细菌全基因组重测序
- 格式:pdf
- 大小:5.30 MB
- 文档页数:1

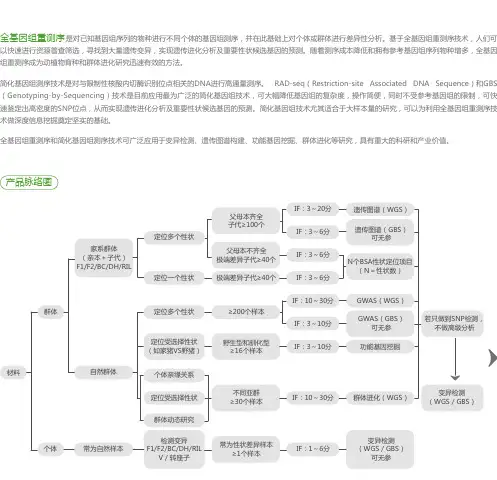
全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。基于全基因组重测序技术,人们可
以快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。随着测序成本降低和拥有参考基因组序列物种增多,全基因
组重测序成为动植物育种和群体进化研究迅速有效的方法。
简化基因组测序技术是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。 RAD-seq(Restriction-site Associated DNA Sequence)和GBS
(Genotyping-by-Sequencing)技术是目前应用最为广泛的简化基因组技术,可大幅降低基因组的复杂度,操作简便,同时不受参考基因组的限制,可快
速鉴定出高密度的SNP位点,从而实现遗传进化分析及重要性状候选基因的预测。简化基因组技术尤其适合于大样本量的研究,可以为利用全基因组重测序技
术做深度信息挖掘奠定坚实的基础。
全基因组重测序和简化基因组测序技术可广泛应用于变异检测、遗传图谱构建、功能基因挖掘、群体进化等研究,具有重大的科研和产业价值。产品脉络图
家系群体(亲本+子代)F1/F2/BC/DH/RIL
检测变异F1/F2/BC/DH/RILV/转座子常为性状差异样本≥1个样本不同亚群≥30个样本野生型和驯化型≥16个样本≥200个样本极端差异子代≥40个父母本不齐全极端差异子代≥40个父母本齐全子代≥100个IF:3~20分遗传图谱(WGS)
GWAS(WGS)
群体进化(WGS)
变异检测(WGS/GBS)可无参变异检测(WGS/GBS)若只做到SNP检测,不做高级分析GWAS(GBS)可无参
功能基因挖掘遗传图谱(GBS)可无参
N个BSA性状定位项目(N=性状数)
IF:10~30分
IF:10~30分IF:3~6分
IF:3~6分
IF:3~6分
IF:1~6分IF:3~10分
IF:3~10分
自然群体群体

高通量测序常用名词汇总
一代测序技术: 即传统的Sanger 测序法,Sanger 法是根据核苷酸在待定序列模板上
的引
物点开始,随机在某一个特定的碱基处终 并且在每个碱基后面进行荧光标
止, 记, 产生以A、
T、C、G结束的四组不同长度的一系列核苷酸,每一次序列测定由一套四个单独的反应构 成,每个反应含有所有四种脱氧核苷酸三磷 (dNTP),并混入限量的一种不同的双脱氧
酸 核
苷三磷酸(ddNTP)。由于ddNTP 缺乏延伸所需要的 3-0H 基团,使延长的寡聚核苷酸
选择
性地在 G、A、T或C处终止,使反应得到一组长几百至几千碱基的链终止产物。它们具有
相比,二代测序技术可以一次对几十万到几百万条核酸分子同时进行序列测定, 从而使得对
一个物种的转录组和基因组进行细致全貌的分析成为可能,所以又被称为深度测序
Deep
sequencing )。NGS 主要的平台有 Roche ( 454 & 454 +) , Illumina ( HiSeq
2000/2500 、
GA IIx 、MiSeq ), ABI SOLiD 等。
是DNA或RNA分子上具有遗传信息的特定核苷酸序列
基因通过复制把遗传信息传递给下一代,使后代出现与亲代相似的性状。
DNA : Deoxyribonucleic acid ,脱氧核糖核酸,一个脱氧核苷酸分子由三部分组成:含氮碱
基、脱氧核糖、磷酸。脱氧核糖核酸通 3',5'- 磷酸二酯键按一定的顺序彼此相连构成长
过 链,
即DNA链,DNA链上特定的核苷酸序列包含有生物的遗传信息, 是绝大部分生物遗传信息
的载体。
RNA : Ribonucleic ,,核糖核酸,一个核糖核苷酸分子由碱基,核糖和磷酸构成。
Acid 核
糖核苷酸经磷酯键缩合而成长链状分子称之
为 RNA链。RNA是存在于生物细胞以及部分病
毒、类病毒中的遗传信息载体。不同种类的 RNA链长不同,行使各式各样的生物功能,如

首页 科技服务 测序指南 基因课堂 市场活动与进展 文章成果 关于我们
随着高通量测序技术不断完善,研究策略也由单一组学逐渐发展为多组学联合分析,为物种生物学问题的研究开启了更多切入点。
Hi-C多组学研究意义
Hi-C多组学技术参数
Hi-C多组学技术路线
Hi-C多组学代表文献
Hi-C多组学案例解析
图1 癌细胞与正常细胞的TAD比较
图2 特有的TAD边界与CNV的联合分析物 种人[1]人[2]哺乳动物[3]果蝇[4]发表时间2016.042014.082014.042013.05期 刊Genome ResearchGenes & DevelopmentNat Rev GenetMolecular CellIF11.35110.04236.97814.018linklinklinklink应用Hi-C和转录组联合分析揭示正常细胞与前列腺癌细胞之间的差异。Hi-C和转录组联合分析揭示激素处理前后的乳腺癌细胞的差异。Hi-C和转录组联合分析揭示蛋白CTCF对转录调控的影响。Hi-C和转录组联合分析揭示绝缘子在空间上的作用。提供领先的基因组学解决方案Leading Edge Genomic Services & Solutions
XX样本XX细胞系转录组测序差异基因分析TAD检测XX样本XX细胞系Hi-C测序全基因组重测序互作分析CNV的检测差异互作区间互作强度分析 差异互作区间表达量差异分析 特有的TAD与CNV联合分析TAD与转录组联合分析Hi-C技术新方向Hi-C多组学
Hi-C技术与转录组联合分析,可以扩宽研究的角度和深度,探索细胞中三维染色质结构变化与基因表达调控之间的关系。1Hi-C技术与全基因组重测序联合分析,可以探究三维染色质结构变化与基因组大结构变异之间的联系,更好的解析疾病产生的原因等生物学问题。2Hi-C技术与全基因组重测序、转录组联合分析能够在DNA、RNA以及三维空间结构上,以多视角的层面揭示各类生物学问题。3

基因组测序技术在微生物分类学中的应用
1. 引言
微生物是地球上最古老、最广泛分布的生物群体之一,对地球生态系统的功能和稳定性具有重要影响。微生物分类学是对微生物进行分类和鉴定的科学,它为我们了解微生物的多样性、功能和进化提供了基础。基因组测序技术作为一种快速、准确、高通量的方法,已经广泛应用于微生物分类学领域。本文将重点介绍基因组测序技术在微生物分类学中的应用。
2. 基因组测序技术概述
基因组测序技术是指对一个个体或一个群体中所有基因组DNA进行高通量测序的方法。目前常用的基因组测序技术包括Sanger测序、454
pyrosequencing、Illumina/Solexa测序和Ion Torrent等。这些技术具有高通量、高准确性和低成本等优点,为微生物分类学研究提供了强有力的工具。
3. 微生物多样性研究
3.1 16S rRNA基因分析
16S rRNA是细菌和古细菌中高度保守的核糖体RNA分子,其在不同种类之间具有一定的序列差异。通过对微生物样品中的16S rRNA基因进行PCR扩增和测序,可以获得微生物的16S rRNA序列信息,从而对微生物进行分类和鉴定。基因组测序技术可以大大提高16S rRNA基因测序的通量和准确性,从而更全面、更准确地了解微生物的多样性。
3.2 全基因组测序
全基因组测序是对一个个体或一个群体中所有基因组DNA进行高通量测序,并获得其完整的DNA序列信息。通过全基因组测序可以获得微生物所有基因的信息,包括编码蛋白质的基因、非编码RNA等。这种方法可以更全面地了解微生物的功能和进化关系,并为微生物分类学研究提供更多有价值的信息。
4. 微生物分类学研究进展
4.1 新种类发现
传统方法对于一些难以培养、形态相似或具有相似代谢特征等特殊群体中新种类或新属类别往往难以鉴定。而利用基因组测序技术,可以通过比较其16S rRNA或全基因组DNA与已知菌株进行比对,从而确定其分类位置。这种方法大大加快了新种类的发现速度,为微生物分类学的发展做出了贡献。