C.;Karle,I.L.J.Am.Chem.Soc.1999,121,6103.
[25]Garcia-Echeverria,C.;Albericio,F.;Giralt,E.;Pons,M.J.Am.Chem.Soc.1993,115,11
663.
[26]Bahrami K,Khodaei,M M,Naali https://www.doczj.com/doc/0318626330.html,d and Highly Effieient Method forthe Synthesis of https://www.doczj.com/doc/0318626330.html,.Chem.,2008,73(17):6835一6837. [27].Du L-H,Wang Y-G A Rapid and Effieient Synthesis of Benzimidazoles Using Hypervalent Iodine as Oxidant.Synthesis,2007,675一678
[28].Carpenter RD,DeBerdt PB,Lam K S,Kurth M J.Carbodiimde-Based Benzimidazole Library https://www.doczj.com/doc/0318626330.html,b.Chem.,2006,8(6):907一91
后过渡金属催化剂综述 1催化剂的意义 催化剂是可以加速化学反应的物质。化学反应若要发生,则反应物分子之间必须有足够能量的发生碰撞以形成活性复合物或过渡态复合物,这个能量就是活化能。而催化剂能够提供一个较低的活化能,因此加速了化学反应的发生。和未添加催化剂的反应的一步实现原理相比,催化反应包含了许多种化合物与过渡态复合物[1]。 催化技术对于目前乃至未来的能源、化学反应、环境工业、石化工业都是至关重要的。原油、煤和天然气向燃料和化学原料的转化,大量石油化工和化学产品的生产,以及CO、NO、碳氢化合物排放物的控制,全都依赖于催化技术。此外,催化剂还是燃料电池电极的必要组分——无论电极使用的是固体氧化物离子还是聚合物质子电解液[2]。催化技术的发展、催化剂的改进和新催化剂的成功开发, 往往会带动已有工艺的改进和新工艺的诞生。据统计,85%以上的化学反应都与催化反应有关。目前工业上采用的催化剂大多为金属、金属盐和金属氧化物等多相催化剂, 其优点是催化性能较稳定, 使用温度广, 容易回收重复使用, 但催化活性较低, 反应常常需要高温、高压条件, 而且副反应较多。最近几十年, 发展了以有机金属络合物为主的均相催化剂, 为化学工业带来革命性进步。这种催化剂分散度高, 活性中心均一, 结构明确, 催化剂活性和选择性都较高, 反应可以在很温和的条件下进行[3]。 2后过渡金属催化剂的性质 聚烯烃工业的发展是一个国家石化工业发展的重要标志。Ziegler - Natta催化剂、茂金属催化剂和后过渡金属催化剂仍然是烯烃聚合催化剂研发的3个主要方向[4]。 90年代,美国北卡罗来纳大学的Brookhart等人[5]报道了利用适当的配体, 可使元素周期表中的第Ⅷ族中Ni和Pd的配合物用来引发烯烃聚合, 从而由单一烯烃可获得高分子量的、有各种支化度的聚合物, 并能实现与极性单体的共聚。他们将这一类催化剂称为烯烃聚合后过渡金属催化剂。后过渡金属催化剂中金属元素的种类涉及到第Ⅷ族中的元素, 目前研究得比较多的为Fe、Co、Ni、Pd4种金属元素[6]。 这类金属配合物的亲氧性相对较弱,对空气和水分不太敏感,特别是催化烯烃以及环烯烃聚合的活性很高[7],而且对比茂金属催化剂, 后过渡金属催化剂具有稳定性好、生产费用低、能生产新品种聚烯烃以及能合成带有官能团的新型聚合物等优点。再加上后过渡金属催化剂合成相对简单, 产率较高,因而其成本远低于茂金属催化剂, 而且聚合时助催化剂用量比较低, 一般与负载的茂金属催化剂相当, 因此成为烯烃聚合用催化剂的新的研究热点[8]。 3 后过渡金属催化剂的种类 后过渡金属烯烃聚合催化剂是指以镍( Ⅱ) 、钯( Ⅱ) 、铁( Ⅱ) 、钴( Ⅱ) 、钌( Ⅱ)等后过渡金属原子为活性中心的一类金属配合物烯烃聚合催化剂。 3.1 镍系 镍系包括双亚胺类、P - O类和N - O类等。双亚胺类镍系烯烃聚合催化剂是指以双亚胺为配体的一类平面型镍(Ⅱ)阳离子配合物。当采用甲基铝氧烷(MAO)作助催化剂时,二溴化双亚胺合镍的衍生物具有很高的催化活性。这类催化剂在Lewis酸如MAO 的作用下形成阳
前过渡金属催化剂的现状及进展 摘要:介绍了非茂前过渡金属催化剂作为高效烯烃聚合催化剂的发展和应用领域。根据催化剂中配位原子的性质将非茂前过渡金属催化剂分为配位原子为0、配位原子为N、硼苯类、类茂类等四大类进行讨论。在分述前过渡金属催化剂类型的同时,详细介绍了各类催化剂的特点,综述了各大聚烯烃公司的研究情况。最后时前过渡金属催化剂当前进展和未来发展趋势进行了总结和展望。 关键词:前过渡金属;非茂;催化剂;进展 纵观聚烯烃工业的发展过程,其进步无不与新型催化剂及工艺技术的开发有关。因此新型催化剂的开发应用是聚烯烃工业中研究的焦点。茂金属催化剂有很多优点,如催化体系具有单活性中心、聚合物相对分子质量可调、聚合活性高等。但茂金属催化剂成本较高,制得树脂的加工性差且专利纠纷不断,致使与茂金属催化剂性能相似,而成本较低的非茂单中心催化剂成为研究开发的新热点。非茂前过渡金属催化剂(简称前过渡金属催化剂)是指不含环戊二烯基,金属中心包括前过渡金属元素有机金属配合物,具有与茂金属催化剂相似的特点,可以根据需要定制聚合物,而且成本较低,专利发展空间相对较大,具有巨大的发展潜力。 1 前过渡金属催化剂分类及进展 1.1 含氧类配体 Kakugo等首先报道了烷氧基钛在MAO助催化作用下对丙烯有较好的聚合催化活性,并发现联二酚类衍生物与钛形成的配合物具有很好的烯烃聚合催化活性,如2,2 硫代双(6 一特丁基一4一甲基苯酚)与钛((TBP)TiCl )的配合物在MAO助催化作用下能获得超高相对分子质量的聚合物,如聚乙烯相对分子质量可达4.2×1O。、聚丙烯则高达8×1O 以上。这类催化剂不仅能够使烯烃均聚,而且能够使a烯烃共聚合。(TBP)TiC1:还可催化苯乙烯间规聚合,所得聚苯乙烯的间规度高达98%以上。这是人们第一次将非茂催化剂成功应用于苯乙烯间规聚合。而且,这种催化剂还能催化苯乙烯与乙烯共聚。 Schavorien等进一步扩展了联二酚类衍生物的研究[2]。他们在考察不同取代基对烯烃聚合的影响时发现,只有硫桥基的联二酚配合物具有高催化活性,而其它桥基或非桥联的联二酚的催化活性较低。该类催化体系对长链烯烃及二烯烃也有很好的催化活性。其后,相继又有β-酮与钛及锆形成的配合物应用于烯烃聚合催化的报道,其中β-二酮-锆配合物在MAO助催化作用下对乙烯聚合具有较高的催化活性。而β-二酮-钛配合物则对苯乙烯聚合有较高的催化活性,所产生的聚苯乙烯具有间规结构,间规度达98%以上,与单茂钛催化剂
第四章金属催化剂及其催化作用 1、金属催化剂的应用及其特性 1)金属催化剂的应用 金属催化剂:指催化剂的活性组分是纯金属或者合金 纯金属催化剂:指活性组分只由一种金属原子组成,这种催化剂可单独使用,也可负载在载体上 合金催化剂:指活性组分由两种或两种以上金属原子组成 2)金属催化剂的特性 常用的金属催化剂的元素是d区元素,即过渡元素(ⅠB、ⅥB、ⅦB、Ⅷ族元素) 金属催化剂可提供的各种各样的高密度吸附反应中心 2、金属催化剂的化学吸附 1)金属的电子组态与气体吸附能力间的关系 (1)金属催化剂化学吸附能力取决于金属和气体分子的化学性质,结构及吸附条件 (2)具有未结合d电子的金属催化剂容易产生化学吸附 (3)价键理论:不同过渡金属元素的未结合d电子数不同,他们产生化学吸附的能力不同,其催化性能也不同(4)配位场理论:金属表面原子核体相原子不同,裸露的表面原子与周围配位的原子数比体相中少,表面原子处于配位价键不饱和状态,他可以利用配位不饱和的杂化轨道与被吸附分子产生化学吸附。(5)吸附条件对进水催化剂的吸附的影响: 低温有利于物理吸附,高温有利于化学吸附 高压有利于物理吸附,也有利于化学吸附 2)金属催化剂的化学吸附与催化性能的关系 (1)金属催化剂的电子逸出功(脱出功) 定义:将电子从金属催化剂汇中移到外界(通常是真空环境中)所需做的最小功,或者说电子脱离金属表面所需要的最低能量 符号:Φ,在金属能带图中表现为最高空能级与能带中最高填充电子能级的能量差 意义:其大小代表金属失去电子的难易程度或说电子脱离金属表面的难易 (2)反应物分子的电离势 定义:指反应物分子将电子从反应物中移到外界所需的最小功,用I表示。 意义:其大小代表反应物分子失去电子的难易程度。 电离能:激发时所需的最小能量 (3)化学吸附键和吸附状态 ①当Φ>I时,电子将从反应物分子向金属催化剂表面专业,反应物分子变成吸附在金属催化剂表面上的正离子。反应物分子与催化剂活性中心吸附形成离子键,它的强弱程度决定于Φ与I的相对值,两者相差越大,离子键越强。这种正离子吸附层可以降低催化剂表面的电子逸出功。随着吸附量的增加,Φ逐渐降低。 ②当Φ 3收稿日期:2008-01-14 作者简介:潘华,博士研究生,从事大气污染控制技术研究;施耀 (通讯作者),教授,博导,从事大气污染控制技术研究,shiyao @https://www.doczj.com/doc/0318626330.html, 。 基金项目:浙江省自然科学基金项目(Y 507720) 文章编号:100926094(2008)0420036206 负载过渡金属催化剂上低碳烃选择催化还原氮氧化物的研究进展3 潘 华,张燕婷,李 伟,施 耀 (浙江大学环境与资源学院环境污染控制技术 研究所,杭州310028) 摘 要:氮氧化物(NO x )是形成酸雨和光化学烟雾的主要物种和引发物,消除氮氧化物污染是环境保护中的重点和难点。目前负载过渡金属催化剂上低碳烃选择催化还原NO x 研究是各国环境研究工作者的研究热点。本文综述了近年来负载过渡金属催化剂上低碳烃选择催化还原氮氧化物的研究进展,着重分析了该反应体系中催化剂的研究状况。探讨了目前比较公认的低碳烃选择催化还原NO x 的反应机理:1)NO 首先被氧化为NO 2;2)含氮有机中间体的生成;3)有机中间物种对NO x 的捕捉和生成N 2。总结了提高该体系中NO x 转化率的方法:1)改进催化剂的制备方法;2)添加助剂;3)等离子体结合催化还原。最后指出了现在研究中存在的主要问题,并提出开发新型催化剂、探索新催化剂制备技术以及引入新实验手段是低碳烃选择还原 NO x 今后的研究方向。 关键词:环境工程;低碳烃;氮氧化物;选择催化还原;过渡金属中图分类号:O643 文献标识码:A 0 引 言 氮氧化物(NO x )是形成酸雨和光化学烟雾的主要物种和引发物,可使人类患发肺气肿和支气管炎等疾病[1,2]。大气中的NO x (包括NO ,NO 2等)主要来自移动源(机动车)和固定源(主要为火力发电厂、工业燃烧装置)2个方面,在发达国家,移动源和固定源对NO x 的贡献约各占50% [3] 。美国学者 S treets 等[4]报道中国1995年NO x 排放总量为112×10-7t ,其 中固定源占76%,移动源占12%,并预测到2020年NO x 排放总量为2166×10 -7 t ,其中固定源占7812%,移动源占1311%。 面对氮氧化物排放量的日益增多以及由此引起对环境与人类生活的严重危害,世界各国政府先后制定了具体的NO x 排放法规[2];企业和科研人员则致力于开发高效率、低成本的脱硝(DeNO x )工艺和技术,其中选择性催化还原NO x 技术 (NO -SCR )已在全世界范围引起了广泛关注。 1 DeN O x 技术的发展 DeNO x 技术可分为燃烧过程控制和尾气控制2大类。燃 烧过程控制主要是通过新型燃烧器的设计和改变炉内燃烧条件而实现,但采用低NO x 燃烧技术最多仅能降低约50%的 NO x 排放[5]。因此目前防治NO x 污染的主要技术是尾气控 制,该法可分为干法和湿法2大类。干法脱硝包括选择催化还原[6,7]、非催化还原法[8]、金属氧化物吸附转化法[9]和等离子法[10,11];湿法脱硝包括酸吸收[12]、碱吸收[12,13]、氧化吸收[13]和化学吸收-生物还原法[14,15]。 目前在国际上仅NH 3的选择催化还原(NH 3-SCR )技术得到了工业化应用[16],该技术转化率高、选择性好、实用性 强。但该技术也存在如下缺点[17] ,1)NH 3是一种有毒腐蚀性气体,存储和输运麻烦,对管路设备要求高,造价昂贵;2)在该过程中,NH 3需要计量控制加入量,容易泄漏或反应不完全而造成二次污染;3)NH 3与烟道气中的S O 2反应,形成腐蚀性的NH 4HS O 4,易使催化剂中毒;4)工作温度范围窄。因此,寻找一种还原剂可以取代NH 3具有十分重要的意义。1990年,日本学者I wam oto 等[6]报道了在含氧气氛下,烯烃在 Cu -ZS M -5催化剂上以高选择性地还原NO 。从此,烃类选 择催化还原NO x 的研究受到了各国学者的广泛关注。英国学者Burch 等[18]介绍了金属氧化物和贵金属铂催化剂上烃类选择还原NO x 的研究进展。国内学者孔科[19]和张涛[20]分别介绍了烃类和甲烷选择还原NO x 的研究进展。在烃类选择还原NO x 的研究中,贵金属催化剂具有活性高和低温特性好的特点,因此成为人们研究的一个热点[21,22],但其产物中含 有较多N 2O (约占产物的50%),对N 2的选择性低[21]。近年来,负载过渡金属(特指第四周期的过渡金属:T i ,V ,Cr ,Mn ,Fe ,C o ,Ni ,Cu 和Zn )催化剂由于活性高、成本较贵金属催化剂低廉而受到了研究者的广泛关注。此外低碳烃(含碳原子数小于3)储量丰富,分布广泛、易得。因此负载过渡金属催化剂上低碳烃选择还原NO x 具有更加广阔的实用前景和经济价值。本文将介绍近几年负载过渡金属催化剂上低碳烃选择还原NO x 的研究进展。 2 负载过渡金属催化剂上低碳烃选择还原N O x 催 化剂的研究进展 近年来关于负载过渡金属催化剂上低碳烃选择还原NO x 的研究有很多。通过SCI 检索统计发现,从2002年到2007年发表的有关负载过渡金属催化剂上低碳烃选择还原NO x 的论文约有90篇(见图1),占这段时期烃类选择催化还原NO x (HC -SCR )论文的约60%,占这段时期选择催化还原NO x (NO -SCR )论文的约20%,充分表明人们对这方面工作关注 的程度。总结10年(尤其近6年) 来用于负载过渡金属催化 图1 2002—2007年间SCI 收录的有关负载金属催化剂 上低碳烃选择还原N O x 的文章 Fig.1 The numbers of documents on N O -SCR with low er hydrocarbon over transition metal b ased catalysts indexed by SCI during 2002-2007 第8卷第4期2008年8月 安全与环境学报Journal of Safety and Environment V ol.8 N o.4 Aug ,2008 《高等无机化学》课程论文文献综述 综述题目后过渡金属催化剂 的研究进展 作者所在系别理学院 作者所在专业无机化学 作者姓名吕海涛 作者学号12S007005 导师姓名唐冬雁 导师职称教授 完成时间2013 年 4 月 哈尔滨工业大学材料化学教研室制 说明 1.文献综述各项内容要实事求是,文字表达要明确、严谨,语言通顺,外来语要同时用原文和中文表达。第一次出现缩写词,须注出全称。 2.学生撰写文献综述,阅读的主要参考文献应在10篇以上。本课程的相关教材也可列为参考资料,但必须注明参考的具体页码。 3.文献综述的撰写格式按撰写规范的要求,字数在2000字左右。 后过渡金属催化剂的研究进展 1 后过渡金属催化剂的进展 后过渡金属催化剂是近年来受到广泛关注的一种新型催化剂,是对聚合催化剂的又一重要革新。它开辟了一个完全崭新的催化领域,将成为继茂金属催化剂之后的又一研究开发热点。后过渡金属( 铁、钴、镍、钯等) 配合物用于烯烃催化研究可追溯至上世纪70年代,其研究结果发展成了SHOP( Shell higher olefin process) 催化体系(1987)[1],被广泛用于工业生产线性A烯烃。然而,由于后过渡金属容易导致B氢消除反应,影响了乙烯聚合催化的发展。直到上世纪90 年代中期,Brookhart研究组发现了A—二亚胺镍、钯配合物能催化乙烯聚合制得高分子量聚乙烯(1995)[2],后过渡金属配合物催化乙烯聚合的重要性才真正为人们所认识。 研究后过渡金属催化剂卓有成效的世界著名大公司有Du Pont、Shell、BP 、BF Goodrich和W.R.Grace 公司等(1996)[3]。他们在该技术领域投人了大量精力,深入研究,取得令人瞩目的成就,其中有的研究已接近于工业化。shell公司于1996年在英国的Carringtion开始运转了一套使用后过渡金属把基络合物催化剂的聚酮装置,生产能力约1.5万t/a ,这种商品名为Carilon的聚酮产品已经销售到了欧洲和美国。该公司目前正对第二套聚酮装置的地点和生产能力进行评估, 准备扩大生产规模。BP公司在英国的Grangemouth也有采用钯基催化剂的CO/烯烃共聚物中试装置运行。 后过渡金属催化烯烃以及环烯烃聚合的研究在近年来取得了重大进展, 已经能够设计合成具有特殊微观结构的聚烯烃;实现了乙烯与极性单体、乙烯与环烯烃的共聚;催化机理的研究也日益完善。这些结果将为新型催化体系的设计及新型功能材料的合成起到一定的指导作用。在后过渡金属烯烃催化剂的合成过程中, 近年来开始出现了一些新的方法和技术。例如高通量筛选方法( high throughput screening, HTS) 的应用(2002)(2003)[4,5],其优点在于, 在相同的时间段内合成和试验数个甚至数十个配体和配合物, 极大地加速了高效催化剂的筛选, 节省了大量时间, 降低了药品的消耗。相信这一技术将大大促进催化剂合成与筛选的速度。 2 后过渡金属催化剂的特点 后过渡金属(铁、钴、镍、钯等)配合物催化剂由于具有稳定性高、易于合成和耐受杂原子和极性基团的能力,具有与前过渡系催化剂明显不同的性能(2009)(2003)[6,7]为烯烃齐聚、聚合及共聚研究提供了新的发展空间。其主要特点有:(l) 聚合活性极高。这种新型络合物均相催化剂无论与传统高效Ziegler催化剂或茂金属催化剂相比, 都显示出异常高的活性, 高达11x106gPE/mol·h。 (2)聚合能力强,聚合单体范围广。可以接受官能化的极性单体,用于全范围的单体聚合及共聚合,合成种类繁多的新型聚烯烃树脂和特种性能树脂等。 过渡金属催化的C-S的合成 摘要:过渡金属催化的C-S交叉偶联反应在有机合成方法学的研究中一直起着不可或缺的作用。这些经过交叉偶联反应所形成的一系列含碳-硫键结构的化合物,在染料、医药、农药、化工以及聚合物的制备中都有广泛的应用。不同过渡金属催化合成硫化物成为当前研究的一个热点。本文简单综述了不同过渡金属催化反应合成含C-S的化合物。 关键词:过渡金属;硫醇;催化;偶联反应;碳一硫键构建 Transition Metal Catalyzed Synthesis of C-S bond Abstract: transition metal catalyzed C-S cross coupling reaction plays an important role in organic synthetic methodology. The compounds synthesized through cross coupling reaction have very good biological activity and wide application in colorant, pharmaceutical, pesticide, and chemical industry , and the preparation of polymer.So transition metal catalytic synthesis of C-S bond becomes a hot issue. In this paper,transition metal-catalyzed reaction was briefly summarized. Key words: transition-metal; thiols; catalyze; coupling reaction;C-S bond formation 许多含硫化合物具有生物活性,包括磺酰胺类抗生素和哮喘药物顺尔宁抗生素等[1-2]。多种含硫化合物的各类构建方法需要深入地研究,碳一硫键的构建和以及进一步的官能团化已经引起科学界的相当关注。硫化物,硫醇及它们的氧化衍生物在有机合成方面有广泛的应用[3-4]。与碳一氧键和碳一氮键的构建方法相比,有机金属试剂催化的碳一硫键的构建方依然是不足的。尽管人们始终认为硫能够毒化金属催化剂,但是金属催化的碳一硫键的构建方法研究有逐渐增强的趋势。 过渡金属催化通过偶联反应构建碳一硫键的各种方法有很多报道,我们接来将介绍不同的过渡金属催化合成碳硫键的这类反应最近进展。 1铜催化 新一代聚烯烃催化剂 ———后过渡金属催化剂 苏 宇 杨海滨(中山大学高分子研究所,广州 510275) 摘 要 本文综述了以α2二亚胺为配体的Ni(Ⅱ)基和Pd(Ⅱ)基、以三吡啶二亚胺为配体的Fe(Ⅱ)基和Co(Ⅱ)基后过渡金属催化剂,包括催化剂的组成、对烯烃聚合及共聚合的性能和聚合机理。 关键词 后过渡金属,镍,钯,烯烃,聚合,催化剂 NOVE L OL EFIN POLYMERIZATION CATALYSTS Su Yu Yang Haibin (Institute of Polymer Science,Zhongshan University,Guangzhou510275) Abstract This paper introduces about Ni(Ⅱ)、Pd(Ⅱ)、Fe(Ⅱ)、Co(Ⅱ)2based novel late transi2 tion metal catalysts,and the composition of catalysts,properties of olefin homopolymerization and copolymerization and mechanism of polymerization reaction are given. K ey w ords late transition metal,Palladium,Nickel,olefin,polymerization,catalyst 全球对聚烯烃的市场需求日益增大。据美国Chem System公司预测[1],到2003年,世界对乙烯的需求量为10500万t,年均增长率为515%;而对丙烯的需求量为6700万t,比1998年增加1600万t。这惊人的数字说明了聚烯烃的生产及改良是市场客观要求的必然。而聚烯烃树脂性能的改进与聚合催化剂密切相关。Ziegler催化剂的开发和改进大大提高了线型聚乙烯的性能,茂金属催化剂的出现使聚烯烃发生了革命性的变革。与此同时,新型非茂金属———后过渡金属催化剂(又称Brookhart催化剂)的开发研究更引人注目,它为制备更宽范围的聚烯烃树脂提供了可能。1995年,Brookhart等人[2]用大体积α2二亚胺配体形成的Ni(Ⅱ)和Pd (Ⅱ)基络合物,成功地实现了促进链增长的目标,成为第一个能够生产高分子量的后过渡金属催化体系。最近2年,伦敦Imperial大学的G ibson研究小组[3]和美国Brookhart研究小组[4]独立发现了Fe (Ⅱ)基和Co(Ⅱ)基催化体系,这种新催化体系不仅在活性和聚合物性能控制方面具有茂金属催化剂的很多优点,而且具有成本低、可生产更宽范围聚合材料的潜力。本文即对后过渡金属催化剂作一综述。 1 后过渡金属催化剂的特点 与传统Ziegler-Natta催化剂及茂金属作比较,后过渡金属催化剂的主要特点是: (1)它选择了Ni、Pd、Fe、Co等后过渡金属,而不是通常茂金属所采用的Ti、Zr等前过渡金属,所制备的催化剂也是单活性中心均相催化剂,因此可按预定目的精确控制聚合物的链结构; (2)聚合能力强,可用于烯烃和极性单体共聚。根据共聚单体特点和反应条件及催化剂种类,极性树脂中共聚单体含量约为013%~12%; (3)用于烯烃均聚(乙烯、丙烯、己烯等)。Ni (Ⅱ)和Pd(Ⅱ)基催化剂可用于生产带支链的聚合 后过渡金属催化剂之前的催化剂的特点 过渡金属催化的碳氢键活化 应用化学,涂适,1132236,135******** 碳-氢(C-H)键的转化和碳-碳(C-C)键的连接是有机化学中最重要、最基础的研究内容之一。作为自然界最基本、最普遍的惰性化学键和结构单元,C-H键广泛存在于各种有机化合物当中(如简单的碳氢化合物、复杂有机分子、生物体内组织,工业多聚物材料等)。而通过直接活化和诱导C-H键形成新的官能团(特别是新的C-C键)无疑是一条极具吸引力的反应策略,它集中体现了原子经济性、步骤经济性、环境友好等特征,近年来已发展成有机化学中最活跃的研究领域之一。 早在上世纪初,人们就发现通过一些特定的方法可以对一些惰性C-H键进行直接的官能团化,但如何在活化过程中对各类形形色色的C-H键进行识别和区分,并有目的性的对特定的位置进行定向官能团衍生,一直是有机合成领域的研究难点。随着过渡金属化学的迅猛发展,一系列新反应、新试剂陆续被发现和合成,并在有机合成中得到了广泛的应用。而过渡金属在C-H活化领域的应用,使得对一些C-H键定向的活化和官能团化成为可能,相关的研究近10年来已取得了令人瞩目的成绩,特别是钌、铑、钯、铱等传统过渡金属催化的一些具有高化学选择性的C-H活化反应已经逐渐发展成熟,并且在有机合成中得到了越来越多的应用。 最近,过渡金属促进的有机反应研究的主要聚焦于两类过渡金属上。一类是以铂、金、银为代表的贵重过渡金属,传统上这几种金属普遍被认为具有惰性及稳定性,通常很少参与有机反应,但近期的研究发现,这些金属往往具有独特的反应性,其在C-H活化领域的应用令人期待。另一类是以铜、铁等为代表的过渡金属化合物,这类金属在地球中储量丰富,具有价格低廉,环境友好等特征。用更经济、更绿色的铜、铁催化剂代替传统稀有过渡金属(如钌、铑、钯等)催化活化碳-氢键实现碳-碳键的构筑是金属有机化学发展的又一热点。 现阶段,各种新的策略和机理被提出并被广泛采用,以下爱是对过渡金属催化的碳氢活化发展及其研究进展的一些介绍。 碳氢活化反应既可以发生在分子内进行,也可以发生在分子间。分子内的碳氢活化反应由于体系对反应位置自由度的限制使得反应具有区域选择性。 分子间碳氢活化反应由于催化剂和C-H键反应的时候具有更大的自由度使得该类型反应有更大的挑战性。影响分子间碳氢活化反应的区域选择性的主要因素有:一、官能团化得芳环的电性(例如,芳基亲电取代发生在供电子基取代的邻对位); 过渡金属催化剂有二大特点:Ⅰ、在反应气氛如H2、O2气下,过渡金属是以金属晶体存在。Ⅱ、最适合用于金属催化剂的活性组份是那些最外层有1~2个S电子,次外层为d电子,d电子为大部分充满状态的元素。 金属催化作用与d电子性质、金属晶体、表面结构有关。 3d带电子填充量为94%,若平均到每个Ni原子上时,d轨道的电子填充量为9.4个电子。即Ni金属晶体中的Ni 原子d轨道中还差0.6个电子就可被完全充满,使d轨道能量或d带能量处于 最低,因而有很强的能力去获得电子,我们把这个电子差额称之为d孔穴。 20、25、36、38、40、42、46、59、61、93-102、104-106、110、144、148、149(很重要)、153、159、172(不错)、173(非常好)、177、180(以及后面连续的几页都比较重要)页有重要信息 几何论和能量匹配论包括两个方面: Ⅰ、吸附物分子与活性位空间结构的几何对应关系。Ⅱ、吸附物分子与活性位之 间的能量对应关系。 EFGH晶面的Miller指数:(100)ABC晶面的Miller指数:(222) 金属结构的应用在于: 形成合金:当原子半径相近,而晶胞结构又相同的一些金属可以相互取代,形成结构不被破坏的合金,如,Pd—Au,Pt—Re,Cu—Ni 等合金催化剂。 低Miller指数晶面上晶格原子排布整齐,高能量的边、角原子少,原子密度高,故其表面剩余能低,稳定性高。 高Miller指数晶面则晶格排布 有不规整的地方,处于高能态的边、角原子多,原子密度低,表面剩余能高,稳定性差。 表面以下面几种方式来降低晶体的总表面能: 1.尽量减少向外暴露的表面积。 2.暴露表面以低表面能的晶面为主。 3.改变金属晶体外露的表面几何结构。 4.增强金属与载体间的相互作用。 改变外露的表面几何结构以减少表面能表面松弛与重构(Relaxation & Reconstruction of 高分子反应研究方法 —二亚胺后过渡金属催化剂催化乙烯聚合的机理与动力学研究 目录 高分子反应研究方法 (1) 一、前言 (1) 1.1 历史 (1) 1.2 催化反应机理与实验 (2) 1.2.1 催化反应机理 (2) 1.2.2 聚乙烯高分子链形态 (3) 二、 -二亚胺Ni/Pd催化乙烯聚合机理研究中使用的研究方法 (4) 三、聚乙烯微结构、构象和形态的Monte Carlo模拟 (4) 3.1 二亚胺Ni催化乙烯聚合产物结构模拟[6] (4) 3.1.1 模型与算法 (4) 3.1.2 结果与分析 (7) 3.2 温度和压力对二亚胺Ni催化乙烯聚合产物微结构影响模拟[7] (7) 3.3 二亚胺Pd催化乙烯聚合产物构象与形态模拟[8] (8) 3.3.1 模型 (9) 3.3.2 结果与讨论 (10) 四、二亚胺Ni催化乙烯聚合反应动力学模型[9] (12) 4.1 模型 (12) 4.1.1 聚合速率模型 (12) 4.1.2 链转移和分子量模型 (13) 4.1.3 分子量分布模型 (14) 4.1.4 链行走和支化密度模型 (15) 4.2 结果与讨论 (19) 参考文献 (21) 一、前言 1.1 历史 1995年Brookhart Maurice 发现了α-二亚胺Ni/Pd 催化剂[1] (图1),这类催化剂可以催化乙烯聚合获得高分子量的聚乙烯,并且其活性比拟甚至超过茂金属催化剂。只需简单地调节温度、压力等参数就可以用乙烯一种原料获得从线性聚乙烯、支化聚乙烯甚至高度支化聚乙烯或超支化聚乙烯。如果说前过渡金属烯烃聚合催化剂最显著的特点是控制立体规整性,那么后过渡金属烯烃聚合催化剂最显著的特点是控制聚合物分子链的形态[2] 和较强的极性单体耐受性。 R 1 R 2 N N R 3 R 3 R 3 M X Y R 3 图1 Brookhart 型二亚胺Ni/Pd 催化剂 事实上,在α -二亚胺Ni/Pd 催化剂发现之前,SHOP 工艺所使用的催化剂与此类似。在 SHOP 工艺所使用催化剂的基础上,利用催化剂结构设计理论,合成出了能够得到高分子量聚乙烯的α-二亚胺Ni/Pd 催化剂。 高分子的形态是指单体单元在高分子中的几何排列,如线性、高度支化、超支化、 树枝状等(图2)。其它的术语如微结构、织构也常用来描述具有不同结构的聚合物。微结构常用来描述高分子局部区域的结构,它不能充分表征高分子的全局形态。而聚合物织构广泛用于描述聚合物结构类型,如星形、嵌段、接枝等不同类型的支化形式和不同整规度的聚合物。而形态(topology )是指构造高分子的单元相同但几何排列不同。例如对于聚乙烯来说,有如下几种形态: 线性聚乙烯 高度支化聚乙烯 超支化聚乙烯树枝状聚乙烯 图2 聚乙烯分子链的常见形态 高分子的形态是决定其物理性质和应用的一个非常重要的参数。人们迫切希望能直接将现有的简单单体聚合来控制所得高分子的形态。链行走催化剂是最吸引人的选择之一。其控 化工与材料工程学院 毕业论文开题报告 钯催化C-H键、C-C键活化反应的研究 1.课题来源及选题意义 人类在新世纪面临俩大危机,一是资源的不断枯竭,而是生态环境的日益恶化。目前世界上工业制造出的化合物的数量大约在2万到3万之间。这些数目巨大的化合物只是由很少数目的原料来制备的,并且碳的来源几乎都是化石物质,即石油,天然气和煤。石油、天然气以及煤中的主要成分是含有惰性的C-H键、C-C键的烷烃类化合物,这些宝贵的化石燃料的利用至今仍主要局限于将其燃烧提供能源。因此,惰性的C-C、C-H键的活化首先可以大幅度的提高资源的利用效率。 早在上个世纪初,人们就发现一些特定的方法可以对一些惰性C-H、C-C键进行直接的官能团的活化,但如何在活化过程中对个形形色色的C-C、C-H键进行识别和区分,并有目的性的对特定位置进行定向官能团的衍生,一直是有机合成领域的一个难点。随着过渡金属化学的迅速发展,一系列新反应、新试剂陆续被发现和合成,并在有机合成中得到广泛的应用。 碳氢(C-H)键的转化和碳碳键的连接是有机化学中最重要、最基础的研究内容之一。作为自然界最简单、最普遍的惰性化学键和结构单元,C-C键与C-H键广泛存在各种有机化合物中(如简单的碳氢化合物、复杂有机分子、生物体内组织,工业多聚物材料等)。而通过活化和诱导C-H键形成新的化合物(特别是新的C-C键)无疑是一条既具有吸引力的反应策略。通过活化C-C键促进芳烃的交叉偶联反应,集中体现了原子经济性、步骤经济性。 C-H键具有较高的电能,并且碳原子和氢原子的电负性相近。因此,C-H键的基本特点是稳定坚固且极性很小,反应活性很小,没有官能团活化的情况下是很难发生化学反应的。所以在C-H键反应过程中遇到的第一个问题是活性,其次的一个问题是反应的选择性。由于大部分情况下是有机分子中含有多个化学性质相似的C-H键,如何对这些形色各异的C-C、C-H键进行识别和催化,并按照预期设想的结果进行反应,就成了催化活化C-C、C-H键最为根本并待于解决的问题。 过渡金属中很多都可以实现C-C、C-H键的活化,而过渡金属的参与为了这一领域的发展带来了无限的机遇,成功的解决了这一类相关研究的难题。 各种过渡金属化合物对于碳氢键、碳碳键的识别和活化的机制各不相同,金属钯是银白色的过渡金属,化学性质不活泼,常温下在空气和潮湿环境中稳定。钯能耐氢氟酸、磷酸、高氯酸、盐酸、硫酸蒸汽的侵蚀。在1803年,英国化学家武拉斯顿从铂矿中发现的。它可以通过催化中环钯化的过程实现对于临近的各类碳氢键的活化。钯催化剂是以钯为主要活性组分,使用钯黑或把钯黑载于氧化铝、沸石等载体上。以钠、镉、铅等的盐为助催化剂。并且,选择钯这种过渡元素可实现有机化合物之间高效,高选择性,且条件温和的转化,是实现绿色化学的重要内容。可实现在环境友好的反应条件下的催化活化,是对于资源的高效的利用。 2.国内外的发展及前景 2.1国外发展状况 第一章催化剂与催化作用基本知识 1、简述催化剂的三个基本特征。 答:①催化剂存在与否不影响△Gθ的数值,只能加速一个热力学上允许的化学反应达到化学平衡状态而不能改变化学平衡;②催化剂加速化学反应是通过改变化学反应历程,降低反应活化能得以实现的;③催化剂对加速反应具有选择性。 2、1-丁烯氧化脱氢制丁二烯所用催化剂为MoO3/BiO3混合氧化物,反应由下列各步组成 (1)CH3-CH2-CH=CH2+2Mo6++O2-→CH2=CH-CH=CH2+2Mo5++H20 (2)2Bi3++2Mo5+→2Bi2++2Mo6+ (3)2Bi2++1/202→2Bi3++02- 总反应为CH3-CH2-CH=CH2+1/202→CH2=CH-CH=CH2+H20 试画出催化循环图。 CH3-CH22 Bi 3、合成氨催化剂中含有Fe3O 4、Al2O3和K20,解释催化剂各组成部分的作用。 答:Fe3O4:主催化剂,催化剂的主要组成,起催化作用的根本性物质 Al2O3:构型助催化剂,减缓微晶增长速度,使催化剂寿命长达数年 K20:调变型助催化剂,使铁催化剂逸出功降低,使其活性提高 第二章催化剂的表面吸附和孔内扩散 1、若混合气体A和B2在表面上发生竞争吸附,其中A为单活性吸附,B2为解离吸附:A+B2+3*→A*+2B*,A 和B2的气相分压分别为p A和p B。吸附平衡常数为k A和k B。 求吸附达到平衡后A的覆盖率θA和B的覆盖率θB。 解:对于气体A:吸附速率v aA=k aA P A(1—θA—θB) ;脱附速率v dA=k dAθA 平衡时:v aA=v dA,即θA=(k aA/k dA)P A(1—θA—θB)=k A·k B(1—θA—θB) 对于气体B:吸附速率v aB=k aB P B(1—θA—θB)2;脱附速率v dB=k dBθB2 平衡时:v aB=v dB ,即θB2= k B P B(1—θA—θB)2 。 m (2)求Al2O3的表面积。(已知:N2分子截面积16.2×10-20m2) 解:P/V a(P0-P)=(1/CV m)[1+(C-1)P/P O]=[(C-1)/CV m](P/P O)+1/CV m 由题目可知:(C-1)/CVm=50.06 1/CVm=0.2723 ∴C=184.84 Vm=0.01987L(标况) n=0.01987/22.4=8.875×10-4S=nN A S O=8.875×10-4×6.023×1023×16.2×10-20=86.60m2 3、多相催化反应一般包括那几个步骤?其中哪几个步骤属于化学过程? 答:多相催化反应包括: 外扩散:反应物分子从气流中向催化剂颗粒表面扩散;(孔)内扩散:反应物分子从颗粒表面向颗粒内表面扩散;化学吸附:反应物分子在催化剂内表面吸附;表面反应:吸附的反应物分子在催化剂表面上反应; 脱附:产物分子自催化剂内表面脱附;(孔)内扩散:产物分子从颗粒内表面向颗粒外表面扩散;外扩散:负载过渡金属催化剂上低碳烃选择催化还原氮氧化物的研究进展
后过渡金属催化剂的研究进展-哈尔滨工业大学教师个人主页
过渡金属催化C-S合成
新型后过渡金属烯烃聚合催化剂—镍系烯烃聚合催化剂
过渡金属催化的碳氢键活化
过渡金属催化理论知识
高分子反应研究方法-后过渡金属催化剂
过渡金属催化
最新工业催化原理—作业汇总(含答案)
相关主题
文本预览