TCGA的乳腺癌RNA-seq数据WGCNA分析示例
- 格式:doc
- 大小:33.50 KB
- 文档页数:10

一文看懂WGCNA分析(2019更新版)发现我这个4年前的WGCNA分析教程可以排在自己最受欢迎的前10个教程里面了,而且直接以我这个授课代码出的SCI文章就有38篇了,当然不排除很多学员使用我的代码却不告知我,也不会致谢我。
不过,我这点战绩根本就算不上什么,其实这个WGCNA包已经是十多年前发表的了,仍然是广受好评及引用量一直在增加,破万也是指日可待。
大家首先可以看到3个教程:•2016-WGCNA-HCC-hub-gene.pdf 中文文章范例)•WGCNA_GBMTutorialHorvath.pdf•WGCNA_YeastTutorialHorvath.pdf其中第一个是我4年前的WGCNA分析教程最主要的参考文献,后面两个是英文教程,我相信你大概率是不会去看的,不过,我还是放在这里了。
(还是需要强调,这两个英文教程完整的展现了WGCNA的全部用法)然后你只需要简单浏览本文档,就可以在rstudio里面打开后缀是proj的文件,打开R代码,一步步跟着店铺!基本概念WGCNA其译为加权基因共表达网络分析。
该分析方法旨在寻找协同表达的基因模块(module),并探索基因网络与关注的表型之间的关联关系,以及网络中的核心基因。
适用于复杂的数据模式,推荐5组(或者15个样品)以上的数据。
一般可应用的研究方向有:不同器官或组织类型发育调控、同一组织不同发育调控、非生物胁迫不同时间点应答、病原菌侵染后不同时间点应答。
基本原理从方法上来讲,WGCNA分为表达量聚类分析和表型关联两部分,主要包括基因之间相关系数计算、基因模块的确定、共表达网络、模块与性状关联四个步骤。
第一步计算任意两个基因之间的相关系数(Person Coefficient)。
为了衡量两个基因是否具有相似表达模式,一般需要设置阈值来筛选,高于阈值的则认为是相似的。
但是这样如果将阈值设为0.8,那么很难说明0.8和0.79两个是有显著差别的。

xgene:WGS,突变与癌,RNA-seq,WES⼈类全基因组测序06SNP(single nucleotide polymorphism):有了10倍以上的覆盖深度以后,来确认SNP信息,就相当可靠了。
⼀个普通黄种⼈的基因组,与hg19这个参考基因组序列相⽐,会有350万个左右的SNP。
⼜有⼤概2万个是落在外显⼦上的,⽽⾮同义的SNP有⼤概9千个。
所谓⾮同义的SNP,就是这些SNP是会引起蛋⽩质的序列变化的。
indel:(insertion & deletion)是指⼩于50个bp以内的微⼩的插⼊、和缺失突变。
⼀个普通黄种⼈的基因组和hg19相⽐,约有50万个Indel。
其中落在外显⼦上的,⼤概在1千个左右。
那么Indel如果⼀旦落在外显⼦区域,它⼀定会引起蛋⽩质序列变化的。
如果它引起的是移码突变,那么在移码位点之后,所有氨基酸序列就和原来的序列完全不同。
如果它(基因)还能保持原来的阅读框,也会引起蛋⽩质中若⼲个氨基酸的增或者减。
SV: structure variation 染⾊体结构变异 1、染⾊体内部的位移2、染⾊体之间的位移3、⼤⽚段的缺失4、⼤⽚段的插⼊5、⼤⽚倍的加倍6、⼤⽚段的倒位 CNV :copy number variation 拷贝数变异,是指染⾊体⽚段的拷贝数变异:包括拷贝数增加,也包括拷贝数减少。
实际上,CNV是和结构变异(也就是SV)紧密相关的。
SV 中的⼤⽚段的增加、和⼤⽚段的缺失,会直接导致CNV的变化。
突变种类与癌症04基因拷贝数异常: 例如:HER2基因,如果HER2基因的拷贝数增加到6个,或者更多,它就⽐较容易引发乳腺癌。
赫赛汀(Herceptin)这个药,可以抑制HER2蛋⽩的活性,所以赫赛洒就对于由HER2基因拷贝数异常增加引发的乳腺癌,有⾮常好的治疗作⽤。
染⾊体结构变异: 强启动⼦替换了弱启动⼦,改变了某个基因在天然条件下的表达量。

TCGA的乳腺癌RNA-seq数据WGCNA分析示例WGCNA(WeightedCorrelationNetworkanalyi)是一个基于基因表达数据,构建基因共表达网络的方法。
WGCNA和差异基因分析(DEG)的差异在于DEG主要分析样本和样本之间的差异,而WGCNA主要分析的是基因和基因之间的关系。
WGCNA通过分析基因之间的关联关系,将基因区分为多个模块。
而最后通过这些模块和样本表型之间的关联性分析,寻找特定表型的分子特征。
网上例子千千万,但是大部分都是从文档翻译而来,要用起来还是有些费劲,要深入的可以移步这里:WGCNA##############etwd('E:/rawData/TCGA_DATA/TCGA-BRCA')ample=read.cv('ClinicalFull_matri某.t某t',ep='\\t',=1)dim(ample)#[1]1003e某pro=read.cv('Merge_matri某.t某t.cv.t某t',ep='\\t',=1)dim(e某pro)#[1]24991100数据读取完成,从上述结果可以看出100个样本,有24991个基因,这么多基因全部用来做WGCNA很显然没有必要,我们只要选择一些具有代表性的基因就够了,这里我们采取的方式是选择在100个样本中方差较大的那些基因(意味着在不同样本中变化较大)继续命令:m.var=apply(e某pro,1,var)e某pro.upper=e某pro[which(m.var>quantile(m.var,prob=eq(0,1,0.25))[4]),]##选择方差最大的前25%个基因作为后续WGCNA的输入数据集通过上述步骤拿到了6248个基因的表达谱作为WGCNA的输入数据集,进一步的我们需要看看样本之间的差异情况datE某pr=a.data.frame(t(e某pro.upper));gg=goodSampleGene(datE某pr,verboe=3);gg$allOK ampleTree=hclut(dit(datE某pr),method='average')plot(ampleTree,main='Samplecluteringtodetec toutlier',ub='',某lab='')从图中可看出大部分样本表现比较相近,而有两个离群样本,对后续的分析可能造成影响,我们需要将其去掉,共得到98个样本clut=cutreeStatic(ampleTree,cutHeight=80000,minSize=10)table(clut )#clut#01#298keepSample=(clut==1)datE某pr=datE某pr[keepSample,]nGene=ncol(datE某pr)nSample=nrow(datE某pr) ave(datE某pr,file='FPKM-01-dataInput.RData')得到最终的数据矩阵之后,我们需要确定软阈值,从代码中可以看出pickSoftThrehold 很简单,就两个参数,其他默认即可power=c(c(1:10),eq(from=12,to=20,by=2))ft=pickSoftThrehold(datE某pr,powerVector=power,verboe=5)##画图##par(mfrow=c(1,2));ce某1=0.9;plot(ft$fitIndice[,1],-ign(ft$fitIndice[,3])某ft$fitIndice[,2],某lab='SoftThrehold(power)',ylab='ScaleFreeTopologyModelFit,ignedR ^2',type='n',main=pate('Scaleindependence'));te某t(ft$fitIndice[,1],-ign(ft$fitIndice[,3])某ft$fitIndice[,2],label=power,ce某=ce某1,col='red');abline(h=0.90,col='red')plot(ft$fitIndice[,1],ft$fitIndice[,5],某lab='SoftThrehold(power)',ylab='MeanConnectivity',type='n', main=pate('Meanconnectivity'))te某t(ft$fitIndice[,1],ft$fitIndice[,5],label=power,ce某=ce某1,col='red')从图中可以看出这个软阈值选择7比较合适,选择软阈值7进行共表达模块挖掘pow=7net=blockwieModule(datE某pr,power=pow,ma某BlockSize=7000, TOMType='unigned',minModuleSize=30,reaignThrehold=0,mergeCutHeight=0.25,numericLabel=TRUE,pamRepectDendro=FALSE,aveTOM=TRUE,aveTOMFileBae='FPKM-TOM',verboe=3)table(net$color)#openagraphicwindow#izeGrWindow(12 ,9)#ConvertlabeltocolorforplottingmergedColor=label2color(net$color)#PlotthedendrogramandthemodulecolorunderneathplotDendroAndCo lor(net$dendrogram[[1]],mergedColor[net$blockGene[[1]]], groupLabel=c('Modulecolor','GS.weight'),dendroLabel=FALSE,ha ng=0.03,addGuide=TRUE,guideHang=0.05)从图中可以看出大部分基因在灰色区域,灰色部分一般认为是没有模块接受的,从这里也可以看出其实咱们选择的这些基因并不是特别好那么做到这一步了基本上共表达模块做完了,每个颜色代表一个共表达模块,统计看看各个模块下的基因个数:那么得到模块之后下一步该做啥呢,或许很多人到这就不知道如何继续分析了这里就需要咱们利用这些模块搞事情了,举个例子如果你是整合的数据(整合lnc与gene),那么同时在某个模块中的基因和lncRNA咱们可以认为是共表达的,这便是lnc-gene共表达关系的获得途径之一了,进一步你可以根据该模块的基因-lnc-基因之间的关系绘制出共表达网络今天咱们这里不讲这个,而是跟表型关联,咱们已经拿到了这98个样本的ER、PR、HER2阳性阴性信息,那么进一步的咱们可以看看哪些共表达模块跟ER、PR、HER2阴性最相关,代码如下:moduleLabelAutomatic= net$colormoduleColorAutomatic=label2color(moduleLabelAutomatic)moduleColorFemale=moduleCol orAutomaticME0=moduleEigengene(datE某pr,moduleColorFemale)$eigengeneMEFemale=orderME(ME0)ample=ample[match((datE某pr),pate0(gub('-','.',(ample)),'.01')),]#匹配98个样本数据trainDt=a.matri某(cbind(ifele(ample[,1]=='Poitive',0,1),#将阴性的样本标记为1ifele(ample[,2]=='Poitive',0,1),#将阴性的样本标记为1ifele(ample[,3]=='Poitive',0,1),#将阴性的样本标记为1ifele(ample[,1]=='Negative'&le[,2]=='Negative'&le[,3]= ='Negative',1,0))#将三阴性的样本标记为1)#得到一个表型的0-1矩阵modTraitCor=cor(MEFemale,trainDt,ue='p')colname(MEFemale)modTraitP=corPvalueStudent(modTraitCor,nSample)te某tMatri某=pate(ignif(modTraitCor,2),'\\n(',ignif(modTraitP,1),')',ep='')d im(te某tMatri某)=dim(modTraitCor)labeledHeatmap(Matri某=modTraitCor,某Label=colname(trainDt),yLabel=name(MEFemale),ySymbol=colname(modlue),colorLabel=FALSE,color=greenWhiteRed (50),te某tMatri某=te某tMatri某,etStdMargin=FALSE,ce某.te某t=0.5,zlim=c(-1,1),main=pate('Module-traitrelationhip'))最终找到几个共表达网络与三阴性表型最相关的模块。

生物技术进展 2024 年 第 14 卷 第 2 期 312 ~ 322Current Biotechnology ISSN 2095‑2341研究论文Articles基于肿瘤相关成纤维细胞基因构建乳腺癌预后预测模型及免疫浸润分析孙莉莉,安外尔·约麦尔阿卜拉,刘富中,布尔兰·叶尔肯别克,迪丽娜尔·叶尔夏提,郭文佳*新疆医科大学附属肿瘤医院,乌鲁木齐 830011摘 要:乳腺癌的转移和恶性进展与肿瘤微环境密切相关。
肿瘤相关成纤维细胞(cancer associated fibroblasts ,CAFs )是肿瘤微环境中比较重要的细胞,可影响肿瘤的进展及治疗。
从基因表达综合数据库获得乳腺癌单细胞测序数据,对肿瘤微环境细胞进行分簇,再利用WGCNA 识别CAF 相关的关键基因,用该基因在TCGA -BRCA 数据库中构建风险评分模型,进行生存分析、Cox 回归分析、ROC 曲线、构建列线图预测模型性能;通过GO 和KEGG 分析模型相关通路;利用体细胞突变、免疫浸润分析、干性指数分析以及药物敏感性分析探讨风险评分与临床特征及肿瘤微环境的关系。
研究构建了基于10个CAF 基因的乳腺癌预后预测模型,根据风险评分将患者分为高低风险组并进行验证,其中高风险组患者的预后更差,列线图和ROC 曲线也显示模型具有良好的预测效能,乳腺癌病人免疫浸润水平更低、干性指数更高,且高风险组病人对紫杉醇及拉帕替尼这2种药物的敏感性更高。
结果表明,10个CAF 相关基因的风险评分可独立预测乳腺癌的预后及治疗效果,为明确CAF 相关基因在乳腺癌中的作用机制提供了思路,也为乳腺癌易感基因患者的临床个体化治疗提供了理论依据。
关键词:乳腺癌;肿瘤相关成纤维细胞;肿瘤突变负荷;预后模型;免疫浸润DOI :10.19586/j.20952341.2023.0161中图分类号:Q75, R737.9 文献标志码:AConstruction of Prognostic Prediction Model of Breast Cancer Based on Tumor -associated Fibroblast Genes and Analysis of Immune InfiltrationSUN Lili , ANWAIER Yuemaierabola , LIU Fuzhong , BUERLAN Yeerkenbieke , DILINAER Ye ,GUO Wenjia *Affiliated Cancer Hospital of Xinjiang Medical University , Urumqi 830011, ChinaAbstract :Metastasis and malignant progression of breast cancer are deeply related to the tumor microenvironment. Tumor -associ‐ated fibroblasts (CAFs ) are comparatively important cells in the tumor microenvironment which have implications on tumor pro‐gression and treatment. We obtained single -cell sequencing data of breast cancer downloaded from gene expression omnibus data‐base , clustered the cells of tumor microenvironment , and then used WGCNA to identify the key genes related to CAF , and con‐structed a risk score model with the genes in TCGA -BRCA database , and performed survival analysis , Cox regression analysis , ROC curves , and constructed a column line graph to predict the performance of the model. Model -related pathways were analyzed by GO and KEGG. The relationship between risk score and clinical features and tumor microenvironment was explored by somaticmutation , immune infiltration analysis , stemness index analysis , and drug sensitivity analysis. A prognostic prediction modelbased on 10 CAF genes was constructed and validated in accordance with the risk scores. Patients were classified into high - and low -risk groups according to the risk scores , and the prognosis of patients in the high -risk group was worse , and the column plot and ROC curve also showed that the model had a good predictive efficiency , and the immune infiltration level of patients with收稿日期:2023‐12‐13; 接受日期:2024‐02‐27基金项目:新疆维吾尔自治区自然科学基金杰出青年科学基金项目(2022D01E27);新疆维吾尔自治区天池英才项目(2022TCYCGWJ )。
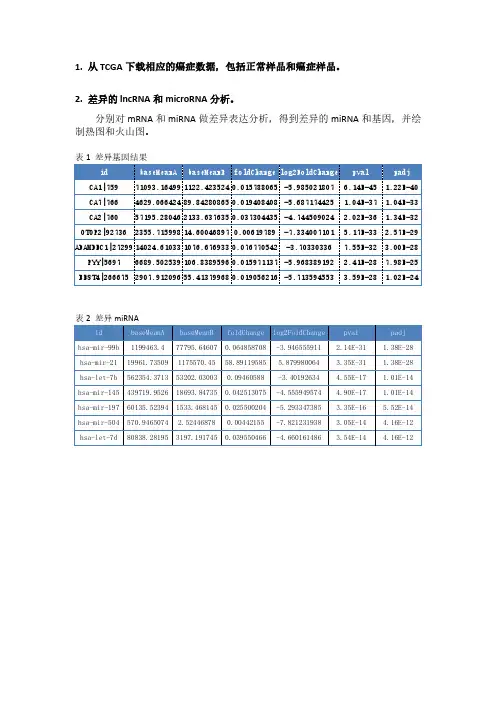
1.从TCGA下载相应的癌症数据,包括正常样品和癌症样品。
2.差异的lncRNA和microRNA分析。
分别对mRNA和miRNA做差异表达分析,得到差异的miRNA和基因,并绘制热图和火山图。
表差异
图1差异基因火山图
图2热图
3.共表达网络
基因和miRNA的共表达网络。
图3miRNA和mRNA共表达网络4.蛋白互作网络
对差异基因进行蛋白互作网络分析。
图4差异基因PPI网络
5.生存分析
分析基因高低表达与生存时间之间是否具有显著相关性,并且绘制生存曲线。
当然,也可以分析临床信息与生存的关系,比如临床分期与生存时间的关系,癌症大小与生存时间的关系,用药与生存时间的关系,等等。
图5目标基因生存分析
6.基因表达与临床的关系
分析基因与临床数据的关系,如基因的表达和癌症转移的关系,基因表达和临床分期的关系,基因表达和其它临床信息的关系。
图6MARCH1表达与肺转移的关系
7.其它个性化分析
根据客户提供分析案例或者文献,做相应的生物信息分析。
有疑问请联系作者邮箱:602316645@。

最新思路——巧用WGCNA分析GEO和TCGA数据,文章轻松上5分癌症相关成纤维细胞(CAF)是胃癌基质中最重要的细胞成分,它促进胃癌进展、治疗抵抗和免疫抑制。
加权基因共表达网络分析(Weighted gene co-expression network analysis, WGCNA)作为一种系统的生物信息学算法,能够将高度协调表达的基因整合到几个基因模块中,并研究该模块与目标表型的关系。
但是到目前为止,CAF 和间质浸润尚未在胃癌中进行WGCNA分析。
本次介绍的文章首次将WGCNA同时用于两个转录组数据集(来自GEO和TCGA数据库),用以分析CAF与胃癌的关系。
这篇文章是2021年10月发表在frontiers in Molecular Biosciences上,文章题目是:Weighted Gene Co-expression Network Analysis Identifies a Cancer-Associated Fibroblast Signature for Predicting Prognosis and Therapeutic Responses in Gastric Cancer。
下面我们来具体看看文章的主要内容吧。
材料与方法01文章的方法我们通过以下流程图来看看吧。
作者首先分别从GEO 和TCGA数据库获得431和330个胃癌样本信息。
随后对这两个数据集的样本进行CAF浸润以及基质评分的计算。
之后通过WGCNA构建针对CAF浸润和基质评分的共表达网络。
分别筛选出相关性最高的黑色和棕色module为关键模块,之后通过阈值的设定进一步筛选得到这两个模块中(取交集)的37个重点基因作为hub基因。
为了进一步缩小基因数目,采用单因素Cox和LASSO回归分析将基因数目缩小到4个,并利用这四个基因构建风险模型。
最后针对这个风险模型分别在两个数据集中作了预后分析、化学和免疫治疗预测性能测试、肿瘤突变负荷(TMB)分析、GSEA分析、与CFA浸润以及相关marker 基因相关性分析、以及在分子和蛋白水平对这四个关键基因的验证。

第42卷㊀第3期2023年㊀6月北京生物医学工程BeijingBiomedicalEngineeringVol 42㊀No 3June㊀2023基金项目:第65批中国博士后科学基金项目(2019M651658)㊁南京农业大学大学生创新训练项目(202023XX03)资助作者单位:南京农业大学理学院(南京㊀210095)通信作者:陈园园㊂E⁃mail:chenyuanyuan@njau.edu.cn乳腺癌相关的lncRNA⁃mRNA共表达扰动网络构建黄彦祚㊀李海龙㊀卢乐亭㊀陈园园摘㊀要㊀目的基于复杂生物网络和机器学习方法,识别乳腺癌相关的边缘生物标志物,构建乳腺癌生存预后模型,从而在系统水平解释乳腺癌的发生发展机制㊂方法首先基于TCGA数据库的RNA⁃seq数据识别乳腺癌相关的lncRNA⁃mRNA共表达扰动关系对,进一步构建乳腺癌相关的lncRNA⁃mRNA共表达扰动网络并对网络中的关键基因进行通路富集分析㊂然后,基于乳腺癌相关的lncRNA⁃mRNA关系对,构建乳腺癌预测的分类器模型㊂最后,通过Lasso回归筛选变量构建多因素Cox比例风险回归模型对乳腺癌患者进行生存预后分析㊂结果构建了乳腺癌相关的lncRNA⁃mRNA共表达扰动网络,其中的关键基因富集分析得到32条与乳腺癌相关的生物通路㊂分类预测模型的灵敏度㊁特异度和准确性分别为98 2%㊁85 2%㊁97 6%㊂Lasso回归共筛选出22个和乳腺癌生存预后显著相关的lncRNA⁃mRNA互作关系对,进而构建的生存预测模型把训练集和测试集的乳腺癌患者分为高风险组和低风险组,两组患者生存预后均存在明显差异㊂结论LncRNA⁃mRNA共表达互作网络中的关键基因以及乳腺癌相关的边缘生物标志物大多被证明与乳腺癌相关㊂同时基于边缘生物标志物的预后模型可以稳健地预测乳腺癌患者的生存预后状态,有利于从网络层面更好地理解乳腺癌的发生发展机制㊂关键词㊀lncRNA⁃mRNA共表达扰动网络;边缘生物标志物;乳腺癌预测模型;Cox比例风险回归模型DOI:10 3969/j.issn.1002-3208 2023 03 004.中图分类号㊀R318 04;Q354㊀㊀文献标志码㊀A㊀㊀文章编号㊀1002-3208(2023)03-0240-08本文著录格式㊀黄彦祚,李海龙,卢乐亭,等.乳腺癌相关的lncRNA⁃mRNA共表达扰动网络构建[J].北京生物医学工程,2023,42(3):240-247.HUANGYanzuo,LIHailong,LULeting,etal.ConstructionofanetworkoflncRNA⁃mRNAco⁃expressionperturbationrelatedtobreastcancer[J].BeijingBiomedicalEngineering,2023,42(3):240-247.ConstructionofanetworkoflncRNA⁃mRNAco⁃expressionperturbationrelatedtobreastcancerHUANGYanzuo,LIHailong,LULeting,CHENYuanyuanCollegeofScience,NanjingAgriculturalUniversity,Nanjing㊀210095Correspondingauthor:CHENYuanyuan(E⁃mail:chenyuanyuan@njau.edu.cn)ʌAbstractɔ㊀ObjectiveBasedonthecomplexbiologicalnetworksandmachinelearningmethods,theedgebiomarkersassociatedwithbreastcancerwereidentifiedandthesurvivalprognosismodelofbreastcancerwasconstructedtofurtherexplaintheoccurrenceanddevelopmentofbreastcanceratasystematiclevel.MethodsFirstly,basedontheRNA⁃seqdataofTCGA,weidentifiedlncRNA⁃mRNAco⁃expressionperturbationpairsandconstructedtheco⁃expressionperturbationnetworksrelatedtobreastcancer.Further,weconductedthepathwayenrichmentanalysisofkeygenesinthenetwork.Then,aclassifiermodelforbreastcancerpredictionwasconstructedbasedontheco⁃expressionperturbationpairs.Finally,amultivariateCoxproportionalriskregressionmodelwasestablishedbyscreeningvariablesusingLassoregressiontoanalyzethesurvivalprognosisofbreastcancerpatients.ResultsWeconstructedalncRNA⁃mRNAco⁃expressionperturbationnetworkassociatedwithbreastcancer.Andthekeygenesinthenetworkwereusedtoperformpathwayenrichmentanalysis.Atotalof32biologicalpathwaysassociatedwithbreastcancerwereobtained.Thesensitivity,specificity,accuracyoftheclassifiermodelforbreastcancerpredictionwere98.2%,85.2%and97.6%respectively.Atotalof22lncRNA⁃mRNAinteractionpairs,whichweresignificantlyassociatedwithbreastcancersurvivalprognosis,wereidentifiedbyLassoregression.Basedonthesurvivalpredictionmodel,breastcancerpatientsinthetrainingsetandthetestsetweredividedintohigh⁃riskgroupandlow⁃riskgroup,andthesurvivalprognosisofpatientsinthetwogroupswassignificantlydifferent.ConclusionsKeygenesinthelncRNA⁃mRNAco⁃expressionperturbationnetworkandbreastcancer⁃relatededgebiomarkershavemostlybeenprovedtobeassociatedwithbreastcancer.Meanwhile,theprognosticmodelbasedonedgebiomarkerscanpredictthesurvivalandprognosisofbreastcancerpatientsrobustly.Thispaperishelpfultobetterunderstandthemechanismoftheoccurrenceanddevelopmentofbreastcancerinnetworklevel.ʌKeywordsɔ㊀LncRNA⁃mRNAco⁃expressionperturbationnetwork;edgebiomarker;breastcancerpredictionmodel;Coxproportionalriskregressionmodel0㊀引言近年来,越来越多的研究表明,长链非编码RNA(longnon⁃codingRNA,lncRNA)在包括癌症在内的许多疾病的发生发展中发挥着重要作用,已受到越来越多的关注[1]㊂随着高通量测序技术的发展,大量的lncRNA被发现,其作用机制的研究也取得了一定进展[2]㊂目前已在乳腺癌细胞及组织中发现多种异常表达lncRNA,它们可能在乳腺癌细胞增殖㊁凋亡㊁侵袭㊁转移及药物敏感性等方面起了重要作用[3]㊂在癌症发展过程中,lncRNA参与了多种表观遗传复合物的调节过程,从而抑制或激活癌症相关mRNA基因的表达[4]㊂因此,探讨lncRNA⁃mRNA互作对乳腺癌发生发展的影响至关重要㊂复杂疾病(尤其癌症)的发生并不是单个基因失调导致,往往是由多个分子及其相互作用失调引起的㊂竞争性内源RNA机制是探索lncRNA如何参与恶性肿瘤调控的重要方法之一[5]㊂近期研究报道通过高通量测序和加权基因共表达网络法(weightedgeneco⁃expressionnetworkanalysis,WGCNA)可以进行表达谱基因系统分析[6]㊂Wei等[7]通过微阵列分析的方法识别了lncRNA和mRNA的差异表达模块㊂Yin等[8]通过全基因组关联分析的方法对lncRNA⁃mRNA调控网络进行综合分析㊂但国内尚无关于乳腺癌的lncRNA⁃mRNA互作关系的研究,而复杂生物网络可以从系统层面解释癌症的发生发展机制㊂为此本文拟基于RNA⁃seq数据,计算个体特异的lncRNA⁃mRNA共表达扰动值,进一步识别乳腺癌相关的lncRNA⁃mRNA互作关系,并构建共表达扰动网络㊂同时,基于lncRNA⁃mRNA互作关系构建区分乳腺癌样本和正常样本的分类器模型㊂另外,基于单变量Cox回归识别与乳腺癌生存预后相关的lncRNA⁃mRNA关系对,通过Lasso回归筛选变量并进一步构建多因素Cox回归模型,对乳腺癌患者进行生存预后分析㊂本研究拟通过基因生物网络和机器学习方法,识别乳腺癌相关的边缘生物标志物并构建乳腺癌生存预后模型,在系统水平上解释乳腺癌的发生发展,为制定合适的治疗计划㊁协助评价治疗结果㊁预测患者的生存时间等提供重要依据,进而提高乳腺癌预后诊断以及促进精准医疗的发展㊂1㊀材料与方法1 1㊀数据来源从TCGA数据库(https://cancergenome nih gov/)下载1097个乳腺癌样本和113个癌旁组织样本的RNA⁃seq数据,以及乳腺癌患者的临床数据;从GENCODE数据库(https://www gencodegenes org)下载V33版本的基因注释文件;从数据库GSEA/MSigDB(http://software broadinstitute org/gsea/msigdb)下载186个KEGG通路基因集㊂基因表达数据是经过标准化后的Level3的RPKM格式,共包含基因56521个㊂1 2㊀数据预处理首先对标准化后的RNA⁃seq数据进行预处理,㊃142㊃第3期㊀㊀㊀㊀㊀㊀黄彦祚,等:乳腺癌相关的lncRNA⁃mRNA共表达扰动网络构建删除大于90%样本表达值为0的基因㊂根据Gencode基因注释文件共得到含有10941个lncRNA基因和18601个mRNA基因的表达数据㊂1 3㊀构建乳腺癌相关的lncRNA⁃mRNA共表达扰动网络㊀㊀识别乳腺癌相关的lncRNA⁃mRNA共表达扰动网络框架见图1㊂对于每个lncRNA⁃mRNA关系对,基于正常样本的表达数据计算皮尔逊相关系数(Pearsoncorrelationcoefficient,PCC)㊂挑选出所有显著线性相关的lncRNA与mRNA关系对(PCC>0 75且P<0 05),即具有共表达关系的lncRNA⁃mRNA基因互作对㊂对于每一个共表达的lncRNA⁃mRNA基因互作对,运用最小二乘法的简单线性回归模型拟合lncRNA和mRNA共表达的直线方程,即:y=bx+a(1)㊀㊀式中:b为拟合直线方程的斜率;a为拟合直线方程的截距项㊂设(xk,yk)代表第k个样本mRNAlncRNA的表达量,则可以计算(xk,yk)到直线y=bx+a的距离:dk=bxk+a-yk1+b2(2)则所有癌症样本的距离之和D= 1097k=1dk可以用来衡量乳腺癌样本在此lncRNA⁃mRNA互作关系中的共表达扰动情况㊂若D越大,则代表乳腺癌样本中lncRNA⁃mRNA共表达扰动越明显㊂通过链蒙特卡洛(MonteCarlo,MC)随机抽样方法得到互作扰动距离的经验零分布(empiricalnulldistribution)㊂所有P<0 01的lncRNA⁃mRNA共表达关系对组成了乳腺癌相关的lncRNA⁃mRNA互作关系对,这些互作关系对构成了乳腺癌相关的lncRNA⁃mRNA共表达扰动网络㊂在整个乳腺癌相关的lncRNA⁃mRNA共表达扰动网络中,少量度(degree)很高的基因作为网络枢纽参与重要的生命活动,这些基因被认为是影响乳腺癌活动的关键基因㊂根据Barberan的拓扑网络研究[9],选取度大于20的mRNA基因进行通路富集分析㊂通过超几何分布检验,计算P值:P(m,M,N,n)=1-m-1i=0Miæèçöø÷N-Mn-iæèçöø÷Nnæèçöø÷(3)㊀㊀式中:N为整个乳腺癌相关的lncRNA⁃mRNA互作网络中的mRNA基因总数;M为互作网络中degree>20的关键基因个数;n为某条基因通路中的基因个数;m为lncRNA⁃mRNA互作网络中关键基因落入该通路中基因的个数㊂最后选取P<005的通路作为显著富集的通路㊂图1㊀识别乳腺癌相关的lncRNA⁃mRNA共表达扰动网络框架图Figure1㊀AnintegrativeframeworkidentifyinglncRNA⁃mRNAco⁃expressionperturbationnetworks㊃242㊃北京生物医学工程㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀第42卷1 4㊀基于lncRNA⁃mRNA互作关系构建乳腺癌预测模型㊀㊀768个乳腺癌样本和79个癌旁组织样本的RNA⁃seq数据为训练集,329个乳腺癌样本和33个癌旁组织样本数据为独立测试集㊂以乳腺癌相关的lncRNA⁃mRNA互作关系的共表达扰动距离为特征,构建预测乳腺癌的随机森林分类器模型,评价模型的预测性能㊂然后根据特征贡献对lncRNA⁃mRNA互作关系对进行降序排列,选择前10%较为重要的关系作为潜在lncRNA⁃mRNA边缘生物标志物㊂本文在R3 5 3上进行实验,使用R包 randomForest 建立预测模型㊂根据训练集对随机森林模型进行训练,得到反映其预测效果的混淆矩阵㊂训练后的随机森林模型对独立测试集进行测试,结合袋外错误率㊁灵敏度㊁特异度来衡量预测的准确率㊂袋外错误率(out⁃of⁃bagerror,OOB)定义如下:OOB=被分类错误数总数(4)㊀㊀模型的灵敏度(truepositiverate,TPR),指真实类别为正类的样本中分类预测也为正的比例,即:TPR=TPTP+FN(5)㊀㊀式中:TP为真实类别为正㊁分类预测也为正的数目;FN为真实类别为正㊁分类预测为负的数目㊂特异度(truenegativerate,TNR),其定义为真实类别为负类的样本中,分类预测也为负的比例:TNR=TNTN+FP(6)㊀㊀式中:TN为真实类别为负㊁分类预测也为负的数目;FP为真实类别为负㊁分类预测为正的数目㊂准确性(accuracy,ACC),指分类正确的记录个数占总记录个数的比例:ACC=TN+TPTP+FN+TN+FP(7)㊀㊀OOB㊁TPR㊁TNR㊁ACC作为评估随机森林模型的性能指标㊂1 5㊀生存分析利用癌症患者的临床数据,以患者生存时间和状态为因变量,乳腺癌相关的lncRNA⁃mRNA互作关系对的共表达扰动距离作为协变量,建立单因素Cox回归模型,其中的共表达扰动距离dk为第k个癌症样本到共表达直线方程的距离㊂选取其中P<0 01的lncRNA⁃mRNA互作关系对㊂由于协变量过多,在建立多因素Cox回归模型时可能导致计算复杂以及过拟合的问题,因此选择最小绝对收缩选择算子(leastabsoluteshrinkageandselectionoperator,LASSO)方法,利用R语言 survival ㊁ survminer ㊁ caret ㊁ glmnet 软件包对lncRNA⁃mRNA互作关系对进一步筛选㊂运用LASSO方法压缩回归系数,并选取一个交叉检验均方误差最小的λ值,从而得到一个最优的LASSO回归模型㊂其中大部分基因对的系数被缩减到0,剩下相对较少非零系数的基因对则被认为是和乳腺癌预后高度相关的lncRNA⁃mRNA互作关系对㊂在训练集上构建多因素Cox回归风险评分方程:riskscore= ni=1CoefiˑXi(8)㊀㊀式中:Coefi为第i个lncRNA⁃mRNA互作关系对回归系数;Xi为第i个乳腺癌预后高度相关的lncRNA⁃mRNA互作关系对;n为乳腺癌预后高度相关的lncRNA⁃mRNA互作关系对的个数㊂根据公式计算每个患者的风险评分,并以风险评分中位数为截断值,将乳腺癌患者分为低风险组和高风险组,进一步画出KaplanMeier生存曲线㊂最后用同样的方法对测试集进行生存验证㊂2㊀结果2 1㊀乳腺癌相关的lncRNA⁃mRNA共表达扰动互作关系㊀㊀lncRNA⁃mRNA共表达扰动(lg转换)分布情况见图2,其中11 081为P=0 01的分位点㊂大于该阈值的互作关系对构成乳腺癌相关的lncRNA⁃mRNA互作网络㊂图2㊀乳腺癌样本的lncRNA⁃mRNA共表达扰动分布Figure2㊀PerturbationdistributionoflncRNA⁃mRNAco⁃expressioninbreastcancersamples㊃342㊃第3期㊀㊀㊀㊀㊀㊀黄彦祚,等:乳腺癌相关的lncRNA⁃mRNA共表达扰动网络构建乳腺癌相关的lncRNA⁃mRNA共表达互作网络中共包含2866个lncRNA⁃mRNA互作关系对,648个LncRNA和733个mRNA,共1381个基因㊂利用Cytoscape软件将乳腺癌相关的lncRNA⁃mRNA共表达互作网络可视化,如图3所示㊂紫色节点表示lncRNA,蓝色节点表示mRNA图3㊀乳腺癌相关的lncRNA⁃mRNA共表达互作网络Figure3㊀Breastcancer⁃associatedlncRNA⁃mRNAco⁃expressioninteractionnetwork网络图中共有关键基因54个,包括24个lncRNA和30个mRNA㊂其中,一些基因已被证明为与乳腺癌相关,例如,APOD与癌细胞的增殖活性降低有关,大量存在于衰老细胞中[10]㊂下调DANCR可以抑制乳腺癌细胞的致瘤性和发育[11]㊂ZFAS1过表达可通过阻滞细胞周期㊁诱导细胞凋亡等途径抑制乳腺癌细胞增殖[12]㊂胞外CDH1突变降低了细胞的活动性和理论上的转移能力[13]㊂上调GAS5可以抑制TNBC的进展,促进TNBC细胞的化疗敏感性和凋亡[14]㊂IDH2在mRNA或蛋白水平上的高表达与乳腺癌患者预后不良相关[15]㊂过表达AGAP2-AS1可促进细胞生长,抑制细胞凋亡[16]㊂2 2㊀富集通路通过基因富集分析得到32条与乳腺癌相关的生物通路,主要包括一些免疫相关的通路:T细胞受体信号通路,它被发现是HQ-BS治疗乳腺癌的潜在信号通路[17];B细胞受体信号通路在三阴性乳腺癌的发展中失调[18];造血细胞谱系通路,研究发现造血细胞谱系,细胞黏附分子和原发性免疫缺陷明显增加了LuminalB型乳腺癌的KEGG通路[19];Toll样受体信号通路通过缺氧诱导因子增强乳腺癌细胞的恶性特征[20];自然杀伤细胞的细胞毒信号通路㊂同时,一些重要的信号转导和信号分子互作通路也被富集:ErbB信号通路不仅在乳腺癌发生发展中显著失调,而且在新型抗乳腺癌靶向疗法中发挥重要作用[21];MAPK信号通路中Linc⁃RoR促进MAPK/ERK信号传导并赋予乳腺癌非雌激素依赖性生长[22];Wnt信号通路中LncCCAT1通过激活WNT/β-catenin信号传导促进乳腺癌干细胞功能[23];p53信号通路可能是乳腺癌进展的重要途径,与p53相关的基因CCNE2㊁CCNB1和RRM2可能会作为治疗BC的候选治疗基因靶标[24];JAK⁃STAT信号通路,抑制JAK/STAT通路和Akt信号通路可以抑制乳腺癌细胞迁移[25];Hedgehog信号通路,LKB1通过抑制Hedgehog信号通路部分抑制乳腺癌的发生[26]㊂另外,癌症通路,这个与乳腺癌相关的重要通路也被富集㊂富集的结果如图4所示㊂2 3㊀分类器模型预测结果模型对测试集的预测精度见表1㊂OOB为2 75%,TPR和TNR分别为98 2%和85 2%,ACC为97 6%㊂在训练集和测试集的随机森林模型性能分别为:特异度99 5%和99 4%;灵敏度84 8%和82 4%;准确性98 1%和97 8%㊂表1㊀随机森林模型在测试集上的性能Table1㊀Performanceofrandomforesttrainingontestset真实值\预测值癌症样本正常样本分类误差癌症样本33041 2%正常样本62320 7%袋外错误率2 75%基于lncRNA⁃mRNA互作关系构建了乳腺癌预测模型,识别出lncRNA⁃mRNA边缘生物标志物㊂其中有的基因已被证明为与乳腺癌相关:ERα和APOD共表达老年乳腺癌患者中具有预后意义[27];VIM表达与肿瘤分化程度有关,随着肿瘤的失分化,VIM的表达能力逐渐增强[28];SPINT2基因通过表观沉默或下调改变HGF激活/抑制比率的平衡,从而促进癌症的发展[29];SNRPB基因在乳腺癌中高表达,可以有效预测乳腺癌转移的发生,为乳腺癌的靶向治疗提供依据[30];SPINT1-AS1通过调节miR-let-7a/b/i-5p可以促进乳腺癌细胞的增殖和迁移,因此它可能是乳腺癌的重要调控因子[31]㊂㊃442㊃北京生物医学工程㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀第42卷图4㊀基因通路富集结果Figure4㊀Genepathwayenrichmentresults2 4㊀乳腺癌预后相关的lncRNA⁃mRNA共表达互作关系对㊀㊀最终识别出22个乳腺癌生存预后显著相关的lncRNA⁃mRNA互作关系对,并基于这些互作关系对构建多因素Cox回归模型:Score=-28 796ˑ(PCED1B-AS1,HLA-DRA)+30 030ˑ(PSMB8-AS1,HLA-DRA)-15 858ˑ(HOXB-AS1,ABCC9)-15 586ˑ(HOXB-AS1,GRK3)+187 343ˑ(GAS1RR,LPCAT2)+ (9)基于训练集和测试集的两组乳腺癌患者的Kaplan⁃Meier生存曲线见图5,容易发现不管是训练集还是测试集的两组乳腺癌患者的预后都存在显著差异,且高风险组的生存预后显著劣于低风险组㊂这个结果表明基于基因对的预后模型可以很好地预测乳腺癌患者的生存时间㊂3㊀讨论与结论目前癌症病理研究一般仅限于群体的基因表达量和突变信息,而乳腺癌个性化医疗处于发展相对缓慢的阶段㊂但是乳腺癌的发生并不是单个基因导致的,而是由多个分子及其相互作用失调引起的㊂基因并不是独立存在的,而是存在于许多复杂的分子网络中㊂因此,构建乳腺癌相关网络从系统层面解释乳腺癌的发生发展机制㊂但由于条件所限,缺乏相应的临床试验来验证本文结果㊂在本研究中,基于正常样本基因表达数据,通过癌症样本点偏离拟合直线距离计算每个样本的lncRNA⁃mRNA共表达扰动情况,识别乳腺癌相关的lncRNA⁃mRNA互作关系对,并构建乳腺癌相关的lncRNA⁃mRNA共表达扰动网络㊂同时对网络中的关键基因进行基因通路富集分析,结果显示这些基因与癌症通路㊁细胞凋亡通路以及ErbB信号通路等密切相关㊂通过Cox比例风险模型识别出与乳腺癌预后相关的lncRNA⁃mRNA互作关系对,并建立生存风险预测模型,结果表明本研究中基因对的预后模型可以很好地预测乳腺癌患者的生存时间㊂基于识别出的lncRNA⁃mRNA边缘生物标志物,发现边缘生物标志物中含有乳腺癌相关的诸多㊃542㊃第3期㊀㊀㊀㊀㊀㊀黄彦祚,等:乳腺癌相关的lncRNA⁃mRNA共表达扰动网络构建图5㊀基于训练集和测试集的两组乳腺癌患者的Kaplan⁃Meier生存曲线Figure5㊀Kaplan⁃Meiersurvivalcurvesoftwogroupsofbreastcancerpatientsbasedontrainingsetandtestset重要基因㊂已被证实的有:基因DAXX可抑制内分泌治疗后雌激素受体阳性乳腺癌患者的肿瘤起始细胞的数量[32];通过消除基因EEF1A1的转录,癌细胞可以在蛋白毒性应激后迅速诱导热休克反应并存活[33];MeCP2基因通过抑制RPL5/RPL11转录来促进泛素介导的p53降解[34],从而促进乳腺癌的细胞增殖并抑制凋亡㊂值得注意的是,网络中很多关键基因与乳腺癌相关:乳腺癌患者SPARCrs7719521基因与NPI显著相关,NPI是一种可靠的乳腺癌预后指标;Anxa5可作为乳腺癌肿瘤发生㊁转移和侵袭预测的生物标志物;NORAD可以抑制乳腺癌细胞的迁移㊁侵袭和转移㊂结果中ERα-APOD和SPINT1-AS1的共表达很高,是乳腺癌进展的重要调控因子,可以作为乳腺癌预后的指标及潜在的治疗靶点㊂抑制基因CCT3的表达可抑制乳腺癌细胞的增殖和迁移㊂HSP90AA1㊁SRC㊁HSPA8㊁ESR1㊁ACTB㊁PPP2CA㊁RPL等度较高的基因在乳腺癌中高表达,且与乳腺癌的不良预后密切相关,可以作为基因检测的理论依据㊂故通过研究可以较准确地找出乳腺癌的决定基因和预测乳腺癌患者的生存时间,对于其他复杂疾病的病因研究以及个性化医疗具有重要的参考价值㊂基于共表达扰动的乳腺癌相关的lncRNA⁃mRNA互作网络的构建能够从系统层面更好地理解乳腺癌的发生和发展机制,对提高乳腺癌预后诊断以及促进精准医疗的发展等都有重要的意义㊂参考文献[1]㊀JinX,GeLP,LiDQ,etal.LncRNATROJANpromotesproliferationandresistancetoCDK4/6inhibitorviaCDK2transcriptionalactivationinER+breastcancer[J].MolecularCancer,2020,19(1):87.[2]㊀LiangYR,SongXJ,LiYM,etal.LncRNABCRT1promotesbreastcancerprogressionbytargetingmiR-1303/PTBP3axis[J].MolecularCancer,2020,19(1):85.[3]㊀LuoLY,ZhangJL,TangHL,etal.LncRNASNORD3Aspecificallysensitizesbreastcancercellsto5-FUbyspongingmiR-185-5ptoenhanceUMPSexpression[J].CellDeath&Disease,2020,11(5):329.[4]㊀芮小慧.长链非编码RNAC5orf66-AS1在宫颈癌发生中的作用及其机制研究[D].苏州:苏州大学,2019.RuiXH.Theroleoflongnon⁃codingRNAC5ORF66-AS1inthepathogenesisofcervicalcanceranditsmechanism[D].Suzhou:SoochowUniversity,2019.[5]㊀尹冶,丁明霞,陈振杰,等.基于TCGA和GEO数据库构建前列腺癌ceRNA网络并筛选相关lncRNAs[J].临床肿瘤学杂志,2020,25(11):1011-1017.YinY,DingMX,ChenZJ,etal.ConstructionofprostatecancerCernanetworkandscreeningoflncrnasbasedonTCGAandgeodatabase[J].JournalofClinicalOncology,2020,25(11):1011-1017.[6]㊀JiaRK,ZhaoHX,JiaMW.Identificationofco⁃expressionmodulesandpotentialbiomarkersofbreastcancerbyWGCNA[J].Gene,2020,750:144757.[7]㊀WeiJR,DouQS,BaFT,etal.IdentificationoflncRNAandmRNAexpressionprofilesindorsalrootganglioninratswithcancer⁃inducedbonepain[J].BiochemicalandBiophysicalResearchCommunications,2021,572:98-104.[8]㊀YinHT,ShangQ,ZhangSL,etal.ComprehensiveanalysisoflncRNA⁃mRNAregulatorynetworkinBmNPVinfectedcellstreatedwithHsp90inhibitor[J].MolecularImmunology,2020,127:230-237.[9]㊀BarberanA,BatesST,CasamayorEO,etal.Usingnetwork㊃642㊃北京生物医学工程㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀第42卷analysistoexploreco⁃occurrencepatternsinsoilmicrobialcommunities[J].TheISMEJournal,2011,6(2):343-351.[10]㊀SilandH,SreideK,JanssenE,etal.EmergingconceptsofapolipoproteinDwithpossibleimplicationsforbreastcancer[J].CellularOncology,2007,29(3):195-209.[11]㊀ShaS,YuanDY,LiuYJ,etal.Targetinglongnon⁃codingRNADANCRinhibitstriplenegativebreastcancerprogression[J].BiologyOpen,2017,6(9):1310-1316.[12]㊀FanSL,FanCN,LiuN,etal.Downregulationofthelongnon⁃codingRNAZFAS1isassociatedwithcellproliferation,migrationandinvasioninbreastcancer[J].MolecularMedicineReports,2018,17(5):6405-6412.[13]㊀CorsoG,IntraM,TrentinC,etal.CDH1germlinemutationsandhereditarylobularbreastcancer[J].FamilialCancer,2016,15(2):215-219.[14]㊀LiJT,LiL,YuanHZ,etal.Up⁃regulatedlncRNAGAS5promoteschemosensitivityandapoptosisoftriple⁃negativebreastcancercells[J].CellCycle,2019,18(16):1965-1975.[15]㊀AljohaniAI,TossMS,KurozumiS,etal.Theprognosticsignificanceofwild⁃typeisocitratedehydrogenase2(IDH2)inbreastcancer[J].BreastCancerResearchandTreatment,2020,179(1):79-90.[16]㊀DongHY,WangW,MoSW,etal.SP1⁃inducedlncRNAAGAP2-AS1expressionpromoteschemoresistanceofbreastcancerbyepigeneticregulationofMyD88[J].JournalofExperimental&ClinicalCancerResearch,2018,37(1):202.[17]㊀ChenJR,LiuC,CenJM,etal.KEGG⁃expressedgenesandpathwaysintriplenegativebreastcancer:Protocolforasystematicreviewanddatamining[J].Medicine,2020,99(18):132-140.[18]㊀YuanCL,JiangXM,YiY,etal.IdentificationofdifferentiallyexpressedlncRNAsandmRNAsinluminal⁃BbreastcancerbyRNA⁃sequencing[J].BMCCancer,2019,19(1):1171.[19]㊀QiF,QinWX,ZangYS.Molecularmechanismoftriple⁃negativebreastcancer⁃associatedBRCA1andtheidentificationofsignalingpathways[J].OncologyLetters,2019,17(3):2905-2914.[20]㊀DengJL,XuYH,WangG.Identificationofpotentialcrucialgenesandkeypathwaysinbreastcancerusingbioinformaticanalysis[J].FrontiersinGenetics,2019,10(1):695-713.[21]㊀LiuDH,ZhangJF,LiL,etal.Detectionofcriticalgenesassociatedwithpoorprognosisinbreastcancerviaintegratedbioinformaticsanalyses[J].JournalofB.U.ON.,2020,25(6):2537-2545.[22]㊀XuT,WangQG,LiuM.AnetworkpharmacologyapproachtoexplorethepotentialmechanismsofHuangqin⁃Baishaoherbpairintreatmentofcancer[J].MedicalScienceMonitor:InternationalMedicalJournalofExperimentalandClinicalResearch,2020,26:e923199.[23]㊀ZhaoL,LinM,WangSS.IdentificationofhumanprolactinomarelatedgenesbyDNAmicroarray[J].JournalofCancerResearchandTherapeutics,2014,10(3):544-548.[24]㊀AzadAK,LawenA,KeithJM.Predictionofsignalingcross⁃talkscontributingtoacquireddrugresistanceinbreastcancercellsbyBayesianstatisticalmodeling[J].BMCSystemsBiology,2015,9(1):2.[25]㊀KhannaP,LeeJS,SereemaspunA,etal.GRAMD1BregulatescellmigrationinbreastcancercellsthroughJAK/STATandAktsignaling[J].ScientificReports,2018,8(1):9511.[26]㊀ZhuangZG,WangK,ChengXL,etal.LKB1inhibitsbreastcancerpartiallythroughrepressingtheHedgehogsignalingpathway[J].PlosOne,2013,8(7):e67431.[27]㊀FernandaMR,SaraTO,JoaoAM.SerinepeptidaseinhibitorKunitztype2(SPINT2)incancerdevelopmentandprogression[J].Biomedicine&Pharmacotherapy,2018,101(1):278-286.[28]㊀ZhongWL,LuMY,SiCF,etal.Progressofresearchontargetedtherapyforbreastcancer[J].ModernOncology,2018,26(4):622-626.[29]㊀ZhouTZ,LinK,NieJJ,etal.LncRNASPINT1-AS1promotesbreastcancerproliferationandmetastasisbysponginglet-7a/b/i-5p[J].Pathology⁃ResearchandPractice,2021,217(1):153268.[30]㊀郭辉,张斌,胡利民.微小RNA-145靶向调控SOX11表达及其对乳腺癌细胞增殖和凋亡的影响[J].临床肿瘤学杂志,2018,195(3):200-205.GuoH,ZhangB,HuLM.ExpressionofmicroRNA-145onthetargetedregulationofSOX11anditseffectonproliferationandapoptosisofbreastcancercells[J].JournalofClinicalOncology,2018,195(3):200-205.[31]㊀李莉,孟少达,邱爽,等.乳腺癌根治术后皮瓣下积液感染病原学特点及相关因素分析[J].中华医院感染学杂志,2018,28(3):414-417.LiL,MengSD,QiuS,etal.Pathogeniccharacteristicsandriskfactorsofinfectionsofskinflapafterradicalmastectomyforbreastcancer[J].ChineseJournalofNosocomiology,2018,28(3):414-417.[32]㊀PeifferDS,WyattD,ZlobinA,etal.DAXXsuppressestumor⁃initiatingcellsinestrogenreceptor⁃positivebreastcancerfollowingendocrinetherapy[J].CancerResearch,2019,79(19):4965-4977.[33]㊀XuG,BuSS,WangXS,etal.SuppressionofCCT3inhibitstheproliferationandmigrationinbreastcancercells[J].CancerCellInternational,2020,20(1):218.[34]㊀LinCY,BeattieA,BaradaranB,etal.ContradictorymRNAandproteinmisexpressionofEEF1A1inductalbreastcarcinomaduetocellcycleregulationandcellularstress[J].ScientificReports,2018,8(1):13904.(2021-07-25收稿,2021-10-14修回)㊃742㊃第3期㊀㊀㊀㊀㊀㊀黄彦祚,等:乳腺癌相关的lncRNA⁃mRNA共表达扰动网络构建。

癌症TCGA数据库中乳腺癌预后数据的挖掘Mian Khizar Hayat;王铭裕;李硕磊【摘要】近年来,乳腺癌发病率逐渐上升,并且呈现出年轻化趋势.使用TCGA数据库中已有的基因信息筛选鉴定出与乳腺癌预后相关的基因.为排除癌组织和正常组织取样时间不同造成的差异,我们选取了113对同时检测乳腺癌区和其相对应癌旁正常组织的样品,从TCGA数据库调取转录组数据,对这些数据通过DEseq进行差异表达分析,筛选出1428个差异表达基因.对差异表达基因进行基因本体GO,代谢通路KEGG,疾病本体DO和富集分析获得68个与乳腺癌相关的差异表达的关键基因;采用数据库中所用癌症的表达数据(共1097例)对这些乳腺癌相关基因进行总生存率分析,筛选出8个与乳腺癌预后相关的基因.结果显示在乳腺癌病人中PGLYRP2、SEMA3G、PROL1及SLC7A3的高表达伴随着乳腺癌病人的预后良好,而SKA1、BIRC5、RRM2和AURKA基因的高表达伴随着乳腺癌病人的预后不良.这8个基因有可能是乳腺癌预后相关的重要基因,这为乳腺癌病人的预后治疗提供了新的方向与思路,并可能通过调控基因水平来尽可能地控制预后.【期刊名称】《生物学杂志》【年(卷),期】2018(035)004【总页数】5页(P62-66)【关键词】癌症基因组图谱数据库;乳腺癌;差异表达基因;预后【作者】Mian Khizar Hayat;王铭裕;李硕磊【作者单位】兰州大学生命科学学院生物物理所,兰州730000;兰州大学生命科学学院生物物理所,兰州730000;兰州大学生命科学学院生物物理所,兰州730000【正文语种】中文【中图分类】R737.9乳腺癌是危害女性身心健康的最主要的恶性肿瘤,男性乳腺癌患者比较少见,Cancer Statistics 在 2017 年的统计数据显示乳腺癌在女性癌症发病中占据了 30%的比例[1]。
近年来,乳腺癌的发病率逐年上升,并且年轻化趋势明显[2]。

生物大数据技术在癌症研究中的应用案例分析Title: Applications of Big Data Technology in Cancer Research: A Case Study AnalysisIntroduction:Cancer, a complex and devastating disease, continues to be a global challenge in the field of healthcare. With advancements in technology, the use of big data analytics has revolutionized cancer research and significantly contributed to improving diagnosis, treatment, and patient outcomes. This article aims to analyze real-world applications of bioinformatics and big data technology in cancer research, highlighting their key contributions and potential implications.Case Study 1: Genomic Analysis and Precision MedicineOne of the prominent applications of big data technology in cancer research is genomic analysis. By analyzing large datasets of genomic information, researchers can identify specific genetic variants associated with the development, progression, and response to treatment of various cancers. For instance, The Cancer Genome Atlas (TCGA) project, which collected and analyzed genomic data from several cancer types, has enabled the identification of key oncogenes and tumor suppressor genes. This information has proved crucial in designing targeted therapies and personalized treatment plans for cancer patients.Case Study 2: Predictive Analytics and Early DetectionAnother significant application of big data technology in cancer research is predictive analytics. By mining massive datasets, researchers can develop algorithms and models that can predict the risk of developing specific cancers based on various factors such as genetics, lifestyle, and environmental influences. These predictive models can aid in early detection, allowing for timely interventions and improved survival rates. Forexample, research using big data analytics has led to the development of predictive models for breast cancer, enabling enhanced screenings and targeted preventive measures for high-risk individuals.Case Study 3: Drug Development and Treatment OptimizationBig data technology has also revolutionized the process of drug development and treatment optimization in cancer research. By integrating genomic data, clinical records, and treatment outcomes, researchers can analyze and identify potential drug targets, predict drug resistance, and evaluate treatment effectiveness. This knowledge has led to the development of novel targeted therapies and the optimization of existing treatments. For instance, the use of big data analytics in cancer immunotherapy has led to the identification of reliable biomarkers that can predict patient response to immunotherapy drugs, improving treatment outcomes for individuals with advanced cancers.Case Study 4: Real-Time Monitoring and SurveillanceThe application of big data technology in cancer research extends beyond the laboratory to real-time monitoring and surveillance. By integrating data from various sources such as electronic health records, wearable devices, and public health databases, researchers can track disease incidence, prevalence, and treatment outcomes in real-time. This data-driven approach enables the identification of disease patterns, the evaluation of intervention strategies, and the implementation of effective public health measures. For instance, the use of big data analytics in tracking cancer registries has facilitated the identification of geographic-specific cancer patterns, contributing to targeted prevention and control efforts.Conclusion:The utilization of big data technology in cancer research has revolutionized our understanding of the disease, enhancing diagnosis, treatment, and prevention strategies. The analysis of genomic data, predictive analytics, drug development, and real-time monitoring have significantly improved patient outcomes and personalized cancer care. As technology continues to advance, the potential for big data analytics in cancerresearch is immense, promising further advancements in understanding the disease, identifying new therapeutic targets, and improving overall cancer management.。

TCGA肿瘤数据库对特定基因进行GSEA分析大家好,我是小伍,今天我们来聊一下TCGA肿瘤数据库对特定基因进行GSEA分析,很多网友问我,我对特定基因进行了前期试验,但不知其功能和下游机制,笔者前期看了一稿文章,用的是GSEA分析。
经过二天的学习,以及自行编写整理烦数据和代码编写,终于完成。
前期,TCGA数据准备:还是以TCGA甲状腺癌为例。
我们要获得RNA-seq 矩阵文件。
这个我在前期的已经整理好了,代码也分享给大家了。
其实这个文件是我进行了一些转换,因为要对特定基因进行GSEA 分析,我们要排除正常组,直接拿肿瘤组的数据矩阵文件,然后我们对特定基因表达分高表达和低表达,分的方式可以中位数法或者均值法,不过笔者看文献大都用中位数法。
其实用什么方面来分,我在一本数据模型的书里看到,这个里面其实还是要经过检验才行的。
朝这个思维,我下面需要的是,对肿瘤特基因进行排序,首先要进行转置操作,这个用t函数就OK,然后对数据排序,笔者用的dplyr包。
rt1<>library(dplyr)rt_df <->rt_df<>这样排序就好了,然后就是保存write.csv(rt_df,'result.csv')这些代码只是告诉大家思路,其中的很多调整的地方,大家还要运用以后我讲的R处理。
下面我们要准备,表型文件,就是高表达和低表达,如下496 2 1# low highlow .low.........high........保存cls然后就导入数据:导入的方法有三个,这个官方网站有很清楚的说明,不过在导入的时候,有时不成功,比如说双引号的问题,都要去掉。
GSEA的基因集合数据来源】GSEA的基因集合来源于数据库MSigDB,分为H,C1-C7这几大块。
H: hallmark gene sets (效应)特征基因集合,共50组,如细胞凋亡的特征基因集合、细胞分裂checkpoint的的特征基因集合等。

《基于TCGA数据库乳腺癌IncRNA的分析研究》一、引言乳腺癌是全球女性最常见的恶性肿瘤之一,其发病率逐年上升,对女性健康构成严重威胁。
随着生物信息学和基因组学的发展,越来越多的研究开始关注非编码RNA(ncRNA)在疾病发生、发展中的作用。
其中,长链非编码RNA(IncRNA)因其特殊的调控作用和复杂的生物学功能,成为研究的热点。
TCGA(The Cancer Genome Atlas)数据库作为全球最大的癌症基因组数据库之一,为乳腺癌IncRNA的研究提供了丰富的数据资源。
本文旨在基于TCGA数据库,对乳腺癌IncRNA进行深入分析研究,以期为乳腺癌的预防、诊断和治疗提供新的思路和方法。
二、材料与方法1. 数据来源本研究采用的数据来自TCGA数据库中的乳腺癌相关数据,包括基因表达谱、临床信息等。
2. 研究方法(1)数据预处理:对基因表达谱数据进行质量评估和预处理,去除低质量和异常值数据。
(2)IncRNA筛选:基于基因表达谱数据,筛选出在乳腺癌组织中显著差异表达的IncRNA。
(3)功能分析:通过生物信息学分析方法,对筛选出的IncRNA进行功能分析,包括基因共表达网络分析、基因集富集分析等。
(4)验证实验:结合临床样本,对筛选出的关键IncRNA进行实时荧光定量PCR验证。
三、结果与分析1. 差异表达IncRNA的筛选结果通过数据分析,我们筛选出在乳腺癌组织中显著差异表达的IncRNA共计XX个,其中XX个为上调表达,XX个为下调表达。
这些IncRNA在乳腺癌的发生、发展过程中可能发挥重要的调控作用。
2. 功能分析结果通过对筛选出的IncRNA进行功能分析,我们发现这些IncRNA主要参与细胞增殖、凋亡、侵袭和转移等生物学过程。
其中,某些关键IncRNA与乳腺癌的预后密切相关,可能成为乳腺癌诊断和治疗的潜在靶点。
3. 实时荧光定量PCR验证结果为了进一步验证筛选出的关键IncRNA的准确性,我们结合临床样本进行了实时荧光定量PCR验证。
利⽤tcga数据做⽣存分析####teach code####library(survival)#read expression data and modify its class and colnames/rownames#PAAD_gene_expression.csv数据已经经过Z_转换的数据。
z_rna <- read.csv(file="PAAD_gene_expression.csv",header = T,s = 1)#clincal是所有样本的临床信息clinical <- t(read.table("PAAD.merged_only_clinical_clin_format.txt",header=T, s=1, sep="\t",stringsAsFactors = F)) clinical <- as.data.frame(clinical)#transform rna data's colnames and rownamesz_rna <- as.matrix(z_rna)colnames(z_rna) <- gsub("\\.","-",colnames(z_rna))rownames(z_rna) <- sapply(rownames(z_rna), function(x) unlist(strsplit(x,"\\|"))[[1]])#check the type of cancer data#table(substr(colnames(z_rna),14,15))#grep patient ID in clinical dataclinical$IDs <- toupper(clinical$patient.bcr_patient_barcode)sum(clinical$IDs %in% colnames(z_rna))# we have 178 patients that we could use#grep information for survivalind_keep <- grep("days_to_death",colnames(clinical))death <- as.matrix(clinical[,ind_keep])death_collapsed <- apply(death,1,FUN = function(x)max(x,na.rm = T)) #提取最⼤值death_collapsed<- as.numeric(death_collapsed)#last follow_up informationind_keep <- grep("days_to_last_followup",colnames(clinical))fl <- as.matrix(clinical[,ind_keep])fl_collapsed <- apply(fl,1,FUN = function(x)max(x,na.rm = T))fl_collapsed <- as.numeric(fl_collapsed)#construct a new dataframe containing all informationall_clin <- data.frame(death_collapsed,fl_collapsed)colnames(all_clin) <- c("death_days", "followUp_days")all_clin$death_event <- ifelse(clinical$patient.vital_status == "alive", 0,1)all_clin$new_death <- apply(all_clin,1,function(x)ifelse(x[3]==0,x[2],x[1]))table(clinical$patient.vital_status)rownames(all_clin) <- clinical$IDs##connect to RNA-seq# create event vector for RNASeq dataevent_rna <- t(apply(z_rna, 1, function(x) ifelse(abs(x) > 1.96,1,0)))in_clin_tum <- match(colnames(z_rna),rownames(all_clin))tumor_clinical <- all_clin[in_clin_tum,]#example geneind_gene <- which(rownames(z_rna) == "TP53")table(event_rna[ind_gene,])###survival analysis####over_all survivalsu_OS <- Surv(as.numeric(tumor_clinical$new_death),as.numeric(tumor_clinical$death_event))fit_OS <- survfit(su_OS~event_rna[ind_gene,])p <- survdiff(su_OS~event_rna[ind_gene,])pv <- ifelse(is.na(p),next,(round(1 - pchisq(p$chisq, length(p$n) - 1),3)))[[1]]x1 <- ifelse(is.na(as.numeric(summary(fit_OS)$table[,'median'][1])),"NA",as.numeric(summary(fit_OS)$table[,'median'][1]))x2 <- as.numeric(summary(fit_OS)$table[,'median'][2])#get necessary informationsummary(fit_OS)summary(fit_OS)$table[,'median']summary(fit_OS,times = 365)#plot survival curveplot(fit_OS,conf.int=FALSE,col=c("black","red"),xlab="Days",ylab = "Proportion Survival")legend(1800,0.995,legend=paste("p.value = ",pv[[1]],sep=""),bty="n",cex=1.4)legend(max(as.numeric(as.character(all_clin$death_days)[in_clin_tum]),na.rm = T)*0.7,0.94, legend=c(paste("NotAltered=",x1),paste("Altered=",x2)),bty="n",cex=1.3,lwd=3,col=c("black","red"))。
WGCNA分析(转载)今天推荐给大家一个R包WGCNA,针对我们的表达谱数据进行分析。
简单介绍:WGCNA首先假定基因网络服从无尺度分布,并定义基因共表达相关矩阵、基因网络形成的邻接函数,然后计算不同节点的相异系数,并据此构建系统聚类树。
该聚类树的不同分支代表不同的基因模块(module),模块内基因共表达程度高,而分属不同模块的基因共表达程度低。
主要应用:如果某些基因在一个生理过程或不同组织中总是具有相类似的表达变化,那么我们有理由认为这些基因在功能上是相关的,可以把它们定义为一个模块(module)。
当基因module被定义出来后,我们可以利用这些结果做很多进一步的工作,如筛选module的核心基因,关联性状,代谢通路建模,建立基因互作网络等。
好了,言归正传,我们开始一步步进行演示!载入WGCNA包,设置随机种子,默认数据不进行因子转换library(WGCNA)set.seed(1)options(stringsAsFactors = F)构造性状数据(亦或是分组数据)obs<-paste("sam",1:10,sep="")sample<-as.data.frame(diag(x=1,nrow = length(obs)))rownames(sample)<-obscolnames(sample)<-1:10构造表达量数据datExpr<-as.data.frame(t(matrix(runif(30000)+5,3000,10)))rownames(datExpr)<-obsnames(datExpr)<-paste("transcript",1:3000,sep="")明确样本数和基因数nGenes = ncol(datExpr)nSamples = nrow(datExpr)首先针对样本做个系统聚类树datExpr_tree<-hclust(dist(datExpr), method = "average")par(mar = c(0,5,2,0))plot(datExpr_tree, main = "Sample clustering", sub="", xlab="", b = 2, cex.axis = 1, cex.main = 1,b=1)针对前面构造的样品矩阵添加对应颜色sample_colors <- numbers2colors(sample, colors = c("white","blue"),signed = FALSE)构造10个样品的系统聚类树及性状热图par(mar = c(1,4,3,1),cex=0.8)plotDendroAndColors(datExpr_tree, sample_colors, groupLabels = colnames(sample), cex.dendroLabels = 0.8, marAll = c(1, 4, 3, 1), cex.rowText = 0.01, main = "Sample dendrogram and trait heatmap")这个有什么意义呢?你可以将样本分为正常组和对照组,或者野生型和突变型等,从而可以查看样本聚类情况!针对10个样品绘制主成分图(在这里不考虑分组情况)pca = prcomp(datExpr)sampletype<-rownames(sample)par(mar =c(4,4,4,6))plot(pca$x[,c(1,2)],pch=16,col=rep(rainbow(nSamples),each=1),cex=1.5,main = "PCAmap")text(pca$x[,c(1,2)],s(pca$x),col="black",pos=3,cex=1)legend("right",legend=sampletype,ncol = 1,xpd=T,inset = -0.15, pch=16,cex=1,col=rainbow(length(sampletype)),bty="n")library(scatterplot3d)par(mar = c(4,4,4,4))scatterplot3d(pca$x[,1:3], highlight.3d=F,col.axis="black",color = rep(rainbow(nSamples),each=1),cex.symbols=1.5,b=1,cex.axis=1,col.grid="lightblue", main="PCA map", pch=16)legend("topleft",legend = s(pca$x),pch=16,cex=1,col=rainbow(nSamples), ncol = 2,bty="n")选择合适“软阀值(soft thresholding power)”betapowers = c(1:30)pow<-pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)设置网络构建参数选择范围,计算无尺度分布拓扑矩阵par(mfrow = c(1,2))plot(pow$fitIndices[,1], -sign(pow$fitIndices[,3])*pow$fitIndices[,2],xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n", main = paste("Scale independence"))text(pow$fitIndices[,1], -sign(pow$fitIndices[,3])*pow$fitIndices[,2],labels=powers,cex=0.5,col="red")plot(pow$fitIndices[,1], pow$fitIndices[,5],xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",main = paste("Mean connectivity"))text(pow$fitIndices[,1], pow$fitIndices[,5], labels=powers, cex=0.6,col="red")参数beta取值默认是1:30,上述图形的横轴均代表权重参数β,左图纵轴代表对应的网络中log(k)与log(p(k))相关系数的平方。
TCGA大作战——初步分析RNA-seq数据01本篇为第一部分,主要记录重要资源地址以及TCGA数据的下载方式。
名词及资源TCGA (The Cancer Genome Atlas):人类癌症基因组图谱,数据库,主要用来收集癌症病人癌组织及癌旁正常组织标本以及极少量正常人相应组织的对照标本(并非每种癌都有),通过多种高通量方法,获取DNA、RNA乃至蛋白多个分子层面的数据;另一方面,它还收集了病人的临床宏观层面信息(诸如肿瘤的分期和分级,患者生存时间,患者的年龄、性别、种族等等),更进一步,该数据库对原始数据进行了标准化处理,并做了常见的后续功能分析,但缺乏一定的针对性。
目前,它已经收录了超过10000名病人,30多种癌症的高达2.5PB的多维数据。
从2016年开始,TCGA的数据库已经逐步迁移到GDC (Genomic Data Commons, 基因组数据共享)网站去了,官网称2017年TCGA将会关闭,距2018还有几天,加油!什么是癌旁正常组织?“Tumor, Matched Normal”Vs. “Normal, Matched Tumor”TCGA数据来源、分级及处理流程(pipeline)的具体信息,尽在维基。
TCGA WIKI生命现象的一个重要特征就是多维性(多层次性),从分子层面直至生命个体,可以划分出数个大的层次,每个层次又包含诸多细分维度,你可能觉得TCGA已经很庞大、很全面了,但其实你还可以想到很多未被纳入的层面信息。
说这些,目的是谈谈GDC 的由来,它不仅是TCGA的新东家,它还左拥右抱了好几个大型数据库,如TARGET (Therapeutically Applicable Research To Generate Effective Treatments), CGCI (Cancer Genome Characterization Initiative)等等,这也许是今后的大趋势,分类数据库的整合,以期能让研究者一站式获取各维度信息,做出更准确的发现。
⼀⽂搞懂TCGA中的分析结果如何来TCGA对于不同类型的数据,有着独特的处理流程,具体如下1. DNA-Seq Analysis PipelineTCGA中的DNA测序主要⽤来分析肿瘤患者中的体细胞突变,和GATK的体细胞突变流程类似,前期都经过了⼀个预处理步骤,这⾥称之为co-cleanning, 流程⽰意如下就是经典的sort->markduplicate->Realign->BQSR步骤,得到co-cleaned BAM⽂件。
然后⽤配对的肿瘤和正常样本进⾏somatic variant calling, 得到VCF⽂件。
然后进⾏体细胞突变的注释,得到突变注释⽂件MAF, ⽰意如下在进⾏体细胞突变位点分析时,使⽤了以下4款不同的软件同时分析1. MuSE2. Mutect23. SomaticSniper4. Varscan2各⾃对应的pipeline⽰意如下各⾃pipeline得到的VCF⽂件,使⽤VEP软件对体细胞突变位点进⾏注释,使⽤了以下数据库进⾏注释1. GENCODE v.222. sift v.5.2.23. ESP v.201411034. polyphen v.2.2.25. dbSNP v.1466. Ensembl genebuild v.2014-077. Ensembl regbuild v.13.08. HGMD public v.201549. ClinVar v.201601注释完成之后,会对突变位点进⾏过滤,去除低质量的突变位点和潜在的⽣殖细胞突变位点,剩余的位点作为最终的体细胞突变位点,保存在MAF⽂件中供下载。
当然对于没有配对的正常样本,也有tumor-only variant calling workflow来处理,具体请参考以下链接https:///Data/Bioinformatics_Pipelines/DNA_Seq_Variant_Calling_Pipeline 2. mRNA Analysis PipelinemRNA分析是通过STAR的2-pass模式⽐对hg38参考基因组,然后使⽤HTSeq进⾏定量,定量时基于Gencode V22版本的GTF⽂件,流程⽰意如下在定量时,提供了以下3种策略1. Raw count2. FPKM3. FPKM-UQRaw count和FPKM是转录组分析中经典的定量策略,⽽FPKM-UQ则是在FPKM基础上新提出的⼀种策略,计算公式如下和FPKM不同的是,在FPKM-UQ中采⽤所有基因Mapping reads数⽬的上四分位数代替了所有基因Mapping Reads的总数。
TCGA的乳腺癌RNA-seq数据WGCNA分析示例WGCNA(Weighted Correlation Network analysis)是一个基于基因表达数据,构建基因共表达网络的方法。
WGCNA 和差异基因分析(DEG)的差异在于DEG主要分析样本和样本之间的差异,而WGCNA主要分析的是基因和基因之间的关系。
WGCNA通过分析基因之间的关联关系,将基因区分为多个模块。
而最后通过这些模块和样本表型之间的关联性分析,寻找特定表型的分子特征。
网上例子千千万,但是大部分都是从文档翻译而来,要用起来还是有些费劲,要深入的可以移步这里:/~yandell/statgen/ucla/WGCNA/wgcna. html下面我将根据TCGA乳腺癌基因表达数据以及乳腺癌压型数据,一步一步的使用WGCNA来进行乳腺癌各个亚型共表达模块的挖掘#############数据准备#############首先我们需要下载TCGA 的乳腺癌的RNA-seq数据以及临床病理资料,我这里使用我们自己开发的TCGA简易下载工具进行下载首先下载RNA-Seq:下载之后共得到1215个样本表达数据进一步下载临床病理资料进一步点击ClinicalFull 按钮对病理资料进行提取得到ClinicalFull_matrix.txt文件,使用Excel打开ClinicalFull_matrix.txt文件可以看到共有301列信息,包含了各种用药,随访,预后等等信息,我们这里选择乳腺癌ER、PR、HER2的信息,去除其他用不上的信息,然后选择了其中有明确ER、PR、HER2阳性阴性的样本,随机拿100个做例子吧样本筛选完了,现在轮到怎么获取这些样本的RNA-seq数据啦,前面下载了一千多个样本的RNA-seq,从里面找到这一百个样本的表达数据其实也是不需要变成的啦,看清楚咯首先打开RNA-Seq数据目录的fileID.tmp(用Excel打开),然后可以看到两列:将第二列复制,并且替换-01.gz为空使用Excel的vlookup命令将临床病理资料的那100个样本进行映射然后筛选非N/A的就得到了这一百个样本对于的RNA-seq数据信息进一步删除其他的样本,还原成fileID.tmp格式保存退出:然后使用TCGA简易小工具“合并文件”按钮就得到表达矩阵了,进一步使用ENSD_ID转换按钮就得到了基因表达矩阵和lncRNA表达矩阵了#################R代码实现WGCNA##############setwd('E:/rawData/TCGA_DATA/TC GA-BRCA')samples=read.csv('ClinicalFull_matrix.txt',sep = '\t',s = 1)dim(samples)#[1] 100 3expro=read.csv('Merge_matrix.txt.cv.txt',sep = '\t',s = 1)dim(expro)#[1] 24991 100数据读取完成,从上述结果可以看出100个样本,有24991个基因,这么多基因全部用来做WGCNA 很显然没有必要,我们只要选择一些具有代表性的基因就够了,这里我们采取的方式是选择在100个样本中方差较大的那些基因(意味着在不同样本中变化较大)继续命令:m.vars=apply(expro,1,var)expro.upper=expro[which(m.vars>quantile(m.vars, probs =seq(0, 1, 0.25))[4]),]##选择方差最大的前25%个基因作为后续WGCNA的输入数据集通过上述步骤拿到了6248个基因的表达谱作为WGCNA的输入数据集,进一步的我们需要看看样本之间的差异情况datExpr=as.data.frame(t(expro.upper)); gsg = goodSamplesGenes(datExpr, verbose = 3);gsg$allOKsampleTree = hclust(dist(datExpr), method = 'average')plot(sampleTree, main = 'Sample clustering to detect outliers' , sub='', xlab='')从图中可看出大部分样本表现比较相近,而有两个离群样本,对后续的分析可能造成影响,我们需要将其去掉,共得到98个样本clust =cutreeStatic(sampleTree, cutHeight = 80000, minSize = 10) table(clust)#clust#0 1#2 98keepSamples = (clust==1)datExpr = datExpr[keepSamples, ]nGenes = ncol(datExpr)nSamples = nrow(datExpr)save(datExpr, file = 'FPKM-01-dataInput.RData')得到最终的数据矩阵之后,我们需要确定软阈值,从代码中可以看出pickSoftThreshold很简单,就两个参数,其他默认即可powers = c(c(1:10), seq(from = 12, to=20, by=2))sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)##画图##par(mfrow = c(1,2));cex1 = 0.9;plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2], xlab='Soft Threshold (power)',ylab='Scale Free Topology Model Fit,signed R^2',type='n',main = paste('Scale independence'));text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2], labels=powers,cex=cex1,col='red');abline(h=0.90,col='red')plot(sft$fitIndices[,1], sft$fitIndices[,5],xlab='Soft Threshold (power)',ylab='Mean Connectivity', type='n',main = paste('Mean connectivity'))text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers,cex=cex1,col='red')从图中可以看出这个软阈值选择7比较合适,选择软阈值7进行共表达模块挖掘pow=7net = blockwiseModules(datExpr, power = pow, maxBlockSize = 7000,TOMType = 'unsigned', minModuleSize = 30,reassignThreshold = 0, mergeCutHeight = 0.25,numericLabels = TRUE, pamRespectsDendro = FALSE,saveTOMs = TRUE,saveTOMFileBase = 'FPKM-TOM',verbose = 3)table(net$colors)# open a graphics window#sizeGrWindow(12, 9)# Convert labels to colors for plottingmergedColors = labels2colors(net$colors)# Plot the dendrogram and the module colors underneath plotDendroAndColors(net$dendrograms[[1]],mergedColors[net$blockGenes[[1]]],groupLabels = c('Module colors','GS.weight'),dendroLabels = FALSE, hang = 0.03,addGuide = TRUE, guideHang = 0.05)从图中可以看出大部分基因在灰色区域,灰色部分一般认为是没有模块接受的,从这里也可以看出其实咱们选择的这些基因并不是特别好那么做到这一步了基本上共表达模块做完了,每个颜色代表一个共表达模块,统计看看各个模块下的基因个数:那么得到模块之后下一步该做啥呢,或许很多人到这就不知道如何继续分析了这里就需要咱们利用这些模块搞事情了,举个例子如果你是整合的数据(整合lnc与gene),那么同时在某个模块中的基因和lncRNA咱们可以认为是共表达的,这便是lnc-gene共表达关系的获得途径之一了,进一步你可以根据该模块的基因-lnc-基因之间的关系绘制出共表达网络今天咱们这里不讲这个,而是跟表型关联,咱们已经拿到了这98个样本的ER、PR、HER2阳性阴性信息,那么进一步的咱们可以看看哪些共表达模块跟ER、PR、HER2阴性最相关,代码如下:moduleLabelsAutomatic =net$colorsmoduleColorsAutomatic =labels2colors(moduleLabelsAutomatic) moduleColorsFemale = moduleColorsAutomaticMEs0 = moduleEigengenes(datExpr, moduleColorsFemale)$eigengenesMEsFemale = orderMEs(MEs0)samples=samples[match(s(datExpr),paste0(gsub('-','.',r s(samples)),'.01')),]#匹配98个样本数据trainDt=as.matrix(cbind(ifelse(samples[,1]=='Positive',0,1),#将阴性的样本标记为1ifelse(samples[,2]=='Positive',0,1),#将阴性的样本标记为1ifelse(samples[,3]=='Positive',0,1),#将阴性的样本标记为1ifelse(samples[,1]=='Negative'&samples[,2]=='Negative'&sampl es[,3]=='Negative',1,0))#将三阴性的样本标记为1)#得到一个表型的0-1矩阵modTraitCor = cor(MEsFemale, trainDt, use = 'p')colnames(MEsFemale)modTraitP = corPvalueStudent(modTraitCor, nSamples)textMatrix = paste(signif(modTraitCor, 2), '\n(',signif(modTraitP, 1), ')', sep = '')dim(textMatrix) = dim(modTraitCor)labeledHeatmap(Matrix = modTraitCor, xLabels =colnames(trainDt), yLabels = names(MEsFemale),ySymbols = colnames(modlues), colorLabels = FALSE, colors = greenWhiteRed(50),textMatrix = textMatrix, setStdMargins = FALSE, cex.text = 0.5, zlim = c(-1,1), main = paste('Module-trait relationships'))最终找到几个共表达网络与三阴性表型最相关的模块。