靶向金黄色色素合成蛋白CrtN的苯并六元含氧脂肪环类抗MRSA候选药物:设计、合成和药理学评价
- 格式:doc
- 大小:14.85 KB
- 文档页数:5
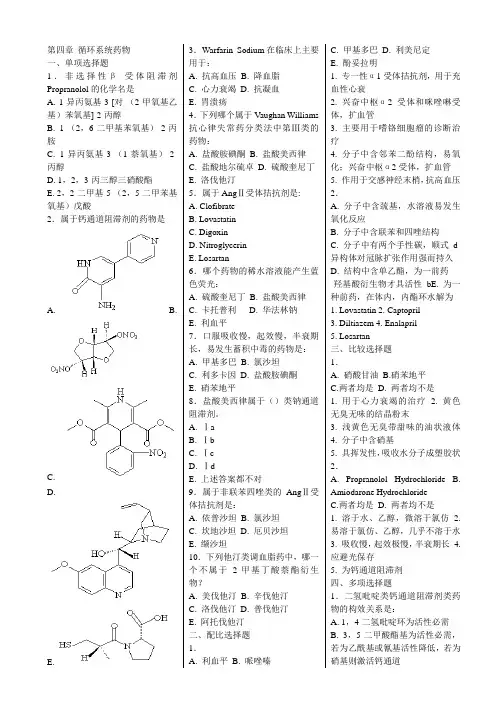
第四章 循环系统药物 一、单项选择题1.非选择性β-受体阻滞剂Propranolol 的化学名是A. 1-异丙氨基-3-[对-(2-甲氧基乙基)苯氧基]-2-丙醇B. 1-(2,6-二甲基苯氧基)-2-丙胺C. 1-异丙氨基-3-(1-萘氧基)-2-丙醇D. 1,2,3-丙三醇三硝酸酯E. 2,2-二甲基-5-(2,5-二甲苯基氧基)戊酸2.属于钙通道阻滞剂的药物是A. B.C.D.E. 3.Warfarin Sodium 在临床上主要用于:A. 抗高血压B. 降血脂C. 心力衰竭D. 抗凝血E. 胃溃疡 4.下列哪个属于Vaughan Williams 抗心律失常药分类法中第Ⅲ类的药物:A. 盐酸胺碘酮B. 盐酸美西律C. 盐酸地尔硫卓D. 硫酸奎尼丁E. 洛伐他汀5.属于Ang Ⅱ受体拮抗剂是: A. Clofibrate B. Lovastatin C. DigoxinD. NitroglycerinE. Losartan6.哪个药物的稀水溶液能产生蓝色荧光:A. 硫酸奎尼丁B. 盐酸美西律C. 卡托普利D. 华法林钠E. 利血平7.口服吸收慢,起效慢,半衰期长,易发生蓄积中毒的药物是: A. 甲基多巴 B. 氯沙坦C. 利多卡因D. 盐酸胺碘酮E. 硝苯地平8.盐酸美西律属于()类钠通道阻滞剂。
A. Ⅰa B. Ⅰb C. Ⅰc D. ⅠdE. 上述答案都不对9.属于非联苯四唑类的Ang Ⅱ受体拮抗剂是:A. 依普沙坦B. 氯沙坦C. 坎地沙坦D. 厄贝沙坦E. 缬沙坦10.下列他汀类调血脂药中,哪一个不属于2-甲基丁酸萘酯衍生物?A. 美伐他汀B. 辛伐他汀C. 洛伐他汀D. 普伐他汀E. 阿托伐他汀 二、配比选择题 1.A. 利血平B. 哌唑嗪C. 甲基多巴D. 利美尼定E. 酚妥拉明1. 专一性α1受体拮抗剂,用于充血性心衰2. 兴奋中枢α2受体和咪唑啉受体,扩血管3. 主要用于嗜铬细胞瘤的诊断治疗4. 分子中含邻苯二酚结构,易氧化;兴奋中枢α2受体,扩血管5. 作用于交感神经末梢,抗高血压 2.A. 分子中含巯基,水溶液易发生氧化反应B. 分子中含联苯和四唑结构C. 分子中有两个手性碳,顺式d-异构体对冠脉扩张作用强而持久D. 结构中含单乙酯,为一前药 -羟基酸衍生物才具活性bE. 为一种前药,在体内,内酯环水解为 1. Lovastatin 2. Captopril 3. Diltiazem 4. Enalapril 5. Losartan三、比较选择题 1.A. 硝酸甘油B.硝苯地平C.两者均是D. 两者均不是1. 用于心力衰竭的治疗2. 黄色无臭无味的结晶粉末3. 浅黄色无臭带甜味的油状液体4. 分子中含硝基5. 具挥发性,吸收水分子成塑胶状 2.A. Propranolol HydrochlorideB. Amiodarone HydrochlorideC.两者均是D. 两者均不是1. 溶于水、乙醇,微溶于氯仿2.易溶于氯仿、乙醇,几乎不溶于水3. 吸收慢,起效极慢,半衰期长4.应避光保存5. 为钙通道阻滞剂四、多项选择题1.二氢吡啶类钙通道阻滞剂类药物的构效关系是:A. 1,4-二氢吡啶环为活性必需B. 3,5-二甲酸酯基为活性必需,若为乙酰基或氰基活性降低,若为硝基则激活钙通道C. 3,5-取代酯基不同,4-位为手性碳,酯基大小对活性影响小,但不对称酯影响作用部位D. 4-位取代基与活性关系(增加):H<甲基<环烷基<苯基或取代苯基E. 4-位取代苯基若邻、间位有吸电子基团取代时活性较佳,对位取代活性下降2.属于选择性β1受体阻滞剂有:A. 阿替洛尔B. 美托洛尔C. 拉贝洛尔D. 吲哚洛尔E. 倍他洛尔3.Quinidine的体内代谢途径包括:A. 喹啉环2`-位发生羟基化B. O-去甲基化C. 奎核碱环8-位羟基化D. 奎核碱环2-位羟基化E. 奎核碱环3-位乙烯基还原4.NO供体药物吗多明在临床上用于:A. 扩血管B. 缓解心绞痛C. 抗血栓D. 哮喘E. 高血脂5.影响血清中胆固醇和甘油三酯代谢的药物是:A.B. C.D.E.6.硝苯地平的合成原料有:A. a-萘酚B. 氨水C. 苯并呋喃D. 邻硝基苯甲醛E. 乙酰乙酸甲酯7.盐酸维拉帕米的体内主要代谢产物是A. N–去烷基化合物B. O-去甲基化合物C. N–去乙基化合物D. N–去甲基化合物E. S-氧化8.下列关于抗血栓药氯吡格雷的描述,正确的是A. 属于噻吩并四氢吡啶衍生物 B. 分子中含一个手性碳C. 不能被碱水解D.是一个抗凝血药E. 属于ADP受体拮抗剂9.作用于神经末梢的降压药有A. 哌唑嗪B. 利血平C. 甲基多巴D. 胍乙啶E. 酚妥拉明10.关于地高辛的说法,错误的是A. 结构中含三个a-D-洋地黄毒糖 B. C17上连接一个六元内酯环C. 属于半合成的天然甙类药物 D. 能抑制磷酸二酯酶活性E. 能抑制Na+/K+-ATP酶活性五、问答题1.以Propranolol为例,分析芳氧丙醇类b-受体阻滞剂的结构特点及构效关系。

可编辑修改精选全文完整版中国多黏菌素类抗菌药物临床合理应用多学科专家共识(2021全文版)多黏菌素类抗菌药物问世于20世纪50年代末,后来因同样有效但更安全的新药不断问世而逐渐淡出临床。
到了20世纪80年代,随着多重耐药革兰阴性菌的增多,此类药物重新受到重视而重返临床。
但由于其上市时间早,至今仍有许多问题给临床造成困惑。
因此,由中国医药教育协会感染疾病专业委员会牵头,联合多学科相关领域著名专家与权威学术组织共同编写本共识,全文以问答的方式展示,分为11个部分,37个问题,10条推荐意见,希望为临床医生合理应用多黏菌素类药物提供切实可行的参考。
多黏菌素类抗菌药物问世于20世纪50年代末,当时主要用于耐药铜绿假单胞菌感染的治疗。
后来因同样有效且安全性更好的新药不断问世,此类药物逐渐淡出临床。
到了20世纪80年代,因多重耐药(multi-drug resistance,MDR)革兰阴性菌的增多,具有特殊抗菌机制的多黏菌素类药物重新受到重视而重返临床。
到目前为止,虽不断有各种新的治疗MDR革兰阴性菌感染的新药产生,但多黏菌素类药物的临床应用价值仍无法取代。
因上市时间早,多黏菌素类药物没有经历过现代药物开发过程中的各种严格验证,至今仍有许多问题给临床医师和药师们造成困惑。
因此,由中国医药教育协会感染疾病专业委员会牵头,联合多学科相关领域著名专家与权威学术组织共同编写本共识,在充分参考国内外此类药品循证医学和临床应用的基础上,历经一年多时间,反复讨论,九易其稿,最终成文。
其主要目的是为临床医生合理应用多黏菌素类药物提供切实可行的参考。
一、共识的背景问题1:多黏菌素类药物为什么会重新回归临床?多黏菌素类抗菌药物主要包括硫酸多黏菌素B(polymyxin B sulfate)、多黏菌素E甲磺酸钠(polymyxin E methanesulfonate sodium)和硫酸多黏菌素E(polymyxin E sulfate)。

实验一TLC铺板、干燥、活化、色谱用硅胶柱的填装1.硅胶薄层色谱板的制备、干燥和活化薄层色谱中的吸附剂是铺在玻璃、塑料或金属片或薄板上的较薄的、均匀的一层细粉状物质,因支持剂的种类、制备方法和选用溶剂的不同,可按吸附、分配或二者结合的方式达到分离化合物的目的。
可以通过比较斑点的R f值,或将未知样品与对照品在同一板上展开至同样高度,对样品进行初步的鉴定。
还可通过比较可见斑点的大小进行半定量的判断。
还可以通过光密度测量法实现定量测定。
TLC中涂布的物质与柱色谱用的吸附剂非常相似,如硅胶、氧化铝、聚酰胺等,只是它们的颗粒更细一些,一般直径为5~40μm。
有些还含有石膏、淀粉等粘合剂以增强涂层与薄板的粘合力。
有时里面还含有荧光指示剂(如硅酸锌等),在254或365nm的紫外光下能显示荧光,可借此对分离的斑点进行检测。
到目前为止,硅胶是最常用的薄层色谱吸附剂。
在涂布吸附剂时,用于排列和放置薄板的排列盘和具有平整表面的薄板是必需的。
而涂布器也很常用,当它从玻璃板上移过时,会在板的表面均匀铺上所需厚度的吸附剂涂层。
(1)实验目的掌握硅胶薄层色谱板的制备方法。
(2)仪器和试剂①玻璃板(5×10cm或10×20cm,洁净且干燥);②薄层色谱用硅胶G;③%羧甲基纤维素钠水溶液;(3)实验步骤①把玻璃板在排列盘中依次相邻放好,置涂布器于其中一端。
②在具塞锥形瓶中把一份硅胶G和2~3份CMC-Na溶液混合,并用力振摇30秒。
③把混好的糊倒入涂布器中,均匀地移动涂布器至排列盘的另一端后,移开涂布器。
④铺好的板静置5分钟,然后把它们面朝上移至一个水平的平面上,阴干。
⑤把阴干后的板在105℃的烘箱中烘30分钟。
⑥待板凉至室温后,置干燥器中保存。
2.色谱用硅胶柱的填装液相柱色谱可以是液-固色谱或液一液色谱。
如果固定相是吸附剂,也称为液相吸附色谱.若为离子交换物质,就称为离子交换色谱;若为非离子的聚合物,如聚苯乙烯或hadex,则称为凝胶渗透色谱、凝胶过滤色谱或分子排阻色谱。

一种新型万古霉素衍生物的合成刘林发布时间:2021-08-31T06:22:25.104Z 来源:《中国科技人才》2021年第15期作者:刘林周金幸李腾飞[导读] 本文设计了一个全新的万古霉素拼接片段的方式,帮助研发万古霉素衍生物药物。
经过制备,化合物的MS表征证明获得的物质为设计目标化合物,能满足抗菌的要求,给万古霉素衍生物合成提供思路。
正大天晴药业集团股份有限公司江苏连云港 222062摘要:本文设计了一个全新的万古霉素拼接片段的方式,帮助研发万古霉素衍生物药物。
经过制备,化合物的MS表征证明获得的物质为设计目标化合物,能满足抗菌的要求,给万古霉素衍生物合成提供思路。
关键词:万古霉素;衍生物;合成引言:糖肽类抗生素是一种高修适度的七肽骨架,通过和细菌细胞壁五肽末端残基结合,可以产生具有较强抗菌活性的抗生素,其中万古霉素是第一个被发现的糖肽类抗生素,由于其具有抑菌作用,并且结构比较特殊,所以成为很多糖肽类抗生素的原型,特别在耐甲氧西林金葡萄引起的感染具有较好的效果[1]。
经过1987年万古霉素耐药菌出现,到2002年万古霉素耐药肠球菌在美国的感染人数增加了30% [2]。
但是万古霉素的结构比较复杂,所以结合其耐药性设计全新的肽骨架比较困难,为了能解决耐药问题,目前采用了对万古霉素末端的氨基和糖环进行修饰,获得万古霉素的衍生物,对解决抗药性问题有良好的效果。
1 万古霉素衍生物合成实验 1.1 仪器和试剂选择实验中使用核磁共振仪,以CDCl3作为溶剂,内标为TMS;使用API2000液相质谱联用仪。
万古霉素是盐酸盐的纯度在98%以上;使用纯度超过99%的双三苯基磷酸氯化钯;其他材料均从市面购买。
1.2 合成过程1.2.1 4-氯联苯甲醛化合物合成在500毫升的三颈瓶中,加入25g对捏苯甲醛和23.3g对氯苯硼酸,然后加入60毫升水和240毫升二甲醚溶解原料,再加入39g固体碳酸钾。
之后对三颈瓶抽真空,然后置换氮气,经过3此置换后,加入1g双三苯基氯化钯,再抽真空,然后置换氮气3次。

第三章填空题1. 反应溶剂直接影响化学反应的反应速度、反应方向、反应深度、产品构型等。
2.水、乙醇、乙酸、乙二胺等属于质子溶剂,乙醚、二氯甲烷、丙酮、吡啶等属于非质子溶剂,甲苯、正己烷、环己烷等属于惰性溶剂。
选择题1.可逆反应属于(A )A. 复杂反应B. 平行反应C. 基元反应D. 简单反应3.在溶剂的作用描述中,不正确的是( B )A.使反应分子能够分布均匀、增加分子间碰撞和接触的机会、有利于传热和散热。
B.溶剂必须是易挥发的C.溶剂必须是惰性的,不能与反应物或生成物反应D.溶剂直接影响化学反应的反应速度、反应方向、反应深度、产品构型等简答题单分子反应,双分子反应,一级反应,二级反应之间的关系?答:在某基元反应过程中若只有一分子参与反应则称为单分子反应。
多数的一级反应为单分子反应。
当相同或不同的两分子碰撞时相互作用而产生的反应称为双子反应,即为二级反应。
化学合成药物工艺研究的主要内容是什么?答:化学合成药物工艺研究的主要内容有7个:配料比、溶剂、温度和压力、催化剂、反应时间及其监控、后处理、产品的纯化和检验。
第四章1.简述天然提取法制备手性药物的一般步骤?答:①酶法分析②酶催化的还原反应③酶催化的氧化反映④酶催化的不对称合成反应⑤酶催化的转氨基化作用2.什么是手性药物,其构型与活性的特点是什么?答:具有药理活性的手性化合物就是手性药物。
①两个对映体具有相同的药理作用。
②两个对映体中的一个有药理活性,另一个则无明显作用。
③两个对映体的药理作用不同,上市后用于不同的适应症。
④在消旋体中增加一种对映体的比例可调整临床疗效。
⑤两个对映体中的一个有治疗价值,另一个则有不良作用。
第五章选择题1、氯霉素抗菌谱广,而最主要的不良反应是(D)A、二重感染B、胃肠道反应C、对肝脏严重损害D、对造血系统的毒性E、影响骨、牙生长2、与氯霉素特点不符的是(A)A、口服难吸收B、易透过血脑屏障C、适用于伤寒的治疗D、骨髓毒性明显E、对早产儿、新生儿可引起灰婴综合症3、氯霉素的下述不良反应中,哪项是与剂量和疗程无关的严重反应?(A)A、不可逆的再生障碍性贫血B、灰婴综合征C、可逆的各类血细胞减少D、溶血性贫血E、出血现象4、可能拮抗氯霉素抗菌作用的药物是(C)A、磺胺类B、庆大霉素C、红霉素D、四环素E、多黏菌素填空题1、氯霉素有2 个手性中心,4 个光学异构体.2、利用氯霉素生产工艺中副产物可以生产除草剂-杀草铵.邻硝基乙苯简答题1、对硝其-α-溴代苯乙酮的制备中水含量有何影响?为什么?答:水分存在时与溴化氢反应,诱导其延长或基本不起反应,因此对硝基苯乙酮溴代反应时,水分存在不利于反应,必段严格控制溶剂的水份第六章选择题:1.具有明显抗癌作用的紫杉醇来源于。

莫西沙星的调研总结莫西沙星莫西沙星是人工合成抗菌药的一种,是由德国Bayer公司研制的第四代氟喹诺酮类广谱抗菌药物,为DNA拓扑异构酶抑制剂;本文主要从产品简介、药理学特点和临床应用研究进展、以及市场分析等方面来做介绍:一、简介1、发展历程莫西沙星为人工合成抗菌药喹诺酮类(喹诺酮类,又称吡酮酸类或吡啶酮酸类,是一类较新的合成抗菌药。
喹诺酮类以细菌的脱氧核糖核酸(DNA)为靶。
妨碍DNA回旋酶,进一步造成细菌DNA的不可逆损害,达到抗菌效果。
本类药物与许多抗菌药物间无交叉耐药性,是理想的抗菌药)的一种,具体发展如下: -第一代: 1962年萘啶酸(nalidixic acid),抗菌谱窄,对大多数G菌有效,但对铜绿假单胞菌无效,抗菌力弱,口服吸收差,血药浓度低,AR多,现已淘汰;第二代:1974年吡哌酸(pipemidic acid),抗菌活性,萘啶酸, 抗菌谱-+由G菌扩大到部分G菌,对铜绿假单胞菌有效,但血药浓度低,主要用于尿道或肠道感染; 第三代:1979年诺氟沙星(norfloxacin)、环丙沙星等口服易吸收,血药浓度大为提高,分布广,扩大和增强了抗菌活性:+G球菌、衣原体、支原体、军团菌及分枝杆菌; 第四代:1999年莫西沙星(moxifloxacin),加替沙星等吸收快,体内分布广,抗菌作用进一步增强:既保留了钱三代抗-+G菌的活性,又明显增强了G菌的活性,特别是增加了对厌氧菌的抗菌活性;临床既用于需氧菌感染,也用于厌氧菌感染,还可用于混合感染,对绝大多数致病菌的综合临床疗效已经达到或超过了β,内酰胺类抗生素;2、基本情况(莫西沙星)通用名:Moxifloxacin商品名:Actira(Bayer)CAS登录号:151096-09-2CAS登录号(盐酸盐):186826-86-8化学名:1-环丙基-6-氟-1,4-二氢-8-甲氧基-7-[(4aS,7aS)-八氢-6H-吡咯并[3,4-b]吡啶-6-基]-4-氧代-3-喹啉羧酸1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6 H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid结构式:分子式(相对分子量):CHFNO(401.43) 212434理化性质(盐酸盐):白色至淡黄色结晶性粉末,熔点324-325? 上市情况:1999年在德国首次上市,同年12月在美国上市;2002年在我国上市; 适应症:细菌感染用法与用量:拜复乐片,一日1次,每次400mg。
靶向金黄色色素合成蛋白CrtN的苯并六元含氧脂肪环类抗MRSA
候选药物:设计、合成和药理学评价
金黄色葡萄球菌是一种致病力及致死率都很高的革兰氏阳性菌,它能在抗生
素环境下快速突变为耐药金黄色葡萄球菌
(methicillin-resistantS.aureus,MRSA),近年来几乎所有上市抗生素都出现了
相应的MRSA菌。为了缓解抗生素耐药造成的公众健康危机,开发一类有别于传统
抗生素的新型治疗策略尤其迫切。
细菌在生长和繁殖过程中会分泌出各种毒力因子,这些毒力因子不仅能随宿
主环境变化进行自适性调节,为细菌营造出更适宜的生长环境,而且能够帮助细
菌躲避宿主的免疫杀伤,更重要的是毒力因子不直接影响细菌的生长和繁殖。因
此以毒力因子为靶点的新型抗细菌感染治疗策略,能够在“不杀菌”的前提下降
低细菌对人体的伤害,并有效避免细菌的耐药性突变。
金黄色色素是金黄色葡萄球菌体内特有的毒力因子,不仅能够帮助细菌抵御
人体的免疫杀伤(ROS杀伤机制),还会加速人体器官和组织的坏死,因此通过阻
断金黄色色素的合成和产生,能高效专一的治疗MRSA引起的感染和损伤。金黄色
色素的合成通路受操纵子crtOPQMN调控,CrtN是其中最重要的色素合成通路之
一。
在课题组之前的研究中,我们通过自建“老药库”发现了已上市抗真菌药物
盐酸萘替芬,能通过靶向CrtN来阻断金黄色色素的合成。我们以盐酸萘替芬为先
导化合物,将其结构中的萘环替换成苯并脂肪环,发展了第一代靶向CrtN的苯并
脂肪环类色素抑制剂,特别是候选药物44表现出了极佳的体内外药效。
但是在随后的研究中,我们发现候选药物44存在水溶性差,心脏hERG毒性强
等成药性缺陷。因此,本论文工作的主要目标就是发现一类具有良好体内外活性,
同时能够克服第一代色素抑制剂缺陷的新型药物小分子。
我们首先以同是苯并脂肪环衍生物的46为先导化合物,分析了化合物水溶
性差的主要根源在于结构中存在疏水性苯并脂肪环,并验证性在46的结构中插
入了一个亲水性氧原子,合成了具有苯并二氢吡喃新骨架衍生物60。60不仅保持
了较好的色素抑制活性(IC50 = 4.6±0.2nM),同时水溶性比46提高了20多倍
(13.2 mg/mL)。
随后以苯并二氢吡喃为母核结构,在新化合物60的其他结构区域进行优化
和改造,在保持其色素抑制活性和水溶性的基础上,改善心脏hERG毒性。在先导
化合物46的烯丙基碳端位(A区域)引入环烷基、取代芳基及杂芳基等,设计合成
了38个新衍生物(47-84),并考察它们对S.aureusNewman的色素抑制能力。
测试结果不仅清晰的展示了化合物结构与抑制活性之间的关系,而且发现了
9个化合物的色素抑制活性达到了个位数纳摩尔级别,通过构效关系结果对化合
物进行初步筛选后,4-二苯基取代基片段的化合物67表现出了最强的色素抑制
活性(IC50 = 3.8±0.1nM)。随后将烯烃端位碳取代固定为对二苯基取代片段,
开展了氮上(R)和链上取代变化(Linker)的考察。
对这两部分的考察共合成了 10个化合物(85-94),根据色素抑制结果可知,
氮上甲基被替换成氢原子、乙基或异丙基后(85-87),色素抑制活性丧失,链上双
键被替换或剔除(89-93)也会丧失色素抑制活性,而引入共轭多烯基团后,色素抑
制活性反而有所上升。色素抑制活性较好的衍生物53、56、57、58、59、60、
63、66、67、81、88、94被优选进行水溶性和心脏hERG毒性方面的筛选,化合
物67和88凭借出色的色素抑制活性、较低的hERG毒性和良好的水溶性,被选中
进行随后的药理筛选实验。
在CrtN酶抑制活性试验中,两个化合物均表现出较好的酶抑制活性
(IC50<300nM)。在MRSA菌株色素抑制试验中,四组MRSA菌,USA400MW2、USA300
LAC、Mu50和NRS271菌株被用来考察化合物的色素抑制活性,结果显示67和88
均表现出良好的MRSA色素抑制活性,但候选药物88在Mu50和NRS271菌株中的
表现更佳(IC50 = 0.36±0.1 nM inMu50和IC50=0.4±0.1 nMinNRS271),我们将
更有成药潜质的88作为候选药物开始进一步的考察。
利用高效液相色谱法测定CrtM的催化产物,证明了候选药物88确实作用在
CrtN色素合成通路。随后设置了四种不同的体外实验,详细考察与候选药物88
共孵育的MRSA菌在免疫清除上的变化,综合各实验结果可知,候选药物88能够在
“不杀菌”的情况下,极大的削弱了细菌的毒力和繁殖能力。
鉴于第一代色素抑制体内药效评价中存在的缺陷,在本论文工作中,通过增
加多种多重耐药MRSA菌株为受试菌、引入万古霉素和利奈唑胺作为阳性对照物、
增加了对给药剂量(0.1mgb.i.d.和0.4mgb.i.d.)和给药模式(前给药模式和正
常给药模式)的考察,全面评价了候选药物88的体内抗毒活性。实验结果表明候
选药物88在S.aureus Newman菌株、中度万古霉素耐药MRSA菌株Mu50和重度
利奈唑胺耐药MRSA菌株NRS271的小鼠感染脓肿实验中均表现出较好的治疗效果,
甚至小剂量给药组表现出好于阳性药组的抑菌能力(部分脏器中抑菌率达到了
99%)。
为了进一步提高化合物水溶性,对88进行了盐型筛选,合成了 10类不同的
盐型,结果发现88磷酸盐具有较好的水溶性(12.9 mg/mL)。以上实验结果都证明
候选药物88可以作为抗毒力因子候选药物进行后续开发。
在苯并二氢吡喃类衍生物设计中,通过在苯并脂肪环结构中引入亲水性氧原
子,显著提高了化合物的水溶性,但是比较遗憾的是,由于在结构优化中引入了疏
水的4-二苯基和共轭双烯,虽极大地改善了hERG毒性,但同时消泯了亲水性氧原
子带来的水溶性提升。在本论文的第二部分工作中,为了进一步改善化合物水溶
性,同时拓展第二代靶向CrtN金黄色色素抑制剂的骨架类型,我们再次以46为先
导化合物,在苯并脂肪环结构中引入两个氧原子,设计并合成一类新颖的,具有苯
并二噁烷骨架的化合物。
苯并二噁烷候选药物的研究策略和发现过程与第一部分工作相同,共有46
个新结构衍生物被合成并测试了S.aureus Newman色素抑制活性,其中有11个位
数纳摩尔抑制活性的化合物被获得。通过构效关系结果筛选,活性较好的A05、
A06、A37、A38(ICs0<3.5nM)被选取测试水溶性、心脏hERG毒性、体外酶活
和其他MRSA菌色素抑制活性,候选药物A37在其中表现出最好的成药潜力,并进
行了体外“不杀菌”免疫清除实验,实验结果可知,A37在“不杀菌”的前提下,
高效的削弱了细菌在免疫环境下的存活能力。
在体内药效实验中,候选药物A37依旧表现出色,实验结果显示大部分A37
给药组显示出了良好的体内药效活性,小剂量给药组的体内活性好于提前给药组,
甚至小剂量正常给药组活性和两个阳性药组相当。所有这些数据都表明候选药物
A37是极具潜力的抗MRSA,VISA和LRSA感染的候选药物。
本论文工作的创新性主要表现在以下3个方面:(1)在苯并脂肪环中引入亲
水性氧原子,形成具有苯并二氢吡喃和苯并二噁烷骨架的第二代靶向CrtN金黄
色葡萄球菌色素抑制剂;(2)克服了苯并脂肪环类色素抑制剂的缺陷,提升了候选
药物的水溶性和改善了心脏hERG毒性;(3)优化了体内药效学评价模型,引入多
重耐药菌株作为受试菌株,同时增加万古霉素和利奈唑胺为阳性药对照组。