世界最佳新药注册程序:欧洲法规与中国相关法规的比较
- 格式:ppt
- 大小:2.97 MB
- 文档页数:59


ind申报材料 中美对比
中美IND申报材料存在一些差异,以下是具体的对比:
1. 申报流程:中国和美国的IND申报流程大致相同,包括临床前研究、临床试验申请、临床试验和新药申请等阶段。然而,在具体细节和执行层面上,两国存在显著的差异。例如,中国需要进行预查验,即检查临床前动物实验的伦理审查以及临床试验申请资料的完整性。美国则有“滚动审查”制度,即申请人可以随时向FDA提交申请资料,FDA进行实时审评。
2. 法规要求:在IND阶段,中美两国都要求提交相关的研究资料以证明药物的安全性和有效性。这些资料包括但不限于药理毒理学研究、药代动力学研究等。然而,两国在某些具体法规要求上存在差异。例如,中国要求申请人提交药品生产工艺和质量控制标准,而美国则要求申请人提交药品的化学成分信息。
3. 文件要求:中美IND申报材料在文件要求方面存在相似之处,例如都需要提交药品的处方、生产工艺、质量标准等信息。但在具体细节上,两国也存在差异。例如,中国要求申请人提交药品的处方组成、工艺描述、质量标准等详细信息,而美国则要求申请人提交药品的合成路线、化学成分、纯度等信息。
4. 通用技术文件格式:中美IND申报材料都需要按照通用技术文件格式(CTD)提交资料。CTD是一种用于药品申报的通用文件格式标准,旨在提高药品申报资料的规范性和可读性。尽管两国都采用CTD格式,但在具体实施上存在一些差异。例如,中国在CTD格式中需要申请人按照特定的格式和要求填写各个部分的内容,而美国则允许申请人根据实际情况和需要进行适当调整。
总的来说,中美IND申报材料在申报流程、法规要求、文件要求和通用技术文件格式等方面存在差异。因此,在进行中美IND申报时,申请人需要充分了解两国的要求和规定,并按照相关要求准备申报材料。同时,对于两国存在的差异,申请人需要进行针对性的调整和补充,以确保申报材料的完整性和准确性。

国内外药品监管模式对比研究及其借鉴意义
随着全球化的不断深入,药品也成为了一个涉及全球社会利益的重要话题。药品作为一种特殊的消费品,其安全和有效性都需要严格监管,而药品监管模式的不同也直接影响了药品的质量和使用效果。本文将探讨国内外药品监管模式的对比研究及其借鉴意义。
一、国内药品监管现状
目前,我国药品监管工作主要由国家药监局负责,其职责主要包括批准药品注册和备案、监管药品生产经营、药品信息发布等。国家药监局是一个行政部门,其主要考虑的是在政策层面上规范药品监管行为和执法。同时,我国还有药品监督管理部门、药品药材检验机构等一系列专业机构,致力于维护药品的质量和安全。
然而,我国药品监管存在一些缺陷。首先,监管部门在药品安全事故发生时,一些监管责任人没有及时履行职责,导致了药品安全问题的进一步扩大。其次,我国药品市场存在复杂的利益关系,监管部门的管理难度较大。例如,一些药品经销商不合规经营,有些生产企业也因为利益而不合规操作。这些都给药品监管部门带来了很大的挑战。
二、国外药品监管现状 国外药品监管机构与我国类似,主要包括FDA(美国食品药品监督管理局)、EMA(欧洲药物评估局)等。这些机构不仅有权对药品从生产到销售的全过程进行监管,还会进行药品的临床试验,以确保药品的安全和有效性。美国FDA甚至对药品的广告宣传活动和市场信息也进行监管,以确保广告信息真实有效、不误导消费者。
相较于我国,国外药品监管更加严格和权威。例如FDA,其审批药品的时间和流程很长,需要对药品的生产过程、药品成分、安全性和有效性等方面进行全面的检查评估。这些步骤虽然繁琐,但能够确保药品的高质量和安全性。此外,国外药品监管机构还会利用先进的技术手段,对药品进行全面监管,以确保消费者的用药安全。
三、对比与借鉴
国内外药品监管机构的对比可以得知,国外的药品监管更加严格和高效。国外监管机构除了对药品生产过程的监管以外,还会对药品从临床试验到销售和宣传的全过程进行监管,以确保消费者的用药安全和合法权益。这一点在我国药品监管机构中尚未得到很好的实践。

我国药品质量管理体系与欧美发达国家药品质量管理体系的异同
摘要:通过对我国药品质量管理体系与欧美发达国家药品管理体系的构成及其要素的介绍,比较他们的异同,促进药品生产企业质量管理水平的普遍提高。说明建立一个全面质量管理体系对保障药品质量的重要性,构建符合新版GMP。
关键词:质量管理体系;新版GMP;
1、国外药品质量管理发展历史和现状
在五六十年代经过了一系列的国际药害事件后,美国政府发现单以抽查检验为药品质量的保障方式存在较大的不确定性,而药品生产过程控制对保证药品质量发挥着重要的作用。正是由于对保障药品质量的认识理念从药品生产结果的抽检到药品生产过程的保证之巨大转变,1962年美国食品药品监督管理局(简称FDA)制定实施了全球首版GMP。FDA工作人员将对国内外的生产企业,进口的各类产品进行跟踪检查,并抽取样品进行检查【1】,另外,FDA对药品上市后的不良反应也非常重视,每年约收到400000份产品不良报告【2】在此之后,欧美各国纷纷建立了各自的药品GMP,1975年WHO也正式向全球公布GMP规范。1982年在国内医药产业不断发展的推动下,参照先进国家药品质量保障的经验和理念,。国际ICH组织分别于2005年11月颁布《Q9药品风险管理》,2008年6月颁布《QlO药品质量体系》,2008年7月欧盟将《Q9药品风险管理》的内容纳入GMP指南中,2009年4月美国FDA参照ICH发布了《符合制药cGMP法规要求的质量系统》指导性文件,正式将风险管理的理念引人药品质量体系建设中。国际上,主要发达国家一系列介绍和推广新质量管理理念的指南文件相继颁布实施,标志着药品行业进入了一个质量管理的新纪元【3—6】。
1.1 欧盟 1994年以设在法国的欧洲药典委员会秘书处为基础成立了欧洲药品质量管理局(EDQM)。EDQM主要功能之一是对上市后的仿制药品的监督管理,其主要监督手段是对产品的符合性签证和对通过欧洲各国官方药品检验所(OMCL)之间的欧洲网络系统来对药品的市场监督【7】.另外,上市后药品监督的另一种方法是不良反应监测【8—9】,例如最近在意大利发生了减肥药西布曲明的死亡事件,中止了该品种的上市销售【10】。
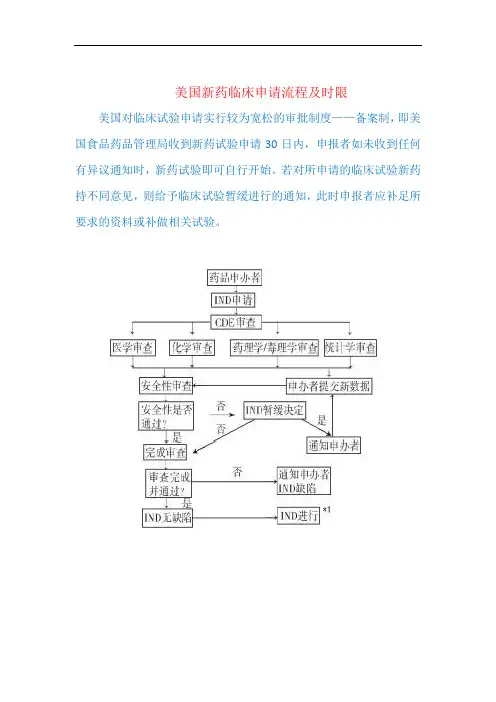
美国新药临床申请流程及时限
美国对临床试验申请实行较为宽松的审批制度——备案制,即美国食品药品管理局收到新药试验申请30日内,申报者如未收到任何有异议通知时,新药试验即可自行开始。若对所申请的临床试验新药持不同意见,则给予临床试验暂缓进行的通知,此时申报者应补足所要求的资料或补做相关试验。
中国新药临床申请流程及时限
我国对临床试验则实行严格的批准制,即要求申请者必须经国家食品药品监督管理局批准后,获得《药物临床试验批件》后方可开展临床试验。严格的审批制度涉及省级食品药品监督管理局、省(自治区、直辖市)药品检验所、药品审评中心、中国药品生物制品检定所、等多个部门,加之某些部门间缺乏有效的沟通协调,一定程度影响了临床试验的评审速度,使得我国临床试验的实际审批时间较美国长很多。
美国新药生产审批时限及流程
新药申请(New Drug Application,NDA):通过三个阶段的临床试验,公司将分析所有的试验数据。如果数据能够成功证明药物的安全性和有效性,公司将向FDA提出新药申请。新药申请必须包括公司所掌握的一切相关科学信息。典型的新药申请有10万页甚至更多。根据法律,FDA审核一份NDA的时限应该为6个月。但是几乎所有案例中的新药申请从首次提交到最终获得FDA批准的过程都超过了这个时限,现在平均大约为2年。
中国新药生产审批时限及流程
按照流程,新药从申报(现场检查)到获批需165~185个工作日,即7.5个月~8.5个月已有标准的药品从申报(现场检查)到获批需145个工作日,即6.6个月特殊药品及疫苗类需60个工作日,即2.7个月。
1.新药申办流程及时限
(一)新药生产审批
注1:特殊药品注册检验60日完成。
注2:技术审评中的120日/100日,120日是指新药审评时限,100日是指实行快速审批品种药品时限。药品审评中心技术审评的40日/25日,40日是指新药审评时限,25日是实行快速审批品种药品时限。