肌营养不良症
- 格式:ppt
- 大小:81.50 KB
- 文档页数:15
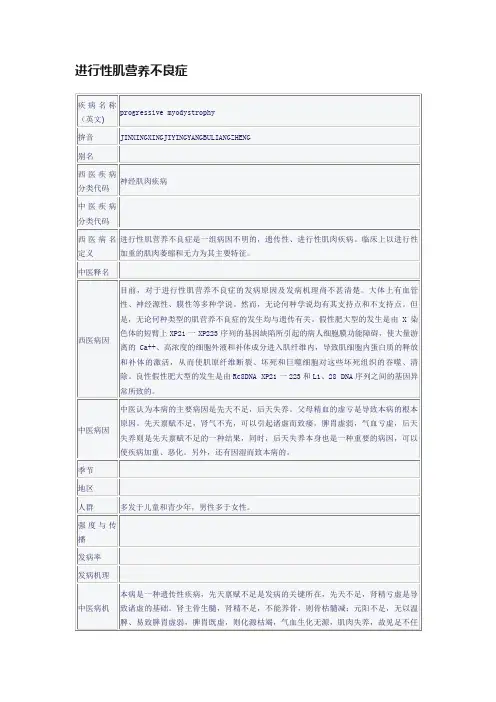




中西医诊治进行性肌营养不良症良方【概述】本组疾病为原发性肌肉变性病,与遗传因素有关,大多有家族史。
临床特征为缓慢进行性加重的对称性肌无力肌肉萎缩,个别类型尚有心肌受累。
根据遗传方式、发病年龄、萎缩肌肉的分布、病程和预后,可分为不同的临床类型。
进行性肌营养不良症在祖国医学中属于痿证的范畴。
痿证指肢体筋脉弛缓,手足痿软无力的一种病证,以下肢不能随意运动及行走者较为多见。
根据其发病原因、部位及临床表现不同,又有皮痿、肌痿、筋痿、肉痿、骨痿“五痿”之称。
多见于温热病中或热病后期,邪热灼伤阴液,筋脉失于濡养;或因湿热浸淫筋脉肌肉,而弛纵不用或因体虚久病,肝肾亏虚,精血不足,不能濡养肌肉筋骨或瘀阻脉络等因而成。
【病因病理】一、西医病因病理本组疾病虽均为遗传性疾病,但遗传方式各不相同。
其中对假肥大型肌营养不良的病因学研究比较深入,近年来有了突破性的进展。
早在80年代初期已确认本病的基因位点在于Xp21上。
以后的研究证明此基因所编码的蛋白系一种细胞骨架蛋白,称为抗肌萎缩蛋白,分布于骨骼肌和心肌的细胞膜上,起支架作用,可保护肌膜抵抗收缩时所产生的力量而不致受损。
患者因基因缺陷而肌细胞内缺乏Dys,造成功能缺失而发病。
二、中医病因病机(一)肺热伤津由于正气不足,感受温热毒邪,高热不退,或病后余邪未尽,低热不解,肺受热灼,津液耗伤筋脉失于濡润,导致手足痿弱不用,而成痿证。
此即《素问·痿论篇》所说:“肺热叶焦,则皮毛虚弱急薄著,则生痿也。
”(二)湿热浸淫久处湿地,或冒雨露,感受外来之湿邪,湿留不去,郁久化热;或饮食不节,如过食肥甘,或嗜酒,或多食辛辣,损伤脾胃,湿从内生,蕴湿积热。
以致湿热浸淫筋脉,影响气血的运行,使筋脉肌肉弛纵不收因而成痿。
如《素问·痿论篇》说:“有渐于湿,以水为事,若有所留,居处相湿,肌肉濡渍,痹而不仁,发为肉痿。
”(三)脾胃虚弱脾胃为后天之本,津液气血资生之源。
如素体脾胃虚弱或因病致虚,脾胃受纳运化功能失常,津液气血生化之源不足,肌肉筋脉失养,渐而成痿。

Duchenne 肌营养不良症(Duchenne musculardystrophy ,DMD )是儿童最常见的X -连锁隐性遗传性肌肉疾病,其临床特点为四肢骨骼肌进行性、对称性肌无力和肌萎缩[1]。
早期受累肌肉表现为弥漫性或局限性水肿,晚期肌肉及肌间隙内可见脂肪浸润,肌纤维变细、减少,最终肌肉相应收缩功能丧失。
患儿多在2~5岁时因血清肌酸磷酸激酶升高而被发现,12岁左右丧失独立行走能力,20~30岁因心肺功能衰竭而死亡[2]。
DOI :10.3969/j.issn.1672-0512.2024.01.019 [基金项目] 深圳市医疗卫生三名工程项目(SZSM202011005);广东省深圳市科创委基金资助项目(JCYJ20230807093815031);深圳市儿童医院疑难疾病精准诊治攻关项目(LCYJ2022092)。
[通信作者] 李志勇,Email :*************。
DTI 评估Duchenne 型肌营养不良症严重程度的初步研究陈太雅1,2,胡颖熠1,2,黄 杨2,3,方雪琳2,3,王景刚4,方佃刚2,路新国5,李志勇21.中国医科大学,辽宁 沈阳 110000;2.广东省深圳市儿童医院放射科,广东 深圳 518038;3.汕头大学医学院,广东 汕头 515041;4.广东省深圳市儿童医院康复科,广东 深圳 518038;5.广东省深圳市儿童医院神经内科,广东 深圳 518038[摘要] 目的:探讨DTI 评估Duchenne 肌营养不良症(DMD )患儿病情严重程度的临床价值。
方法:28例DMD 患儿均行大腿肌肉常规MRI 和DTI 检查,测量右侧大腿14块肌肉(臀大肌、阔筋膜张肌、股外侧肌、股中间肌、股内侧肌、股直肌、缝匠肌、长收肌、大收肌、股薄肌、半膜肌、半腱肌、股二头肌长头、股二头肌短头)的各向异性分数(FA )值、ADC 值。
根据运动功能评估量表(MFM )评价患儿运动功能,并分析各肌肉FA 值、ADC 值和14块肌肉平均FA 值、ADC 值与MFM 总评分的相关性。


肌营养不良症疾病概述肌营养不良症是由遗传因素所致的以进行性骨骼肌无力为特征的一组原发性骨骼肌坏死性疾病,临床上主要表现为不同程度和分布的进行性加重的骨骼肌萎缩和无力。
也可累及心肌。
发病机制本病病因是遗传异常,在不同的类型中可以不同的方式进行,但遗传因素通过何种机制最终造成肌肉变性,则始终未明。
临床表现按照典型的遗传形式和主要临床表现,可将肌营养不良症分为下列类型:1、假肥大型:属X-连锁隐性遗传,是最常见的类型,根据临床表现,又可分为Duchenne 型和Becker。
1)Duchenne型营养不良症(DMD):也称严重性假肥大型营养不良症,几乎仅见于男孩,母亲若为基因携带者,50%男性子代发病,常起病于2-8岁,初期感走路苯拙,易于跌倒,不能奔跑及登楼,站立时脊髓前凸,腹部挺出,两足撇开,步行缓慢摇摆,呈特殊的“鸭步”步态,当由仰卧走立时非常困难,必先翻身俯卧,再双手攀缘两膝,逐渐向上支撑起立(Gower 征)。
亦可见于肢近端肌肉、股四头肌及臂肌。
2)Becker型(BMD):也称良性假肥大型肌营养不良症,常在10岁以后起病,首发症状为骨盆带及股部肌肉力弱,进展缓慢,病程长,出现症状后25年或25年以上才不能行走,多数在30-40岁时仍不发生瘫痪,预后较好。
2、面肩-肱型肌营养不良症:男女均有,青年期起病,首先面肌无力,常不对称,不能露齿,突唇.闭眼及皱眉,口轮匝肌可有假性肥大,以致口唇肥厚而致突唇,有的肩、肱部肌群首先受累,以致两臂不能上举而成垂肩,上臂肌肉萎缩,但前臂及手部肌肉不被侵犯。
病程进展极慢,常有顿挫或缓解。
3、肢带型肌营养不良症:两性均见,起病于儿童或青年,首先影响骨盆带肌群及腰大肌,行走困难,不能登楼,步态摇摆,常跌倒,有的则只累及股四头肌。
病程进展极慢。
4、其它类型:股四头肌型、远端型、进行性眼外肌麻痹型、眼肌-咽肌型等,极少见。
辅助检查实验室检查血清酶测定:1、血清肌酸磷酸激酶(CPK):CPK增高是诊断本病重要而敏感的指标,可在出生后或出现临床症状之前已有增高,当病程迁延时活力逐渐下降。


