Facile Synthesis of Hierarchically Ordered Porous Carbon via in Situ
- 格式:pdf
- 大小:3.48 MB
- 文档页数:8
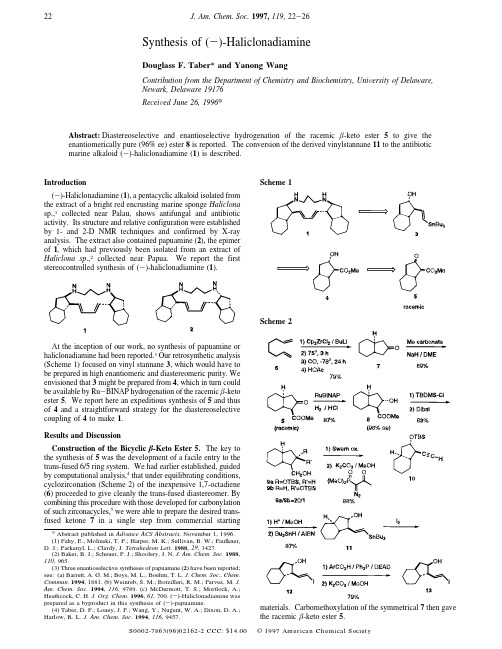
Synthesis of (-)-HaliclonadiamineDouglass F.Taber*and Yanong WangContribution from the Department of Chemistry and Biochemistry,Uni V ersity of Delaware,Newark,Delaware 19176Recei V ed June 26,1996XAbstract:Diastereoselective and enantioselective hydrogenation of the racemic -keto ester 5to give the enantiomerically pure (96%ee)ester 8is reported.The conversion of the derived vinylstannane 11to the antibiotic marine alkaloid (-)-haliclonadiamine (1)is described.Introduction(-)-Haliclonadiamine (1),a pentacyclic alkaloid isolated from the extract of a bright red encrusting marine sponge Haliclona sp.,1collected near Palau,shows antifungal and antibiotic activity.Its structure and relative configuration were established by 1-and 2-D NMR techniques and confirmed by X-ray analysis.The extract also contained papuamine (2),the epimer of 1,which had previously been isolated from an extract of Haliclona sp .,2collected near Papua.We report the first stereocontrolled synthesis of (-)-haliclonadiamine (1).At the inception of our work,no synthesis of papuamine or haliclonadiamine had been reported.3Our retrosynthetic analysis (Scheme 1)focused on vinyl stannane 3,which would have to be prepared in high enantiomeric and diastereomeric purity.We envisioned that 3might be prepared from 4,which in turn could be available by Ru -BINAP hydrogenation of the racemic -keto ester 5.We report here an expeditious synthesis of 5and thus of 4and a straightforward strategy for the diastereoselective coupling of 4to make 1.Results and DiscussionConstruction of the Bicyclic -Keto Ester 5.The key to the synthesis of 5was the development of a facile entry to the trans-fused 6/5ring system.We had earlier established,guided by computational analysis,4that under equilibrating conditions,cyclozirconation (Scheme 2)of the inexpensive 1,7-octadiene (6)proceeded to give cleanly the trans-fused diastereomer.By combining this procedure with those developed for carbonylation of such zirconacycles,5we were able to prepare the desired trans-fused ketone 7in a single step from commercial startingmaterials.Carbomethoxylation of the symmetrical 7then gave the racemic -keto ester 5.XAbstract published in Ad V ance ACS Abstracts,November 1,1996.(1)Fahy,E.;Molinski,T.F.;Harper,M.K.;Sullivan,B.W.;Faulkner,D.J.;Parkanyl,L.;Clardy,J.Tetrahedron Lett.1988,29,3427.(2)Baker,B.J.;Scheuer,P.J.;Shoolery,J.N.J.Am.Chem.Soc.1988,110,965.(3)Three enantioselective syntheses of papuamine (2)have been reported;see:(a)Barrett,A.G.M.;Boys,M.L.;Boehm,T.L.J.Chem.Soc.,mun.1994,1881.(b)Weinreb,S.M.;Borzilleri,R.M.;Parvez,M.J.Am.Chem.Soc.1994,116,9789.(c)McDermott,T.S.;Mortlock,A.;Heathcock,.Chem .1996,61,700.(-)-Haliclonadiamine was prepared as a byproduct in this synthesis of (-)-papuamine.(4)Taber,D.F.;Louey,J.P.;Wang,Y.;Nugent,W.A.;Dixon,D.A.;Harlow,R.L.J.Am.Chem.Soc .1994,116,9457.Scheme 1Scheme 222J.Am.Chem.Soc.1997,119,22-26S0002-7863(96)02162-2CCC:$14.00©1997American Chemical SocietyKinetic Resolution by Catalytic Hydrogenation.With the racemic -keto ester5in hand,we were then faced with the problem of carrying it on to the enantiomerically pure and diastereomerically defined -hydroxy ester4.We thought that this might be accomplished by kinetic resolution under the conditions of Ru-BINAP hydrogenation.6,7The reduction was known6,7to give predominantly the trans- -hydroxy ester when applied to a cyclic -keto ester.This reduction is thought to proceed by complexation of the intermediate Ru-H species with the ester carbonyl with subse-quent delivery of the nucleophilic H to the ketone.As one enantiomer of the -keto ester would fit the twist of the BINAP ligand and the other would not,we thought that one enantiomer of the ketone might reduce much more rapidly than the other. Kinetic resolution in this manner had been reported8but not with good diastereocontrol.In the event(Table1),we found that the racemic -keto ester 5only hydrogenated smoothly in the presence of added HCl.8 By optimizing the amount of HCl added,we could control the proportion of total ketone reduced.We were pleased to observe that we could convert nearly90%of the“matched”ketone without significant reduction of the other enantiomer.It is interesting that if too much HCl is added initially,the(S)-BINAP-RuCl2will reduce the mismatched enantiomer also, to a mixture of diastereomers.The catalyst turnover reported in these studies is lower by an order of magnitude than that previously observed.6-8We do not believe that this limitation is inherent.The reactions were optimized originally at the half millimole scale,and the amount of catalyst used was convenient at that scale.The diastereoselectivity of the hydrogenation depends on the particular acid promoter8employed.With HCl,the trans diastereomer was dominant.With acetic acid as the promoter, the reaction was still fast,but the major product from the reduction was the cis diastereomer.Establishment of the Enantiomeric Purity of9a.Silylation of the hydroxy ester4followed by reduction gave the primary alcohols9a,b.The minor amount of9b,from the cis alcohol produced in the reduction,was removed at this stage.To establish the enantiomeric purity of9a,we prepared the camphorsulfonate15.The camphorsulfonate prepared from the racemic trans alcohol(from sodium borohydride reduction of the starting -keto ester)showed nice resolution of the oxygen-ated carbons in the13C NMR(δ74.0/73.8and70.0/69.9).By this measurement,9a was a98:2ratio of enantiomers. Construction of the Iodo Stannane18.Following this procedure,we were able to prepare alcohol9a in high enantiomeric and diastereomeric purity in just five steps from 1,7-octadiene(6).To move on to the haliclonadiamine precursor 18,we had to homologate the primary alcohol of9a to the(E)-vinylstannane.We also had to construct the N,N′-diaminopro-pane from two equivalents of the secondary diol,with inversion of absolute configuration in one case and with retention (probably double inversion)in the other.To effect homologation,alcohol9a was oxidized to the aldehyde,which was subjected without purification to the Ohira procedure9to give the alkyne10.Our experience has been that this simple procedure,which can be effected at ambient temperature,uses easily available reagents,and is not sensitive to stoichiometry,is in general the method of choice for this common transformation.Deprotection of10followed by radical addition of tributyltin hydride10then provided an87%yield of the(E)-vinylstannane11.We next faced the difficulty of inverting the hindered secondary alcohol of11.To this end,we converted a portion of the stannane11that we had prepared to the iodide12.As expected,Mitsunobu coupling12with4-nitrobenzoic acid initially was sluggish.We were pleased to observe that substitution of benzene for THF as the reaction medium solved this problem. Mild transesterification then liberated the desired cis alcohol 13,in75%overall yield from the iodide12.The final challenge that we faced was construction of the N,N′-dialkyl1,3-diaminopropane of18(Scheme3).It seemed plausible that Mitsunobu coupling could again be efficacious. The key to this approach was the selection of an activating group that would make the N-H sufficiently acidic to participate in the Mistsunobu reaction while leaving the anion sufficiently(5)(a)Rousset,C.J.;Swanson,D.R.;Lamaty,F.;Negichi,E.-i. Tetrahedron Lett.1989,30,5105.(b)Akita,M.;Yasuda,H.;Yamamoto,H.;Nakamura,A.Polyhedron1991,10,1.(6)For the first report of the reduction of Ru-BINAP-mediated hydrogenation of -keto esters to provide the -hydroxy esters with excellent yield and enantioselectivity,see:Noyori,R.;Ohkuma,T.;Kitamura,M. J.Am.Chem.Soc.1987,109,5856.(7)For further studies of Ru-BINAP-mediated hydrogenation of -keto esters,see:(a)Cesarotti, E.;Mauri, A.;Pallavicini,M.;Villa,L. Tetrahedron Lett.1991,32,4381.(b)Kitamura,M.;Tokunaga,M.;Ohkuma, T.;Noyori,R.Tetrahedron Lett.1991,32,4163.(c)Kitamura,M.; Tokunaga,M.;Noyori,.Chem.1992,57,4053.(d)Taber,D.F.; Silverberg,L.J.Tetrahedron Lett.1991,32,4227.(e)King,S. A.; Thompson,A.S.;King,A.O.;Verhoeven,.Chem.1992,57, 6689.(f)Kitamura,M.;Tokunaga,M.;Noyori,R.J.Am.Chem.Soc.1993, 115,144.(g)Genet,J.P.;Pinel,C.;Ratovelomanana-Vidal,V.;Mallart, S.;Pfister,X.;Cano De Andrade,M.C.;Laffitte,J.A.Tetrahedron1994, 5,665.(h)Mashima,K.;Kusano,K.;Sato,N.;Matsumura,Y.;Nozaki, K.;Kumobayashi,H.;Sayo,N.;Hori,Y.;Ishizaki,T.;Akutagawa,S.;Takaya,.Chem.1994,59,3064.(i)Burk,M.J.;Harper,T.G.P.; Kalberg,C.S.J.Am.Chem.Soc.1995,117,4423.(8)(a)For the role of added acid in Ru-BINAP hydrogenation,see ref 7d,e.(b)The procedure described here represents a significant improvement over that previously described for kinetic resolution of a bicyclic -keto ester:Kitamura,M.;Ohkuma,T.;Tokunaga,M.;Noyori,R.Tetrahedron: Asymmetry1990,1,1.(9)Ohira,mun.1989,19,561.(10)(a)Stille,J.K.Angew.Chem.,Int.Ed.Engl.1986,25,508.(b) Zhang,H.X.;Guibe,F.;Balavoine,.Chem.1990,55,1857.(11)Hanson,R.N.;El-Wakil,.Chem.1987,52,3687.(12)(a)Martin,S.F.;Dodge,J.A.Tetrahedron Lett.1991,32,3017.(b)Caine,D.;Kotian,.Chem.1992,57,6587.Table1.Hydrogenation of -Keto Ester5aHCl (mol%)conversion(%)(of racemate)e of desireddiastereomer(%)13.098.444.79.543.5968.133.01006.229.9100a Hydrogenation pressure,52psi;temperature,80-85°C;time,14h;HCl,methanolic HCl;RuCl2-BINAP,1.2mol%.Synthesis of(-)-Haliclonadiamine J.Am.Chem.Soc.,Vol.119,No.1,199723nucleophilic.The activating group should also be easily removed.We found that the N ,N ′-bistriflamide nicely met these criteria.13While the Mitsunobu coupling again worked poorly in THF,reaction of 11in benzene with two equivalents of N ,N ′-bistriflyl-1,3-diaminopropane (16)gave clean conversion to the monoalkylated diamide 17.The final Mitsunobu coupling of 17with 13also proceeded smoothly in benzene,to give the desired N ,N ′-dialkylated diamide 18.Synthesis of (-)-Haliclonadiamine (1).With the iodo stannane 18in hand,the final assembly of (-)-haliclonadiamine (1)was straightforward (Scheme 3).Thus,following the report by Barrett 3a in the synthesis of (+)-papuamine,slow addition of 18to 20mol %of Pd(PPh 3)4at 100-105°C in toluene followed by 36h at reflux gave the cyclized product 19in 43%yield.Deprotection of the bistriflamide 19with LiAlH 4in ether 14(LiAlH 4in THF was not effective)then gave the free base of (-)-haliclonadiamine (1).The 1H NMR,13C NMR,and MS of synthetic 1were identical with the spectra of natural (-)-haliclonadiamine.1The 1H NMR was also identical with that recorded for a synthetic sample.3c ConclusionDuring the course of this total synthesis of the unusual marine alkaloid (-)-haliclonadiamine (1),an efficient diastereoselective preparation of the -keto ester 5via intramolecular diene cyclozirconation and carbonylation was developed.The dias-tereoselective and enantioselective Ru -BINAP-mediated hy-drogenation of -keto ester 5to the -hydroxy ester 4was also achieved.Finally,the sequential Mitsunobu coupling with bistriflamide 16as the nucleophile proved very efficient even with the sterically hindered secondary alcohols 11and 13.Experimental Section 15-Keto Ester 5.To 1,7-octadiene (11.52g,0.105mol)and zirconocene dichloride (36.7g,0.125mol)in toluene (250mL)was added n -BuLi (1.99M,126mL)via syringe at -78°C over 30min under argon.The resulting light yellow mixture was stirred at room temperature and then warmed to 75-80°C for 3h.After cooling to room temperature,the dark brown reaction mixture was further cooledwith liquid N 2and evacuated to about 0.001mmHg.The solidified reaction mixture was maintained at -78°C,and CO was charged into the reaction container so that the pressure of CO was kept at 1.1-1.2atm for 1h,until no obvious CO absorbance was observed.The reaction mixture was warmed to room temperature and stirred 12h.HOAc (25.1mL)was added to the dark reaction mixture.The resulting yellow solution was stirred at room temperature for 30min;then the reaction was quenched with 10%aqueous H 2SO 4.The mixture was extracted with 20%EtOAc/petroleum ether.The combined organic extract was dried (Na 2SO 4)and concentrated.The residue was distilled to give 11.5g (79%yield)of the ketone 7:bp 0.05mmHg )80-90°C (pot);1H NMR δ2.44-2.28(m,2H),2.05-1.02(m,12H);13C NMR δ218.3(C),45.6(CH 2),43.9(CH),31.3(CH 2),26.1(CH 2).To a suspension of NaH (60%in mineral oil,579mg,14.5mmol)in DME (8mL)containing a trace of methanol was added dimethyl carbonate (978mg,10.9mmol).The mixture was heated to reflux for 10min,then the bicyclic cyclopentanone 7(500mg,3.62mmol)was added over two h.The reaction mixture at maintained at reflux overnight,then was quenched with HOAc (1.0mL)and partitioned between H 2O and CH 2Cl 2.The combined organic extract was dried (Na 2SO 4),concentrated and chromatographed to give 489mg (55%yield from 1,7-octadiene)of the -keto ester 5:TLC R f )0.34(10%EtOAc/petroleum ether);1H NMR δ3.72(s,3H),2.84(d,J )12.4Hz,1H),2.42(dd,J )17.9,6.7Hz,1H),2.03-1.16(m,11H);13C NMR δu 210.1,169.5,44.9,31.1,30.2,26.0,25.8;d 61.7,52.2,47.3,41.0;IR (neat)(cm -1)1759,1728,1436;MS (CI)m /z 196(M +,11),168(58),94(100).HRMS:m /e calcd for C 11H 17O 3(MH +),197.1178;found,197.1177.BINAP -RuCl 2.A 5mL reactivial equipped with a stir vane was charged with (RuCl 2-cyclooctadiene)n (39mg),(S )-(-)-2,2′-bis-(diphenylphosphino)-1,1′-binaphthyl (100mg),triethylamine (200mg),and toluene (4mL)in the dry box.The vial was capped securely and then heated in a 140-145°C oil bath for 3h,until a clear red solution was achieved.The contents of the vial were transferred with toluene to a 100mL round-bottom flask under nitrogen.The reaction mixture was concentrated in V acuo under the Schlenk line,and the residue was taken up in THF (10mL)in the dry box.The brown THF solution was divided into 10×1mL portions,which were stored in stoppered vials under N 2.Hydroxy Ester 8.Under N 2,a modified Parr bottle equipped with a stir bar was charged with racemic bicyclic -keto ester 5(250mg,1.28mmol),methanol (1.65mL),BINAP -RuCl 2as prepared above (0.5mL,from 5mg of BINAP),and 1.35mL (0.162mmol)of a 0.12N HCl -methanol solution (prepared by dissolving 1.0mL of concen-trated aqueous HCl in 99mL of methanol).The bottle was flushed with hydrogen for 5min,and hydrogenation was then carried out at 50-52psi of H 2for 14h in an 80-85°C oil bath.The reaction mixture was concentrated in V acuo and chromatographed to give 132mg of residual -keto ester and 110mg (87%of theory)of the product hydroxy ester 8as a colorless oil,which subsequent analysis showed to be 96%ee:TLC R f )0.19(10%EtOAc/petroleum ether);1H NMR δ4.48(m,1H),3.71(s,3H),2.75(s,br,1H),2.33(dd,J )11.1,5.1Hz,1H),1.95-0.98(m,12H);13C NMR δu 175.3,41.0,31.4,30.5,25.9(2);d 75.0,59.9,51.6,49.2,43.8;IR (neat)(cm -1)3445,1733;MS (CI)m /z 199(M ++H,1),170(80),74(100).HRMS:m /e calcd for C 11H 19O 3(MH +),199.1334;found,199.1328.Protected Diol 9.A mixture of the hydroxy ester (581mg,2.93mmol),imidazole (999mg,14.7mmol),and tert -butyldimethylsilyl chloride solution (2.0M in CH 2Cl 2,2.2mL,4.40mmol)in dry CH 2-Cl 2(14mL)was stirred at room temperature overnight.The reaction mixture was partitioned between saturated aqueous NaHCO 3and CH 2-Cl 2.The combined organic extract was dried (Na 2SO 4),concentrated,and chromatographed to give 920mg of the silyl ether ester (99%)as a colorless oil:TLC R f )0.80(10%EtOAc/petroleum ether);1H NMR δ4.42(m,1H),3.85(s,3H),2.30(dd,1H),2.90-1.42(m,6H),1.35(m,4H),0.90(s,9H),-0.02(s,6H);13C NMR δ175.75;IR (neat)(cm -1)2927,2855,2361,1735;MS (EI)m /z 312(M +,1),255(69),89(100).HRMS:m /e calcd for C 17H 31O 3Si (M +),312.2042;found,312.2056.Dibal-H (1.0M solution in CH 2Cl 2,1.64mL)was added dropwise via syringe to the solution of the silyl ether ester (170mg,0.545mmol)in CH 2Cl 2(5mL)at -78°C over 15min.The reaction mixture was(13)After this work was completed,an independent report of efficient Mitsunobu coupling of an alkyl triflamide appeared;see:Bell,K.E.;Knight,D.W.;Gravestock,M.B.Tetrahedron Lett.1995,36,8681.(14)(a)Hendrickson,J.B.;Bergeron,R.Tetrahedron Lett .1973,3839.(b)Hendrickson,J.B.;Bergeron,R.;Sternbach,D.J.Am.Chem.Soc.1973,95,3412.(15)For general experimental procedures,see:Taber,D.F.;Houze,.Chem.1994,59,4004.Scheme 324J.Am.Chem.Soc.,Vol.119,No.1,1997Taber and Wangstirred at-78°C for1h and then partitioned between saturated aqueous NaHCO3and CH2Cl2.The combined organic extract was dried(Na2-SO4),concentrated,and chromatographed to give117mg of trans-monoprotected diol9a(76%yield)and6mg(3.9%)of cismonopro-tected diol9b.9a:TLC R f)0.53(10%EtOAc/petroleum ether);1H NMRδ4.08 (m,1H),3.75(dd,J)10.5,4.6Hz,1H),3.65(m,1H),1.86-0.91 (m,13H),0.88(s,9H),0.059(s,3H),0.055(s,3H);13C NMRδu 63.7,41.4,31.7,30.4,26.3,26.2,18.0;d75.8,56.9,46.5,43.9,25.8, -4.4,-4.8;IR(neat)(cm-1)3359,1471,1446;MS(EI)m/z269(M+ -Me,1),135(90),75(100);HRMS:m/e calcd for C16H33O2Si(MH+), 285.2250;found,285.2255.9b:TLC R f)0.65(10%EtOAc/petroleum ether);1H NMRδ4.38 (dd,J)13.4,7.1Hz,1H),3.85(ddd,J)3.0,2.9,11.5Hz,1H), 3.61(m,1H),2.90(dd,J)9.6,3.3Hz,1H),2.20-2.10(m,1H), 1.92-0.98(m,11H),0.88(s,9H),0.059(s,3H),0.055(s,3H).Camphorsulfonate Derivative15.To the trans monoprotected diol 9a(5mg,0.0176mmol)and triethylamine(5.3mg,0.0528mmol)in CH2Cl2(1mL)was added(1S)-camphorsulfonyl chloride(6.6mg, 0.0264mmol).The mixture was stirred at room temperature for24h and then concentrated.The residue was chromatographed to give8.5 mg(99%)of product15:TLC R f)0.72(5%petroleum ether/tert-butyl methyl ether/CH2Cl2)8/2/1).Racemic derivative:1H NMRδ4.31(m,2H),4.06(m,1H), 3.07(d,J)15.0Hz,1H),2.95(d,J)15.0Hz,1H),2.54-2.8(m, 1H),2.12-0.90(m,19H),1.14(s,3H),0.88(s,9H),0.059(s,3H), 0.055(s,3H);13C NMRδu214.4,70.0+69.9(1:1),57.9,47.9, 46.6,42.6,41.6,31.5,30.2,26.9,26.2,26.1,24.9(CH2),19.9,17.9;d 74.0+73.8(1:1),54.5,46.8,43.6,42.8,25.8,19.9,19.7,-4.8,-4.8.Enantiomerically enriched derivative:1H NMRδ4.34(dd,J) 9.7,4.6Hz,1H),4.25(dd,J)9.7,5.1Hz,1H),3.07(d,J)15.0 Hz,1H),2.95(d,J)15.0Hz,1H),2.54-2.8(m,1H),2.12-0.90 (m,19H),1.14(s,3H),0.88(s,9H),0.059(s,3H),0.055(s,3H);13C NMRδu214.4,70.0+69.9(98:2),57.9,47.9,46.6,42.6,41.6,31.5, 30.2,26.9,26.2,26.1,24.9(CH2),19.9,17.9;d74.0+73.8(98:2), 54.5,46.8,43.6,42.8,25.8,19.9,19.7,-4.8,-4.8;IR(neat)(cm-1) 1750,1700,1653,1558.Protected Alkyne10.DMSO(441mg,5.65mmol)in dry CH2Cl2 (2mL)was added dropwise to a solution of oxalyl chloride(388mg, 3.06mmol)in CH2Cl2(3mL)at-78°C.After10min,the alcohol 9a(668mg,2.35mmol)in CH2Cl2(2mL)was added.The reaction mixture was stirred at-78°C for1h and then warmed to-50°C. Triethylamine(1.19g,11.76mmol)was added,the ice bath was removed,and the mixture was allowed to warm to room temperature for5h.The reaction mixture was partitioned between saturated aqueous NaHCO3and CH2Cl2.The combined organic extract was dried (Na2SO4)and concentrated in V acuo to provide a light yellow syrup.To the crude aldehyde in anhydrous methanol(10mL)were added dimethyl(1-diazo-2-oxopropyl)phosphonate(903mg,4.70mmol)and K2CO3(811mg,5.88mmol)successively at0°C.The reaction mixture was gradually warmed to room temperature overnight and then partitioned between saturated aqueous NaHCO3solution and CH2Cl2. The combined organic extract was dried(Na2SO4),concentrated,and chromatographed to give553mg(85%from alcohol9a)of the alkyne product10.TLC R f)0.52(10%EtOAc/petroleum ether);1H NMR δ4.23(m,1H),2.09(d,J)1.8Hz,1H),2.18-0.98(m,13H),1.04 (s,9H),0.06(s,3H),0.05(s,3H);13C NMRδu86.6,69.4,41.8, 31.5,30.0,26.0,25.9,18.1;d78.9,72.3,51.9,46.8,43.4,25.9,-4.5, -4.8;MS(EI)m/z278(1),221(100),145(27).HRMS m/z calcd for C17H30OSi,278.2066;found,278.2059.Vinylstannane11.The mixture of starting material10(180mg, 0.79mmol)and Dowex-50sulfonic acid resin(1.0g)in methanol(5 mL)was stirred at room temperature overnight.The reaction mixture was filtered,and the solid was washed thoroughly with methanol.The filtrate was concentrated in V acuo,and the residue was chromatographed to give122mg(95%yield)of the deprotected hydroxy alkyne:TLC R f)0.52(10%EtOAc/petroleum ether);1H NMRδ4.30(m,1H), 2.12-0.91(m,16H).To a solution of the hydroxy alkyne(188mg,1.15mmol)and AIBN (10mg)in toluene(3mL)was added Bu3SnH at room temperature under argon.The reaction mixture was heated at100-105°C for3h and then stirred at room temperature overnight.The reaction mixture was concentrated and chromatographed to give425mg(87%yield)of the vinylstannane product11as a mixture of isomers(E:Z)16:1): TLC R f)0.57(10%EtOAc/petroleum ether);[R]D)5.4°(c)0.0075, CH2Cl2);1H NMRδ5.92(d,J)18.8Hz,1H),5.79(dd,J)18.8, 7.1Hz,1H),4.05(m,1H),2.02-0.77(m,40H);13C NMRδu40.8, 31.8,30.0,29.1,27.2,26.4,26.3,9.5;d150.3,128.8,77.7,64.4,50.8, 43.8,13.6;IR(neat)(cm-1)3332,2595;MS(EI)m/z455(1),399 (100),177(25).HRMS:m/e calcd for C19H35OSn(M+-Bu), 399.1709.found,399.1717.Vinyl Iodide12.To a solution of the vinylstannane11(135mg, 0.30mmol)in Et2O(2mL)was added iodine(83mg,0.33mmol). After30min,the reaction mixture was partitioned between Et2O(20 mL)and10%aqueous Na2S2O3.The combined organic extract was dried(Na2SO4),concentrated and chromatographed to give83mg(96% yield)of the vinyl iodide12as a colorless oil.TLC R f)0.29(5% EtOAc/petroleum ether);[R]D)-36.4°(c)0.006,CH2Cl2);1H NMR δ6.41(dd,J)13.9,9.0Hz,1H),6.06(d,J)13.9Hz,1H),4.06 (m,1H),2.02-1.94(m,1H),1.91-0.88(m,12H);13C NMRδu 40.8,31.6,29.7,26.1,26.0;d147.6,143.1,76.8,62.5,50.3,43.4;IR (neat)(cm-1)3332,1602,1446;MS(EI)m/z292(3),165(15),147 (42),76(100).HRMS:m/e calcd for C11H17O1I(M+),292.0324.found, 292.0358.Inverted Hydroxy Vinyl Iodide13:To the vinyl iodide12(45 mg,0.15mmol),triphenylphosphine(201mg,0.77mmol),and 4-nitrobenzoic acid(113mg,0.68mmol)in benzene(3mL)was added DEAD(134mg,0.77mmol)dropwise via syringe.The reaction mixture was stirred at room temperature for14h and then concentrated and chromatographed to give55mg(82%)of the4-nitrobenzoate product as a white foam:TLC R f)0.75(5%EtOAc/petroleum ether); 1H NMRδ8.28(d,J)9.0Hz,2H),8.14(d,J)9.0Hz,2H),6.52 (dd,J)14.5,8.9Hz,1H),6.11(d,J)14.5,1H),5.45(dt,J)7.4, 4.5Hz,1H),2.56-2.45(m,1H),2.36-2.35(m,1H),1.92-1.77 (m,4H),1.54-0.86(m,7H);13C NMRδu165.1,135.9,129.0,39.2, 31.6,29.8,26.2,26.0;d143.5,130.6,130.5,129.0,123.6,76.9,54.5,48.6,44.0.A mixture of4-nitrobenzoate(40mg,0.091mmol)and anhydrous K2CO3(0.182mmol)in methanol(4mL)was stirred for24h.The reaction mixture was concentrated,then partitioned between50% EtOAc/petroleum ether and water.The combined organic extract was dried(Na2SO4)and concentrated.The residue was chromatographed to give25mg(96%)of the desired vinyl iodide13as a white foam: TLC R f)0.43(10%EtOAc/petroleum ether).[R]D)-72.0°(c) 0.0058,CH2Cl2);1H NMRδ6.58(dd,J)14.5,9.2Hz,1H),6.05(d, J)14.5Hz,1H),4.26(m,1H),2.30(m,1H),2.04-1.97(m,1H), 1.85-1.71(m,4H),1.58-0.80(m,7H);13C NMRδu41.5,32.7,29.9, 26.4,26.1;d145.1,128.3,73.1,57.9,47.6,44.3;MS(EI)m/z292 (M+,1),147(40),76(100).HRMS:m/e calcd for C11H17O1I(M+), 292.0324;found,292.0348.N,N′-Bistriflyl-1,3-diaminopropane(16).To the mixture of1,3-diaminopropane(200mg,2.70mmol)and Et3N(1.09g,10.8mmol) in CH2Cl2(5mL)was added triflic anhydride(1.60g,5.68mmol)at -78°C over5min.The mixture was stirred at-78°C for1h;then the reaction was quenched with saturated aqueous NaHCO3and the mixture partitioned between CH2Cl2and water.The combined organic extract was dried(Na2SO4),concentrated,and chromatographed to give 674mg of16:TLC R f)0.12(10%EtOAc/petroleum ether);1H NMR δ5.45(bs,2H),3.45(t,J)6.8Hz,4H),1.90(m,2H);13C NMR δ119.6(CF3,J)318Hz),u40.5,31.1;MS(EI)m/z338(1),162 (55),69(100).Vinylstannane Bistriflamide17.To the vinylstannane11(133mg, 0.29mmol),N,N′-bistriflyl-1,3-diaminopropane(16)(194mg,0.58 mmol),and triphenylphosphine(191mg,0.73mmol)in benzene(3 mL)was added DEAD(127mg,0.73mmol)via syringe dropwise at room temperature.The reaction mixture was stirred at room temper-ature for20h and then concentrated.The residual syrup was taken up in ether(5mL),and the suspension was filtered to remove the white precipitate.The filtrate was concentrated and chromatographed to give 74mg(66%)of the desired product17as a colorless oil:TLC R f) 0.69(10%EtOAc/petroleum ether);[R]D)15.2°(c)0.0025,CH2-Cl2);1H NMRδ6.07(d,J)19.0Hz,1H),5.86(dd,J)7.0,19.0 Hz,1H),5.03(m,1H),4.31(m,1H),3.54-3.13(m,6H),2.38(m, 1H),2.20-0.80(m,39H);13C NMRδ119.5(CF3,J)325Hz);uSynthesis of(-)-Haliclonadiamine J.Am.Chem.Soc.,Vol.119,No.1,19972541.7,31.2,30.8,30.6,29.3,29.1,29.0,28.9,27.3,26.1,26.0,9.4;d 145,133.6,50.4,43.2,13.6;MS(EI)m/z455(1),399(100),285(17). HRMS:m/e calcd for C19H35OSn(M+-Bu);397.1704;found, 397.1704.Iodo Stannane18.To the mixture of vinylstannane17(20mg, 0.026mmol),inverted iodo alcohol13(7.5mg,0.026mmol),and triphenylphosphine(8.5mg,0.032mmol)in benzene(0.75mL)was added DEAD(5.6mg,0.032mmol)dropwise via syringe at room temperature.The reaction mixture was stirred for28h and then concentrated and chromatographed to give 2.5mg(13%)of the recovered vinyl stannane starting material and15mg(56%yield based on starting material not recovered)of product18as a white foam:TLC R f)0.82(10%EtOAc/petroleum ether);[R]D)9.2°(c)0.004,CH2-Cl2);1H NMRδ6.31(dd,J)14.6,8.63Hz,1H),6.15(d,J)14.6 Hz,1H),6.08(d,J)19.0Hz,1H),5.85(dd,J)19.0,6.82Hz,1 H),4.36(m,1H),3.42-2.82(m,6H),2.40-0.92(m,44H),0.87(t, J)7.20Hz,9H);13C NMRδ119.5(CF3,J)320Hz);u60.4,43.4, 31.5,31.2,30.7,29.4,29.1,29.0,27.3,26.9,26.2,26.0,25.9,25.6, 21.0,9.5;d145.5,133.2,115.8,64.0,62.6,60.4,56.4,54.9,50.3,49.1, 44.8,43.4,43.2,13.7;IR(neat)(cm-1)1653,1457,1383;MS(EI) m/z1049(M+,1),993(100),179(64).Bistriflylhaliclonadiamine19.To a solution of(PPh3)4Pd(8.4mg, 0.0082mmol)in toluene(20mL)under argon was added one-twentieth of iodo stannane18(36mg,0.036mmol)in toluene(5.0mL),and the mixture was heated to100-105°C in the dark.The remaining solution of18was added over6h via syringe pump.The reaction mixture was maintained at100-105°C for2h,and additional(PPh3)4Pd(4.2 mg,0.0041mmol)was added.The resulting mixture was maintained at100-105°C for14h in the dark and then cooled to room temperature.The mixture was filtered through Celite,washing thoroughly with EtOAc.The combined filtrate was concentrated and chromatographed to give9.5mg(43%)of the cyclized product19as a white foam:TLC R f)0.57(4%EtOAc/petroleum ether);[R]D) -7.8°(c)0.005,CH2Cl2);1H NMRδ6.19(d,J)14.7Hz,2H),5.51-5.45(m,2H),5.22(m,1H),4.36(m,1H),3.37-3.11(m,4H),2.59-2.52(m,2H),2.15-0.80(m,26H);13C NMRδ120.2(CF3, J)320Hz);u65.3,64.0,39.7,34.4,31.9,31.5,31.0,30.5,29.7, 29.5,29.4,28.4,26.4;d1345,127.5,125.2,124.2,65.2,52.2,49.5, 47.6,43.8,43.0,42.9,30.0;MS(EI)m/z632(20),499(95),191(100).(-)-Haliclonadiamine(1).In a1mL reactivial,LiAlH4(1.0M in Et2O,0.5mL,0.5mmol)was added to a solution of19(3.0mg,3.1µmol)in Et2O(0.2mL),and the mixture was heated(sealed tube,70°C)for4days.The reaction mixture was cooled to0°C,and then saturated aqueous sodium potassium tartrate solution(2drops)was added.The reaction mixture was heated to reflux for1h and then was cooled to room temperature and filtered with EtOAc.The combined organic was concentrated in V acuo.The residue was purified by reverse phase HPLC to give0.6mg(34%yield)of1:TLC R f) 0.30(CH2Cl2/MeOH/35%NH4OH)87/12/1);1H NMRδ6.25-6.18 (m,1H),6.14-6.06(m,1H),5.58-5.50(m,1H),5.35-5.28(m,1 H),3.20-2.90(m,2H),2.65-1.90(m,6H),1.90-0.98(m,26H); 13C NMRδ134.0,133.9,132.5,132.0,68.2,62.6,60.1,51.5,51.2, 49.6,48.7,47.1,44.8,44.0,41.0,37.0,32.25,32.19,31.6,30.5,30.1, 26.8,26.5,26.34,26.31;MS(CI)m/z369(12),279(4),185(100). HRMS(CI):m/e calcd for C25H40N2(M+):368.3191;found,368.3177. Acknowledgment.We thank the donors of the Petroleum Research Fund,administered by the American Chemical Society, for support of this work.We also thank Professor Clayton Heathcock for providing spectra of both natural and synthetic (-)-haliclonadiamine.This article is dedicated to John Alex-ander Oates,mentor and friend.JA962162U26J.Am.Chem.Soc.,Vol.119,No.1,1997Taber and Wang。

研究生科技论文写作考试试题2011一写出下列有关SCI论文中一些英文缩写的中文和英文意思(5题,每题2分,共10分)SCI:ISI:SSCI:ISTP:IF:二写出下列单词或短语的中文意思(10题,每题1分,共10分)(1)intraparticle(2)specific surface area(3)photocatalytic activity(4)complete mineralization(5)biomemitic photocatalysis(6)hierarchically bimodal macro-/mesoporous structures(7)a simple and environmentally benign method(8)average crystallite size(9)X-ray photoelectron spectroscopy(10)photoluminescence spectra三判断下列说法是否正确,正确打√,错误的打×(5题,每题2分,共10分)影响因子=(该刊前两年发表论文在统计当年被引用的总次数)÷(该刊前两年发表论文总数)SCI影响因子是衡量一个学术期刊影响力的重要指标。
SCI影响因子由Eugene Garfield在20世纪50年代创立。
中国学者的论文不易被高影响因子的SCI期刊接受,主要是语言问题。
实验结果的描写一般采用现在时。
四下列观点是否正确,正确打√,错误的打×(10题,每题2分,共20分)1. 论文题目一般是一个句子。
2. 论文标题和课题名称的区别在于,前者主要是讲你发现了什么,后者主要是讲你将研究什么。
3. 摘要一般具有独立性和自明性,并包含论文的主要内容。
4. 引言的作用是揭示论文的主题、目的和总纲,便于读者了解论文所述内容的来龙去脉。
5. 实验内容的描述一般用现在完成时。
6. 研究结果是一篇论文的核心。

铜和对苯二甲酸金属有机骨架材料绿色合成那立艳;张伟;华瑞年;宁桂玲【摘要】采用以水为唯一溶剂的绿色路线合成了金属有机骨架(MOFs)材料。
通过将羧酸类配体转化为水溶性铵盐配体,在水溶液中探索了金属有机骨架材料的绿色合成。
以醋酸铜(Cu(OAc)2)和对苯二甲酸(H2BDC)甲胺盐在水溶液中反应,获得了化合物CuBDC‐H2O 。
同时作为参照,以Cu(OAc)2和H2 BDC在N ,N‐二甲基甲酰胺(DM F)和水的混合溶液中,获得了化合物CuBDC‐DMF 。
通过粉末XRD、IR、SEM、TGA以及N2吸附测试等手段对两种产物进行了表征,结果显示经140℃真空热处理10 h后,化合物CuBDC‐DM F 具有和在水溶液中直接获得的产物CuBDC‐H2 O相同的晶体结构及相似的性质,而且以水作为反应介质的新的合成路线具有便捷、快速、低成本及节约能源等特点。
%A green process for the synthesis of metal‐organic frameworks (MOFs) particles that uses water as the only solvent is presented . The strategy is to convert carboxylic acids ligands into carboxylate ammonium salts ligands which have high solubility in water ,thus allowing the synthesis to be performed in water .CuBDC‐H2O (BDC=1 ,4‐benzenedicarboxylate) is synthesized by mixing aqueous solutions of Cu (OAc)2 and the methylammonium salt of H2 BDC .As a reference ,CuBDC‐DMF is obtained by mixing water solution of Cu (OAc)2 with DMF solution of H2BDC .The two compounds are characterized by powderXRD ,IR ,SEM ,TGA and N2 and adsorption measurements . The experimental results reveal that after evacuation at 140 ℃ for 10 h ,CuBDC‐DMF has the same crystal structure and properties with CuBDC‐H2 O ,andthe new synthetic route performed in water solution has the advantages of being straight forward ,rapid ,cheap and energy‐saving .【期刊名称】《大连理工大学学报》【年(卷),期】2013(000)004【总页数】5页(P474-478)【关键词】金属有机骨架;绿色合成;孔材料【作者】那立艳;张伟;华瑞年;宁桂玲【作者单位】大连理工大学精细化工国家重点实验室,辽宁大连 116024; 大连民族学院生命科学学院,辽宁大连 116600;大连民族学院生命科学学院,辽宁大连116600;大连民族学院生命科学学院,辽宁大连 116600;大连理工大学精细化工国家重点实验室,辽宁大连 116024【正文语种】中文【中图分类】O6410 引言金属有机骨架(MOFs)材料是在微孔晶体材料上发展起来的一种新型功能材料,由于其在气体存储与分离、催化、药物传递和传感器等方面的潜在应用而备受关注[1-5].目前用于合成MOFs材料的方法有溶剂诱导沉淀法[6]、溶剂热合成法[7]、反相微乳液法[8]、微波辅助合成法[9]、超声法[10]等.然而这些合成方法往往存在反应时间长、反应温度高、大量使用有机溶剂或复杂的反应设备等缺点,对其实际应用造成了制约,因而开发快速、简便、节约能源和环境友好的合成路线具有重要意义.DMF由于具有较高的沸点(140℃)和对羧酸类配体良好的溶解能力,是合成MOFs材料经常使用的溶剂.DMF分子中的氧原子通常以配体或客体分子的形式填充到孔道当中,因而,为了获得金属活性中心和开放的骨架结构,通常要对合成的产物进行热处理,以除去配位及孔道中的DMF分子.如果能在反应中以水代替DMF 溶剂,那么不仅能够节约成本,而且在热处理过程中,只需将水分子除去即可,可以明显缩短热处理过程的时间,降低热处理温度,进而有效地节约能源. 本文的合成策略是先将非水溶性的配体对苯二甲酸与甲胺反应,生成水溶性的甲胺羧酸盐;然后利用水溶性配体与金属盐在水溶液中完成聚合反应.由于CuBDC具有独特的磁性、优良的气体分离性能及催化活性[11],本文分别在水溶液及DMF溶液中进行CuBDC 的合成探索,通过对比两种反应体系的合成条件及产物性能,探索羧酸类配体构筑的MOFs材料的快速、高效、低能耗合成.1 实验部分1.1 试剂与仪器原料:试剂均为市售分析纯.仪器:日本岛津XRD-6000 衍射仪;Nicolet FT-IR 740红外光谱仪;日本日立S-4800高分辨电子显微镜;Micromeritics ASAP 2020表面分析仪;美国Perkin-Elmer SDTA 851热失重分析仪.1.2 合成1.2.1 水溶性配体前驱体((NH3CH3)2BDC)的合成取1.667g(10mmol)对苯二甲酸,搅拌下滴加20%~30%的甲胺水溶液至溶液澄清,于真空干燥箱中20℃下减压至溶液黏稠,室温挥发得白色晶体.将上述晶体用水转移至100 mL 容量瓶中,配制成0.1mol/L甲胺盐水溶液,反应方程式如下:1.2.2 CuBDC-H2O的合成称取0.03 g Cu(OAc)2·H2O溶于7 mL 水中,搅拌下滴入0.1mol/L(NH3CH3)2BDC 溶液3 mL,马上产生浅蓝色沉淀,搅拌2h后离心.然后将产物经乙醇洗涤—超声分散—离心两次,自然干燥.1.2.3 CuBDC-DMF的合成称取0.03 g Cu(OAc)2·H2O溶于4mL水中,再称取0.04g H2BDC溶于6 mL DMF 中,两种溶液充分溶解后,搅拌下将配体溶液滴入金属盐水溶液中,马上产生蓝色沉淀,搅拌2h后,静置2h,离心.然后将产物经乙醇洗涤—超声分散—离心两次,自然干燥.2 结果与讨论2.1 XRD 表征两种产物及经140℃真空热处理10h 后CuBDC-DMF的粉末XRD 谱图见图1.由图可知有机溶液中获得的CuBDC-DMF 与水溶液中获得的CuBDC-H2O 峰位明显不同(图1曲线a、c),说明两者结构不同.CuBDC-DMF 是一种已知结构的化合物[11-12](与本文合成方法不同),具有开放的骨架结构,结构中的金属中心铜为五配位,4个氧原子来自于BDC基团,另外一个氧原子来自于DMF分子(图2(a)).当CuBDC-DMF 在140℃真空热处理10h脱除了DMF 基团后,其XRD 谱图(图1曲线b)与CuBDC-H2O(图1 曲线c)相同.这说明CuBDC-DMF高温脱除溶剂分子后,与在水溶液中一步获得的产物CuBDCH2O 具有相同的结构,图2(b)是CuBDC-H2O 的可能结构.图1 粉末XRD 谱图Fig.1 Powder X-ray diffraction(PXRD)patterns图2 晶体结构Fig.2 Crystal structures2.2 红外表征用KBr压片法测定了CuBDC-H2O、CuBDCDMF及经140℃真空热处理10h 后的CuBDCDMF红外光谱,见图3.通过对比吸收峰的位置,发现CuBDC-DMF经热处理后的红外谱图(图3曲线b)与CuBDC-H2O(图3曲线c)基本一致,两个谱图中,1 575cm-1与1 398cm-1处的吸收峰归属为BDC配体中羧酸基团的非对称伸缩振动和对称伸缩振动;1 690~1 730cm-1没有出现羰基的吸收峰,说明配体上的羧酸基团已全部去质子化.而在CuBDC-DMF 的红外谱图(图3曲线a)中,1 688cm-1处的吸收峰为客体DMF中羰基的吸收峰,1 628cm-1处的吸收峰可归属为与金属中心配位的DMF 中羰基的吸收峰,氧原子与金属中心配位作用的存在,使得吸收峰向短波数方向移动.1 292cm-1处的吸收峰为酰胺中C—N 键的伸缩振动.以上3个与DMF相关的特征吸收峰在真空热处理后消失,说明通过加热,已成功去除客体DMF 分子及配位的DMF基团,并且其结构与水溶液中一步获得的产物CuBDC-H2O 相同.图3 红外谱图Fig.3 IR spectra2.3 SEM 表征图4(a)和图4(b)分别是CuBDC-DMF 和CuBDC-H2O 的扫描电镜照片,可以看出,两种合成条件下获得的产物均为方形片状晶体.CuBDCDMF晶体长和宽约为400nm,厚度在50nm 左右,与文献报道该化合物的形貌基本一致[12];CuBDC-H2O 晶体长为10μm,宽为4μm,厚度在1μm 左右.由于在水溶液中进行反应时,配体已经提前完成了去质子化的过程,因而反应速度比较快,所以CuBDC-H2O的尺寸明显大于CuBDC-DMF.图4 扫描电镜照片Fig.4 SEM images2.4 吸附测试采用-196℃下氮气吸附的方法对两种不同条件下获得的化合物进行了氮气吸附等温测试.样品量约为0.2g,测试前均在140℃真空干燥10h,采用BET 方法在p/p0=0.05~0.33获得比表面积.从图5可以看出,两种化合物在相对低的压力范围内(p/p0<0.1),都有一个明显的台阶存在,然后曲线趋于平缓,吸附等温曲线为I型,说明了样品中微孔孔道的存在.CuBDC-DMF和CuBDC-H2O 在低压区(p/p0=0.01)时,氮气的吸附量分别为65cm3/g和68cm3/g,随着压力的升高,吸附量略有增加,在高压区(p/p0=1.0)时,氮气的吸附总量分别为89cm3/g和80cm3/g,BET 比表面积分别为407m2/g和439m2/g.从以上结果可以看出,在CuBDC-DMF脱除客体及配位的DMF基团后,与在水溶液中获得的产物CuBDC-H2O 具有相似的比表面及孔结构信息.图5 氮气吸附等温曲线Fig.5 N2adsorption isotherms采用巨正则蒙特卡罗法(grand canonical Monte Carlo)模拟了CuBDC-DMF 在77K时的氮气吸附曲线[13],见图5.分子模型是通过CuBDC-DMF的实验XRD 晶体学数据建立的.采用通用力场(UFF)描述MOF骨架原子[14],N2力场参数为σ=0.331nm,ε=36 K.利用Music code软件进行模拟[15],并用BET 方法对模拟吸附等温线的线性段(p/p0=0.001~0.015)进行拟合,进而得到BET 方程参数及比表面积,结果如图6所示.模拟吸附曲线及理论比表面积均与实验结果接近.图6 BET 法比表面积计算Fig.6 Specific surface area calculation by the BET method2.5 热稳定性测试对CuBDC-DMF、CuBDC-H2O 及经140℃真空热处理10h后CuBDC-DMF 进行了热失重分析,温度区间为30~500℃,氮气气氛.CuBDCDMF热失重曲线(图7 曲线a)显示该化合物在90~250℃失重值约为27%,100℃左右的失重可归属为水分子的脱除,在140~250℃的失重可归属为配体DMF的失去,温度超过300℃,化合物的骨架开始塌陷.图7中曲线c是CuBDC-H2O的热失重曲线,可以看出化合物在200℃之前基本没有失重,骨架结构可稳定存在至300℃.图7中曲线b 是CuBDC-DMF 经真空热处理后的热失重情况,热失重曲线与CuBDC-H2O 的基本相同,说明热处理过程除去了配位的DMF 分子后,CuBDC-DMF与CuBDC-H2O 拥有相同的骨架热稳定性能.图7 热失重曲线Fig.7 TGA curves3 结论(1)通过水溶性羧酸甲胺盐配体的合成,开发出一种一步绿色合成金属有机骨架材料的新方法.(2)对比有机体系和水体系中CuBDC 合成过程及产物性质,发现有机体系下获得的配合物在真空热处理除去配位的溶剂分子后,与在水溶液中一步获得的产物具有相同的结构和相似的性质.但以水溶液代替有机溶剂使用,不仅降低了合成成本,减少了污染,同时缩短了真空热处理过程的时间,降低了热处理温度.(3)本文开发的水溶液合成路线具有简便、快速、节能和环保的特点,该绿色合成路线具有可推广性,可用于更多以羧酸类配体构筑的金属有机骨架材料的合成.【相关文献】[1]Murray L J,Dinca M,Long J R.Hydrogen storage in metal-organic frameworks [J].Chemical Society Reviews,2009,38(5):1294-1314.[2]Li J R,Kuppler R J,Zhou H C.Selective gas adsorption and separation in metal-organic frameworks[J].Chemical Society Reviews,2009,38(5):1477-1504.[3]WU Chuan-de,HU Ai-guo,ZHANG Lin,et al.A homochiral porous metal-organic framework for highly enantioselective heterogeneous asymmetric catalysis [J].Journal of the American Chemical Society,2005,127(25):8940-8941.[4]Horcajada P,Serre C,Vallet-Regi M,et al.Metalorganic frameworks as efficient materials for drug delivery [J].Angewandte Chemie International Edition,2006,45(36):5974-5978.[5]CHEN Bang-lin,WANG Liang-bo,XIAO Yunqing,et al.A luminescent metal-organic framework with Lewis basic pyridyl sites for the sensing of metal ions[J].Angewandte Chemie International Edition,2009,48(3):500-503.[6]Oh M,Mirkin C A.Chemically tailorable colloidal particles from infinite coordinationpolymers [J].Nature,2005,438(7068):651-654.[7]Jung S,Oh M.Monitoring shape transformation from nanowires to nanocubes and size-controlled formation of coordination polymer particles [J].Angewandte Chemie International Edition,2008,47(11):2049-2051.[8]Rieter W J,Taylor K M,An H,et al.Nanoscale metal-organic frameworks as potential multimodal contrast enhancing agents [J].Journal of the American Chemical Society,2006,128(28):9024-9025.[9]Ni Z,Masel R I.Rapid production of metal-organic frameworks via microwave-assisted solvothermal synthesis[J].Journal of the American Chemical Society,2006,128(38):12394-12395.[10]QIU Ling-guang,LI Zong-qun,WU Yun,et al.Facile synthesis of nanocrystals of a microporous metal-organic framework by an ultrasonic method and selective sensing of organoamines[J].Chemical Communications,2008(31):3642-3644. [11]Carson C G,Hardcastle K,Schwartz J,et al.Synthesis and structure characterization of copper terephthalate metal-organic frameworks [J].European Journal of Inorganic Chemistry,2009(16):2338-2343.[12]XIN Zhi-feng,BAI Jun-feng,SHEN Yong-ming,et al.Hierarchically micro-and mesoporous coordination polymer nanostructures with high adsorption performance [J].Crystal Growth &Design,2010,10(6):2451-2454.[13]Walton K S,Snurr R Q.Applicability of the BET method for determining surface areas of microporous metal-organic frameworks [J].Journal of the American Chemical Society,2007,129(27):8552-8556.[14]Karra J R,Walton K S.Effect of open metal sites on adsorption of polar and nonpolar molecules in metal-organic framework Cu-BTC [J].Langmuir,2008,24(16):8620-8626.[15]Gupta A,Chempath S,Sanborn M J,et al.Objectoriented programming paradigms for molecular modeling[J].Molecular Simulation,2003,29(1):29-46.。

简述caci2法制备感受态细胞流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!简述 CaCl2 法制备感受态细胞流程一、准备工作阶段。

MWCNT/TTO的制备及其自然光下降解甲基橙染料的性能研究李荡#,韦国兰,董伟,汤宏(凯里学院大健康学院,贵州凯里556011)摘要:以多壁碳纳米管(MWCNT)为基核,钛酸丁酯为前驱体,采用简单的水热法制备了MWCNT/TCO复合材料。
利用了差热-热重(TG)、透射电子显微镜!TEM)、X-射线衍射(XRD)及紫外-可见光谱!UV-VT)等分析手段对材料进行了表征。
以甲基橙为目标降解物,考察了不同含量MWCNT/TC0复合材料在自然光下的光催化活性。
结果表明:MWCNT的加入可有效地防止TC0粒子团聚而不会影响TC0的晶型。
且由于MWCNT的加入可减小TC0的禁带宽度,降低光生电子和空穴的复合率,MWCNT/TC0复合材料表现出比纯TC0更好的可见光吸收率。
光催化研究表明,MWCNT/TCO复合材料的光催化性能与MWCNT含量关系密切,当MWCNT加入量为0.04g时,MWCNT/TCO的复合材料的光催化活性最强,自然光下照射反应5h后,甲基橙降解率可达到85%。
关键词:水热法;MWCNT/TCO复合材料;甲基橙;催化降解机制;光催化活性中图分类号:X788;TB33文献标识码:A文章编号:1008-021X(2021)05-0039-03Preparation of MWCNT^TiO2and Its Performance inNatrral Light Degradation of Methyl Orange DyeLi Dang*,Wei Guolan,Dong Wei,Tang Hong(SchooeoiLoieand Heaeih Scoence,KaoeoUnoeeesoiy,Kaoeo556011,Chona)Abshrach:TheMWCNTS LToO2composoiewaspeepaeed bysompeehydeoiheemaemeihod woih MWCNTS(MWCNTS)asihebasoc nuceeusand buiyeioianaieasihepeecuesoe.Themaieeoaesweeechaeacieeozed bydo i e eenioaeiheemogeaeomeiey(TG),ieansmossoon eeecieon moceoscopy(TEM),X-eaydo i eacioon(XRD)and UV-VIS.ThephoiocaiaeyiocacioeoiyoiMWCNTLToO2composoiewoih do i e eeniconieniundeenaiueaeeoghiwasoneesiogaied woih meihyeoeangeasiheiaegeidegeadaioon.Theeesueisshow ihaiihe addition of MWCNT can ePec—ve—prevent T1T2particles from a/glomera—on without affecting the crystal—nity of T1T2.Moreover, becauseiheaddoioon oiMWCNTS can eeduceiheband gap wodih oiToO2and eeduceihephoiogeneeaied eeecieon and hoee composite rate,the MWCNTS/TiT2composite material shows a bd—r visible light absorption rate than pure T1T2.Pho—camlytic siudosshowAd ihaiihAphoiocaiaeyiocpAeioemancAoiMWCNTLToO2composoi wasceosAeyeeaid ioihAconinioiMWCNT. WhAn ehAamouneoiMWCNTwas0.04g,ehAphoeocaeaeyeocaceoeoeyoiMWCNTLToO2composoe wasehAseeongAse,and ehA dAgeadaeoon eae oimAehyeoeangAeachAd85%aiee5houesoio e adoaeoon undAenaeueaeeoghe.K&y words:hydeoehAemae mAehod;MWCNTLToO2composoe;mAehye oeangA;caeaeyeoc dAgeadaeoon mAchanosm; phoeocaeaeyeocaceoeoey随着全球工业化推进,人类科技飞速发展,同时带来的环境问题也日趋严峻。

Facile synthesis of SAPO-34with small crystal size for conversion of methanol to ole finsJianqing Li,Zhuo Li,Dezhi Han ⁎,Jinhu Wu ⁎Key Laboratory of Biofuels,Qingdao Institute of Bioenergy and Bioprocess Technology,Chinese Academy of Sciences,Qingdao 266101,PR Chinaa b s t r a c ta r t i c l e i n f o Article history:Received 23January 2014Received in revised form 10April 2014Accepted 21April 2014Available online 29April 2014Keywords:Dry gel liquid-phase transport SAPO-34Methanol to ole fins (MTO)MorpholineTetraethylammonium hydroxideSAPO-34molecular sieves were fabricated by a dry gel liquid-phase transport method using morpholine (Mor)or morpholine –tetraethylammonium hydroxide (Mor –TEAOH)as the structure-directing agent (SDA).The prepared samples were characterized by scanning electron microscope (SEM),nitrogen adsorption –desorption,X-ray diffraction (XRD),ammonia temperature-programmed desorption (NH 3-TPD),thermo-gravimetric analy-sis (TG)and Fourier transform infrared spectroscopy (FT-IR).The results indicated that the dry gel liquid-phase transport method is very effective to fabricate SAPO-34molecular sieves with average crystal size about 0.6μm using Mor –TEAOH as SDA and 3μm using Mor as SDA,respectively.Moreover,the addition of TEAOH is bene ficial to produce SAPO-34with small crystal size.Methanol-to-ole fins (MTO)reaction was chosen to test the catalytic performance of as-prepared SAPO-34catalysts.The prepared SAPO-34catalyst using Mor –TEAOH as SDA exhib-ited the excellent catalytic performance in MTO process owing to its small crystal size and high crystallinity.The conversion of methanol and selectivity of ethylene and propylene are up to 100%and 87.9%,respectively.©2014Elsevier B.V.All rights reserved.1.IntroductionSilicoaluminophosphate (SAPO)molecular sieves were firstly invented by Lok et al.in 1984[1,2],as a new family of molecular sieves.The development of SAPO series provides a broad range of dif-ferent frameworks and different compositions,some are analogous to available silica-dominant zeolites,such as SAPO-34and SSZ-13.Among all SAPOs,SAPO-34with small pore mouth (3.72Å)is well known as the commercial and industrial catalyst for the application of methanol-to-ole fins (MTO),which is mainly attributed to its mild acid-ity and shape selective catalysis by small pore entrance [3].In terms of SAPO-34synthesis,the nature and amount of the selected structure-directing agent (SDA)play a signi ficant role in incorporating silicon into the aluminophosphate framework and the purity of structure and crystallinity of SAPO-34.Hydrothermal method,as the conventional strategy,has been applied in the preparation of SAPO-34by numerous researchers [4–15].However,separation between solvents and solid products is required by filtration or centrifugation,and partial organic SDAs are inevitably preserved in the liquid phase,resulting in the wastewater pollutants and downstream pu-ri fication system installation [16].To overcome the current problems originating from hydrothermal method,dry gel conversion (DGC)meth-od is alternative and effective towards the preparation of SAPO-34[17].DGC can be classi fied into two types:vapor-phase transport (VPT)and steam-assisted conversion (SAC).In fact,the VPT technique [18]involvesthe crystallization of a dry silicoaluminophosphate gel in a vapor phase containing volatile SDA and water.The dry gel is placed in a sample holder located in the middle of an autoclave while a very small amount of the aqueous solution of SDA and deionized water is added to the bottom of the autoclave,the aqueous solution is physically separated from the dry gel powder,and the dry gel is converted to a zeolite in evaporated steam.Smaller amount of SDA is required in VPT method than that used in hydrothermal method [19].The SAC approach is similar to VPT except that the SDA is contained in the initial dry gel,instead of the aqueous so-lution containing SDA,pure water is placed at the bottom of the autoclave as the steam source.However,the DGC method is more complex and has a high requirement towards the equipment,and the space ef ficiency of reactors is reduced.Therefore,the synthesis of zeolites by a simple and environmentally friendly method has been always a challenge.Herein,we present a facile method to synthesize SAPO-34via a dry gel liquid-phase transport method.The prepared samples were well charac-terized and their catalytic properties were evaluated by MTO reaction.Moreover,the relevant characterization and the catalytic performance comparison of samples fabricated by different synthetic methods were also reported.2.Experimental2.1.Synthesis of SAPO-34samplesPseudoboehmite (70wt.%Al 2O 3,Chinalco,China),silicon sol (28wt.%SiO 2,Xi'an Chem.,China)and orthophosphoric acid (85wt.%,Bodi Chem.,China)were used as sources of Al,Si and P,respectively.Powder Technology 262(2014)177–182⁎Corresponding authors.Tel.:+8653280662763;fax:+8653280662761.E-mail addresses:handz@ (D.Han),wujh@ (J.Wu)./10.1016/j.powtec.2014.04.0820032-5910/©2014Elsevier B.V.All rightsreserved.Contents lists available at ScienceDirectPowder Technologyj o u r n a l h o me p a g e :w w w.e l s e v i e r.c o m /l o c a t e /p o w t e cMorpholine(Mor)(99wt.%,Sinopharm Chemical Reagent Co.Ltd,China) and tetraethylammonium hydroxide(TEAOH)(25wt.%,Sinopharm Chemical Reagent Co.Ltd,China)were used as the SDA in the preparation of SAPO-34.SAPO-34samples were synthesized using the dry gel liquid-phase transport method with a molar composition of1.0SiO2: 1.5Al2O3:1.5P2O5:45H2O:xR,where R and x represent SDA and the molar ratio of SDA,respectively.In a typical synthesis,34.59g of H3PO4was mixed with53.82g of deionized water to form an ortho-phosphoric acid solution.21.86g of pseudoboehmite was then added to the above solution.After vigorous stirring,a homogenous solution was obtained.Subsequently,21.43g of silicon sol was added and the resulting mixture was stirred for another2h to get a homogeneous gel,and then followed by holding the gel at80°C with stirring until dry gel was formed.20g of as-prepared dry gel was ground and then added to the mixture of50mL of SDA and50mL of distilled water under stirring,the resulting solution was then transferred to a stainless-steel autoclave without Teflon,then heated and crystallized at180°C under autogenic pressure for48h.Finally,the products werefiltered, washed with distilled water,and followed by drying at120°C for4h and calcination at550°C in air atmosphere for5h to remove the SDA. The obtained SAPO-34samples in the presence of Mor and the mixture of75%(volume)Mor-25%TEAOH were denoted as S34-L-M and S34-L-M-T,respectively.For comparison,SAPO-34was also prepared by hydrothermal meth-od and VPT method using morpholine as SDA according to the proce-dures of Brent M.Lok and Hualan Zhou[2,20],the obtained samples were designated as S34-H-M and S34-V-M,respectively.2.2.CharacterizationThe powder X-ray diffraction(XRD)patterns of as-synthesized samples were obtained on a Rigaku D/max-3C X-ray diffractometer with Cu Kαradiations under instrumental setting of35kV and 40mA.The spectra were recorded from5°to40°with a step size of 0.02°.N2adsorption–desorption was performed at−196°C using a Quantachrome AUTOSORB-1C instrument.Before the measurement, samples were degassed under vacuum at300°C for at least10h.The specific surface area(S BET)and the micropore volume were calculated according to Brunauer–Emmett–Teller(BET)equation and t-plot meth-od,respectively.And the average pore diameter was estimated from the adsorption branches of the isotherms by the BJH method.Scanning elec-tron microscope(SEM)images were recorded on a HITACHI S-570scan-ning electron microscope.Prior to the image capture,each sample was placed onto a carbon membrane and Au sputtering coating was operat-ed to reduce charging effects.The temperature-programmed desorp-tion of ammonia(NH3-TPD)was performed on a Micromeritics ASAP 2020C instrument.0.1g of sample was pretreated at450°C for2h in an Arflow of20mL min−1.After cooling to100°C,the sample was sat-urated with10vol.%NH3/Ar.After that,the sample was purged with Ar for1h to eliminate the physically absorbed NH3.Desorption of NH3was carried out from100to650°C at a heating rate of5°C min−1.Fourier transform infrared spectroscopy(FT-IR)spectra of samples in the re-gion of framework stretching vibrations(400–4000cm−1)were measured using KBr containing pellets on a Bruker TENSOR27spec-trophotometer.Thermo-gravimetric(TG)measurement was per-formed on Mettler Toledo TGA/SDTA851e instrument under an air flow of100mL min−1at a ramp of10°C min−1from100°C to700°C.2.3.Catalytic evaluationThe catalytic tests of SAPO-34for MTO reaction were carried out using an integralfixed-bed reactor(φ32mm×650mm)at atmosphere pressure.Catalyst(2g,40–60mesh)was loaded into the stainless steel reactor.The sample wasfirst pretreated in N2at500°C for1h and then the temperature of reactor was adjusted to380°C to start the MTO reaction.The weight hourly space velocity(WHSV)was4.8h−1for the feedstock of methanol.The reaction products were continuously an-alyzed using gas chromatograph(WenlingFuli GC-9790)equipped with aflame ionization(FID)detector and an ATSE-30capillary column.3.Results and discussion3.1.Crystalline structure and morphological featuresFig.1shows the wide-angle XRD patterns of the as-synthesized SAPO-34molecular sieves.It can be seen that the representative diffrac-tion peaks at9.5°,13°,16.2°,20.7°,26°and31°are observed in the XRD of all samples,corresponding to pure SAPO-34with CHA-structure[19]. High peak intensity of XRD lines and absence of baseline drift indicate the high crystallinity without impurity for all samples.Of the four pre-pared samples,the intensity of these characteristic diffraction peaks for S34-L-M-T is the highest,indicating that the crystallinity and the phase purity of S34-L-M-T are better than that of the other three sam-ples.Furthermore,the width of XRD diffraction peaks for S34-L-M-T is wider,illustrating that the crystallite size of S34-L-M-T is smaller.SEM images of prepared SAPO-34molecular sieves are shown in Fig.2.Well-distributed cubic structures,typical for SAPO-34crystal were observed.The average crystal size of S34-L-M-T sample is about 0.6μm.While,S34-L-M,S34-H-M and S34-V-M samples have the simi-lar average crystal size of about3μm.Thisfinding is very consistent with XRD data.This is probably due to the addition of TEAOH during the synthesis of S34-L-M-T sample,and this is consistent with previous study showing that the crystals with larger size are obtained when only morpholine is used as the SDA[21].The addition of TEAOH is ben-eficial to synthesize molecular sieves with small crystal size in the dry gel liquid-phase transport process.When the TEAOH is used as SDA, the TEAOH reacts easily with the pseudoboehmite,silica sol and ortho-phosphoric acid,therefore more nucleation centers are formed,and more molecular sieves with small crystal size are generated.3.2.Nitrogen adsorption–desorptionThe nitrogen adsorption–desorption isotherms and the pore struc-ture characteristics of prepared SAPO-34samples are illustrated in Fig.3and Table1,respectively.All SAPO-34molecular sieves display a type I isotherm with hysteresis loops originating from the mesopores formed between the crystal particles[22].S34-L-M-T has the largest surface area and the smallest micropore pore volume among the four prepared samples,while the BET surface areas for S34-L-M,S34-H-M and S34-V-M samples using Mor as SDA are somewhat smaller because of their larger pore,and the micropore pore volumes of the three samples are approximately the same and in accordance with previous report[13]. The average pore diameters of S34-H-M and S34-V-M samples arelarger Fig.1.XRD patterns of the prepared SAPO-34samples.178J.Li et al./Powder Technology262(2014)177–182than that of the S34-L-M-T and S34-L-M samples because of the existence of more mesopores formed between their crystal particles.3.3.NH 3-TPDThe NH 3-TPD pro files of the prepared SAPO-34molecular sieves are presented in Fig.4.Two desorption peaks in the TPD spectrum at 100–250°C and 250–525°C are primarily observed.In general,the desorption temperature represents the strength of acid sites,that is,the lowtemperature desorption peak at 100–250°C is assigned to the weak acid sites,which corresponds to P \OH hydroxyl groups that are not fully coordinated by AlO 4tetrahedra [6].And the high temperature de-sorption peak at 250–525°C is ascribed to the strong acidic sites which are generated by the incorporation of silicon into the framework of SAPO-34molecular sieve [8].As shown in Fig.4,S34-L-M-T contains two temperature peaks centered at 176°C and 428°C,corresponding to the weak and strong acid sites,pared with the S34-L-M-T,the peaks corresponding to weak acid sites of the S34-L-M and S34-V-M shift to the lower temperature,however,the peak of weak acid sites shifts to higher temperature for the S34-H-M.In terms of peaks at higher temperatures,the corresponding peaks of the S34-L-M,S34-H-M and S34-V-M all shift to the lower temperature.The desorption peak areas of the weak and strong acid sites for both S34-L-M-T and S34-L-M samples are almost the same,implying that they have the similar acid concentration for the weak and strong acid sites.S34-H-M contains more weak acidity and less strong acid acidity comparing to S34-L-M-T and S34-L-M samples.Regarding S34-V-M sample,the least acidity is ob-served in both weak acid sites and strong acid sites among the four pre-pared samples,and its intensity of strong acid sites is the weakest.According to the NH 3-TPD results,the catalysts synthesized by the dry gel liquid phase transport method have similar acid properties both in acid site concentration and strength no matter if the SDA is Mor or a mix-ture of Mor and TEAOH.The differences in the acid properties including acid site concentration and strength are observed when different synthe-sis methods are used with the same SDA.These properties will cause a dif-ferent performance on the MTOprocess.Fig.2.SEM images of prepared SAPO-34samples:(a)S34-L-M-T,(b)S34-L-M,(c)S34-H-M and (d)S34-V-M.Fig.3.The nitrogen adsorption –desorption isotherm of prepared samples.179J.Li et al./Powder Technology 262(2014)177–1823.4.FT-IR spectroscopyThe FT-IR spectra of the prepared samples are given in Fig.5.All characteristic peaks of the four molecular sieves are identical to those reported in the literature [10],according to which the basic framework vibration frequency region are as follows:the absorption peaks at 490cm −1and 632cm −1are T \O bending bands of SiO 4and D-6rings,respectively,and the bands at 730cm −1and 1095cm −1are the symmetric stretch vibration of P \O (or Al \O)and asymmetric stretch vibration of O \P \O,respectively.Moreover,the peak at 1651cm −1corresponds to the vibration of hydroxyl groups from adsorbed water on samples.In addition,the broad peak centered at 3445cm −1corre-sponds to Si \OH and P \OH groups interacting through H-bonds [15].In comparison of SAPO-34-H-M and SAPO-34-V-M samples,the intensi-ty of all the vibration bands for SAPO-34-L-M-T and SAPO-34-L-M is stronger.3.5.TG/DTG analysisTG/DTG curves of the prepared SAPO-34samples before calcinations are shown in Fig.6.Four weight loss peaks are observed for S34-L-M-T precursor.It has two small weight loss peaks in the range of 100–243°C and 243–322°C [23],centered at 117°C and 281°C,respectively,which are attributed to the desorption of water from the molecular sieve with a weight loss of 0.34wt.%and 0.46wt.%,respectively,corresponding to desorption of physically and chemically adsorbed water.One signi ficant weight loss (5.12wt.%)in the temperature range of 322–490°C cen-tered at 432°C is detected and could be related to the oxidative decom-position of morpholine.Meanwhile,another relatively gradual weight loss of 0.78wt.%at 529°C in the temperature range of 490–600°C is ob-served and corresponds to the oxidative decomposition of TEAOH.The addition of TEAOH is more useful to form protonated molecule with the negative charge of molecular sieve skeleton,resulting in a higher de-composition temperature [23].From Fig.6(b)and (c)we can see that the TG/DTG curves for the S34-L-M and S34-H-M precursors are basically the same,and two weight loss peaks are observed.The first gradual small weight losses of 0.81wt.%and 0.78wt.%centered at about 140°C ap-peared below 250°C,respectively,this is due to the removal of adsorbedwater.The obvious weight loss peaks in the temperature range of 230–600°C and 250–600°C centered at 467°C and 465°C for S34-L-M and S34-H-M,respectively,are attributed to the oxidative decomposition of morpholine with a weight loss of 12.48wt.%and 12.28wt.%,respectively.For the TG/DTG curves of S34-V-M precursor,it is observed that two grad-ual small weight losses of 0.03wt.%and 0.06wt.%centered at 130°C and 240°C appeared below 250°C,respectively,both of them can be ascribed to the physically and chemically adsorbed water associated with SAPO-34molecular sieve.Another signi ficant weight loss (1.26wt.%)in the tem-perature ranges of 250–600°C,centered at 443°C,is detected and can be related to the oxidative decomposition of morpholine.3.6.MTO performanceFig.7shows the MTO performance of prepared samples.For S34-L-M-T sample,the methanol conversion and the total selectivity for ethylene and propylene increase with temperature increase,with a maximum of 100%at 400°C and 87.9%at 440°C,respectively.As temperatures increase,the selectivity for ethylene decreases before 340°C,but then increases and retains 45.88%at 420°C.While the selec-tivity for propylene shows the opposite trend with an initial increase be-fore 360°C and a subsequent decrease,and a constant of 41.38%at 420°C.In the case of S34-L-M,S34-H-M and S34-V-M,the methanol conversions are similar and reach to 100%after 400°C.As temperature rises,the selectivity for ethylene increases,and the selectivity for pro-pylene increases first and then decreases for all the three samples.The highest selectivity for ethylene is 45.72%,52.43%and 45.7%at 440°C,re-spectively.Moreover,the maximum of the selectivity for propylene on S34-L-M is 47.57%at 360°C,while the selectivity on S34-H-M and S34-V-M is 44.47%at 340°C and 45.1%at 360°C,respectively.With temperature increases,the total selectivity for ethylene and propylene increases slowly first,reaches the maximum and then decreases,show-ing the same trend for the three samples.The highest total selectivity on S34-L-M is 84.37%at 400°C,and that on S34-H-M and S34-V-M is 84.53%at 400°C and 80.95%at 420°C,respectively.For all prepared samples,the S34-L-M-T shows the most excellent performance for MTO process,this difference may arise from their different crystal mor-phologies and acid properties.The smaller the crystal size of S34-L-M-T is,the shorter the diffusion path is.The ethylene and propylene can dif-fuse out of the SAPO-34pores easily before being converted to other ad-ditives such as higher ole fins,aromatics and coke,therefore,the higher selectivity for ethylene and propylene is obtained.However,in terms of S34-L-M,S34-H-M and S34-V-M samples with larger crystal size,the diffusion path is longer,this presumably results in a second reaction along with a lower light ole fin selectivity.The characteristic that both the intensity and amount of strong acid sites for S34-L-M-T are relative-ly high is another reason for the excellent performance in MTO process.It has been reported that the conversion of methanol to dimethyl etherTable 1The pore structure characteristics of prepared SAPO-34samples.Sample BET surface area (m 2g −1)Micropore volume (cm 3g −1)Average pore diameter (nm)S34-L-M-T 546.60.260.440S34-L-M 463.20.270.435S34-H-M 447.30.280.466S34-V-M425.30.280.473Fig.4.NH 3-TPD pro files of prepared SAPO-34samples.Fig.5.FT-IR spectra of prepared SAPO-34samples.180J.Li et al./Powder Technology 262(2014)177–182Fig.6.TG/DTG curves of SAPO-34samples:(a)S34-L-M-T,(b)S34-L-M,(c)S34-H-M and (d)S34-V-M.Fig.7.MTO performance over SAPO-34catalysts at different reaction temperatures:(a)S34-L-M-T,(b)S34-L-M,(c)S34-H-M and (d)S34-V-M.(1)Methanol conversion.(2)Total selec-tivity for ethylene and propylene.(3)Selectivity for propylene.(4)Selectivity for ethylene.Reaction conditions:WHSV (MeOH)=4.8h −1,P =0.1Mpa.181J.Li et al./Powder Technology 262(2014)177–182(DME)occurs mainly on the weak acid sites while the conversion of DME and methanol to light ole fins occurs mainly on the strong acid sites [24].It is worthy of noting that for all prepared samples,the selectiv-ity for propylene is higher than that for ethylene at lower temperatures,and then decreases,while the selectivity for ethylene approaches the same at high temperatures.This may be ascribed to the change of pore size caused by coke deposition.When temperature is higher than 400°C,the methanol conversion reaches 100%,the coke formation rate increases because more molecules are involved in the reactions,causing the reduction of the SAPO-34pore size.The methanol conversion and DME selectivity in products with time on stream over prepared SAPO-34catalysts are shown in Fig.8.S34-L-M and S34-V-M samples have 100%conversion of methanol at the initial stage and keep this activity without DME intermediate in products for 180min and 150min,respectively,and then DME increases rapidly.In the case of S34-H-M,only trace amounts of DME with a selectivity of 0.36%is detected after 180min but then increases rapidly.Meanwhile,S34-L-M-T shows the longest catalyst longevity (the time when DME was observed from the beginning)for 240min.This is mainly due to its smaller crystal size and more number of acid sites.4.ConclusionsA dry gel liquid-phase transport method has been developed for syn-thesizing SAPO-34molecular sieve.By using Mor and Mor –TEAOH as structure directing agents,SAPO-34catalysts with regular cubic morphol-ogy and various crystal sizes were synthesized.The addition of TEAOH renders SAPO-34with small crystal pared to SAPO-34prepared by conventional hydrothermal and VPT method,the SAPO-34catalyst fabricated by dry gel liquid-phase transport method exhibits outstanding catalytic performance for MTO reaction.Among all catalysts,the S34-L-M-T catalyst displayed the best catalytic performance owing to its smaller crystal size and higher crystallinity.The conversion of methanol and se-lectivity to both ethylene and propylene reach 100%and 87.9%,respec-tively.The dry gel liquid-phase transport method could be extended to synthesize other molecular sieves due to its advantages,including the re-usability of expensive structure directing agent,enhancing the ef ficiency of synthesis reactors,higher molecular sieve yield and reducing the use of structure directing agent.AcknowledgmentsThe authors greatly thank the financial support from the Ministry of Science and Technology of the People's Republic of China (Grant No.2011BAD22B06),Chinese Academy of Sciences (XDA07070301and Y2010022)and Postdoctoral Science Foundation of Qingdao,China.The authors also thank Dr.Zhanquan Zhang for his fruitful advices.References[1] B.M.Lok,C.A.Messina,R.L.Patton,R.T.Gajek,T.R.Cannan,E.M.Flanigen,Crystallinesilicoaluminophosphates,US Patent 4,440,871(1982).(to Union Carbide Corporation).[2] B.M.Lok,C.A.Messina,R.L.Patton,R.T.Gajek,T.R.Cannan,E.M.Flanigen,Silicoaluminophosphate molecular sieves:another new class of microporous crystalline inorganic solids,J.Am.Chem.Soc.106(1984)6092–6093.[3]S.Wilson,P.Barger,The characteristics of SAPO-34which in fluence the conversionof methanol to light ole fins,Microporous Mesoporous Mater.29(1999)117–126.[4]M.Salmasi,S.Fatemi,A.T.Najafabadi,Improvement of light ole fins selectivity andcatalyst lifetime in MTO reaction;using Ni and Mg-modi fied SAPO-34synthesized by combination of two templates,J.Ind.Eng.Chem.17(2011)755–761.[5]Q.Wang,L.Wang,H.Wang,Z.Li,H.Wu,G.Li,X.Zhang,S.Zhang,Synthesis,characterization and catalytic performance of SAPO-34molecular sieves for methanol-to-ole fin (MTO)reaction,Asia Pac.J.Chem.Eng.6(2011)596–605.[6]L.Ye,F.Cao,W.Ying,D.Fang,Q.Sun,Effect of different TEAOH/DEA combinations onSAPO-34's synthesis and catalytic performance,J.Porous.Mater.18(2011)225–232.[7]T.Álvaro-Muñoz,C.Márquez-Álvarez,E.Sastre,Use of different templates on SAPO-34synthesis:effect on the acidity and catalytic activity in the MTO reaction,Catal.Today 179(2012)27–34.[8]P.Wang,A.Lv,J.Hu,J.Xu,G.Lu,The synthesis of SAPO-34with mixed template andits catalytic performance for methanol to ole fins reaction,Microporous Mesoporous Mater.152(2012)178–184.[9]T.L.Chew,A.L.Ahmad,S.Bhatia,Rapid synthesis of thin SAPO-34membranes usingmicrowave heating,J.Porous.Mater.18(2011)355–360.[10]J.Tan,Z.Liu,X.Bao,X.Liu,X.Han,C.He,R.Zhai,Crystallization and Si incorporationmechanisms of SAPO-34,Microporous Mesoporous Mater.53(2002)97–108.[11]W.Dai,G.Wu,L.Li,N.Guan,M.Hunger,Mechanisms of the deactivation of SAPO-34materials with different crystal sizes applied as MTO catalysts,ACS Catal.3(2013)588–596.[12] F.M.Shalmani,R.Halladj,S.Askari,Effect of contributing factors on microwave-assisted hydrothermal synthesis of nanosized SAPO-34molecular sieves,Powder Technol.221(2012)395–402.[13]Y.J.Lee,S.C.Baek,K.W.Jun,Methanol conversion on SAPO-34catalysts prepared bymixed template method,Appl.Catal.A 329(2007)130–136.[14]Y.Hirota,K.Murata,M.Miyamoto,Y.Egashira,N.Nishiyama,Light ole fins synthesisfrom methanol and dimethylether over SAPO-34nanocrystals,Catal.Lett.140(2010)22–26.[15]Z.Li,J.Martínez-Triguero,P.Concepción,J.Yu,A.Corma,Methanol to ole fins:activ-ity and stability of nanosized SAPO-34molecular sieves and control of selectivity by silicon distribution,Phys.Chem.Chem.Phys.15(2013)14670–14680.[16]H.Zhou,J.Gu,Y.Wu,J.Wang,Synthesis of SUZ-4zeolite by a dry gel conversionmethod,J.Porous.Mater.20(2013)523–530.[17] B.Chen,Y.Huang,Dry gel conversion synthesis of SAPO-and CoAPO-based molec-ular sieves by using structurally related preformed AlPO precursors as the starting materials,Microporous Mesoporous Mater.123(2009)71–77.[18]H.Shao,X.Chen,B.Wang,J.Zhong,C.Yang,Vapor-phase transport synthesis ofMnSAPO-34and its catalytic properties,China Pet.Process.Petrochem.Technol.14(2012)68–74.[19]P.R.Hari Prasad Rao,M.Matsukata,Dry-gel conversion technique for synthesis ofzeolite BEA,mun.12(1996)1441–1442.[20]Z.You,G.Liu,L.Wang,X.Zhang,Binderless nano-HZSM-5zeolite coatings preparedthrough combining washcoating and dry-gel conversion (DGC)methods,Micropo-rous Mesoporous Mater.170(2013)235–242.[21] A.Izadbakhsh,F.Farhadi,F.Khorasheh,S.Sahebdelfar,M.Asadi,Y.Z.Feng,Effect ofSAPO-34's composition on its physico-chemical properties and deactivation in MTO process,Appl.Catal.A 364(2009)48–56.[22]R.Xu,W.Pang,J.Yu,Q.Huo,J.Chen,Chemistry-zeolites and Porous Materials,Sci-ence Press,Beijing,2004.pp.147–148.[23]L.Zhang,J.N.Primera-Pedrozo,A.J.Hernández-Maldonado,Thermal detemplation ofNa-SAPO-34:effect on Sr 2+ion exchange and CO 2adsorption,J.Phys.Chem.C 114(2010)14755–14762.[24]L.Travalloni,A.C.L.Gornes,A.B.Gaspar,M.A.P.da Silva,Methanol conversion overacid solid catalysts,Catal.Today 133–135(2008)406–412.Fig.8.Methanol conversion and DME selectivity in products over SAPO-34catalysts.Reac-tion conditions:T =440°C,P =0.1Mpa,WHSV (MeOH)=4.8h −1.182J.Li et al./Powder Technology 262(2014)177–182。
One-step hydrothermal synthesis of ordered mesostructured carbonaceous monoliths with hierarchical porosities wYan Huang,a Huaqiang Cai,a Dan Feng,a Dong Gu,a Yonghui Deng,a Bo Tu,a Huangting Wang,b Paul A.Webley b and Dongyuan Zhao*abReceived (in Cambridge,UK)19th March 2008,Accepted 2nd April 2008First published as an Advance Article on the web 9th May 2008DOI:10.1039/b804716bHierarchical carbonaceous monoliths with ordered 2-D hexa-gonal mesostructures have been successfully synthesized by using phenolic resols as precursors and mixed triblock copolymers as templates via a one-step hydrothermal approach.Among the various nanostructured materials,porous carbon monoliths have attracted great interest in many fields,such as electrochemistry,energy storage,separation and chromato-graphy,mainly due to their high thermal and chemical stabi-lities,large surface areas and substantial advantages concerning mass transport.1–3Since the hierarchical structure of multi-level pores can greatly benefit mass flow transporta-tion,great interest has been focused on the design and synth-esis of carbon monoliths that have tailored porosity and pore interconnectivity.4Various approaches towards the synthesis of hierarchically-structured carbon monoliths with ordered mesopore channels have been conducted using the ‘‘nanocast-ing’’strategy,which involves multiple steps:first,the synthesis of mesoporous silica monolith templates with hierarchical porosities by a surfactant self-assembly process,then replica-tion of the silica template with carbon sources,and finally removal of the silica template.5–7However,this nanocasting strategy is somewhat fussy and costly given its complicated multiple step synthetic procedures,and sacrificial use of both surfactants and monolithic silica templates.Recently,ordered mesoporous carbon materials with var-ious symmetries have been successfully synthesized by an organic–organic self-assembly strategy via a solvent evapora-tion-induced self-assembly (EISA)method,8–10or by common aqueous reaction routes under atmospheric pressure at rela-tively low temperatures.11,12However,these methods are problematic for large-scale industrial production because of long processing times and the large vessels used for the inter-face assembly of mesostructures.Hydrothermal synthesis is a more efficient approach under controlled temperature and pressure that has been extensively exploited in a variety of inorganic syntheses of zeolites,nanomaterials,catalysts and ion-conductors.13–15In addition,this powerful method isfaster and more energy efficient than conventional aqueous chemical processing conditions.Herein,we report a facile,one-step,hydrothermal synthesis of carbon monoliths with hierarchical porosities,built up from ordered mesopores and interconnected macropores.Mixed triblock poly(propylene oxide)–poly(ethylene oxide)–poly-(propylene oxide)(PEO–PPO–PEO)copolymers of Pluronic F127and P123were employed as templates,and phenolic resols were used as carbon precursors (Fig.1).The synthesiswas carried out under hydrothermal conditions,in the pre-sence of a base catalyst,in an aqueous medium,at 1001C,for 10h.The resultant carbonaceous monoliths were formed in high yield (around 90wt%based on the carbon precursor),and were thermally stable and crack-free.It is interesting that the monoliths showed hierarchically porous structures,with interconnected macropores of around 3m m,ordered 2-D hexagonally-arranged mesopores of around 3nm and micro-pores in the mesopore walls.Microspherical and body-cen-tered cubic mesoporous carbons with irregular shapes could also be obtained by only employing F127as the template (see ESI w ).Our results also show that the high temperature synth-esis approach could be useful for the large-scale industrial production of mesoporous carbonaceous materials.The synthesis of monoliths was carried out in an autoclave with an aqueous solution containing the triblock copolymer templates.Typically,1.0g of phenol and 3.5mL of a formalde-hyde solution (37wt%)were dissolved in 5mL of a 0.5M NaOH solution and stirred at 701C for 0.5h.Then,an aqueous solution of mixed PEO–PPO–PEO (0.75g P123and 1.25g F127in 10mL of water)was added to the above solution,and the mixture continuously stirred at 701C for an additional 3h.After that,the solution was poured into an autoclave and transferred into an oven at 1001C for 10h.The polymeric monolith wasFig.1Schematic representation of the hydrothermal synthesis route towards carbon monoliths with hierarchical porosities.aDepartment of Chemistry,Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials,Advanced Materials Laboratory,Fudan University,Shanghai,200433,P.R.China.E-mail:dyzhao@;Fax:+8621-6564-1740;Tel:+8621-5566-4194bDepartment of Chemical Engineering,Monash University,VIC 3800,Australiaw Electronic supplementary information (ESI)available:Character-isation data and procedures.See DOI:10.1039/b804716bThis journal iscThe Royal Society of Chemistry mun.,2008,2641–2643|2641COMMUNICATION /chemcomm |ChemCommD o w n l o a d e d b yE a s t C h i n a U n i v e r s i t y o f S c i e n c e & T e c h n o l o g y o n 15 M a r c h 2012P u b l i s h e d o n 09 M a y 2008 o n h t t p ://p u b s .r s c .o r g | d o i :10.1039/B 804716BView Online / Journal Homepage / Table of Contents for this issuecollected by filtration,washed with water anddried in air.The obtained monolith sample was calcined at 6001C for 3h in a nitrogen flow to obtain mesoporous carbonaceous monoliths.To obtain carbon spheres,2.0g of F127was used as the template in the hydrothermal synthesis,and the other processes were per-formed according to the above procedure.To obtain ordered 3-D body-centered cubic mesostructured carbon,2.0g of F127was used as the template,and the autoclave was placed in a 1001C oil bath with vigorous magnetic stirring.A detailed characterization of the products is given in the ESI.wPhotographs of our typical,as-synthesized polymeric mono-liths and calcined carbon materials show a good bulk macro-scopic appearance (Fig.2),and both are very stable and crack-free.The color of the as-synthesized polymeric monolith is buff,and it turns to black after calcination at 6001C.The sizes of the monoliths can be easily adjusted by choosing autoclaves with different inner diameters.A typical,as-synthesized monolith in Fig.2a is large,with a length of B 1.0cm and a diameter of B 1.4cm.The high yield of carbonaceous monoliths and the relatively rapid speed of the procedure suggest that this high temperature synthetic approach could be useful for the large-scale industrial production of mesoporous carbonaceous materials.Mesoporous carbon monoliths obtained after carbonization at 6001C for 3h under a nitrogen atmosphere exhibit a black color and a similar column-like shape (0.9cm in diameter and 0.6cm in height),with comparably smaller sizes to the polymeric monolith.The volume shrinkage was calculated to be about 75%during the carboniza-tion process.A preliminary mechanical strength test showed that the carbon monolith is quite rigid and does not crack under a pressure of 0.2MPa.The mesostructure obtained from the organic–organic self-assembly method possesses an integral fra-mework and is mechanically stable,as shown by Meng et al .10Scanning electron microscopy (SEM)images (Fig.3a and b)show that the structures of the as-synthesized monoliths are constructed from interconnected particles.These particles build up a 3-D disordered macroporous framework,with sizes in the range of 2–5m m.After carbonization at 6001C,the macropore size slightly increases,although the structure contracts to about 75%.The macroporosity could also be revealed by Hg porosi-metry (Fig.4).The carbon monolith after calcination at 6001C showed an average macropore size of 3m m,with a relatively narrow size distribution and a correspondingly high total macro-pore volume of 1.14cm 3g À1.Transmission electron microscopy (TEM)images of the calcined sample (Fig.3c and d)reveal that these building blocks are of an ordered 2-D hexagonal meso-structure.The cell parameter,a ,is estimated from the TEM images to be B 10.0nm.The ordered periodic arrangement of mesopores could be further confirmed by their small angle X-ray scattering (SAXS)patterns (Fig.5a).The pattern of the as-synthesized carbonaceous monolith exhibits two scattering peaks.After carbonization at 6001C,the SAXS pattern becomes more resolved.Three resolved scattering peaks are observed,which can be indexed as 10,11and 20reflections associated with a 2-D hexagonal mesostructure (space group of p 6mm ).The cell parameter,a ,is calculated to be 10.0nm,which is in good agreement with TEM observations.Shrinkage of the meso-porous framework is about 24%,which is much less than the macroscopic contraction.N 2adsorption–desorption isotherms of mesoporous carbon monoliths (Fig.5b)show typical type-IV curves,with a clear condensation step at P /P 0=0.3–0.5,suggesting a uniform mesopore.The specific BET surface area is calculated to be as high as 620m 2g À1.The micropore area is 360m 2g À1.The micropore and mesopore volumes are 0.16and 0.20cm 3g À1,respectively.The Barrett–Joyner–Halenda (BJH)mesopore size distribution,calculated from the adsorption branch,is quite narrow,with a mean value of 3.0nm.Fig.2Representative photos of (a)the as-synthesized polymeric monolith and (b)the carbon monolith after calcination at 6001C under a nitrogen atmosphere for 3h.Fig.3SEM images of the (a)as-synthesized and (b)calcined mono-lith samples.TEM images of the hierarchical carbon monolith with its ordered hexagonal mesostructure viewed from the (c)[10]and (d)[11]directions.Fig.4Hg porosimetry curves of mesoporous carbon monoliths after carbonization at 6001C under a nitrogen atmosphere.2642|mun.,2008,2641–2643This journal iscThe Royal Society of Chemistry 2008D o w n l o a d e d b yE a s t C h i n a U n i v e r s i t y o f S c i e n c e & T e c h n o l o g y o n 15 M a r c h 2012P u b l i s h e d o n 09 M a y 2008 o n h t t p ://p u b s .r s c .o r g | d o i :10.1039/B 804716BFig.1illustrates the hydrothermal synthetic process for preparing carbonaceous monoliths with hierarchical porosities through the organic–organic self-assembly of triblock copoly-mers and phenolic precursors.In the first stage,the polymeric,water-soluble resol precursors are involved in a supramolecu-lar templating process with triblock copolymers in an aqueous medium.The phenolic oligomers show more compatibility with the PEO blocks of amphiphilic PEO–PPO–PEO tem-plates compared to the aqueous environment,and can be self-assembled into ordered mesostructures.Fast polymerization under hydrothermal conditions causes the macro-domains to be divided into a phenolic resin/PEO–PPO–PEO-rich phase and a water-rich phase.In the mixed F127–P123/resin/water system,the resin domains partially aggregate and further polymerize to form a rigid co-continuous structure,where both the resin and aqueous phases are continuous and highly interconnected.After drying in air,the aqueous phase do-mains can turn to the interconnected macropore system.16When only F127is used as the template,it tends to form dispersed,less ordered microspheres in 80%yield (ESI Figs.S1and S2w ).Our results show that the diameters of the microspheres are not very uniform,around 8m m,and vary with F127content and the hydrothermal treatment time.The spherical morphology implies that the higher EO/PO ratio of F127can effectively stabilize the resin domain by greatly reducing the interface free energy.While under vigorous stirring,the SAXS,SEM and TEM image results (ESI Figs.S3and S4w )show that the products have an ordered body-centered cubic mesostructure with an irregular shape.The mesopore size is uniform,with a mean value of 3.8nm.This can be explained by a uniform templating effect and a higher surface free energy caused by shearing forces.The removal of triblock copolymer templates leads to the formation of mesopores.It has also been found that mesopor-ous carbonaceous materials templated by the triblock copoly-mers inherently possess microporosity within the phenolic walls,due to a strong affinity between the PEO block and the precursors.17,18With the cross-linking of the phenolic resins,the PEO segments are embedded into the pore walls and then removed by calcination,resulting in the formation of micropores within the carbonaceous walls.The resultantcarbon monoliths have a unique hierarchical construction of interconnected macropores,ordered mesopores and micro-pores in the pore walls after removal of the template.In summary,we have demonstrated a simple one-step hydro-thermal method for the synthesis of carbonaceous monoliths with hierarchical porosities,by employing F127–P123mixed triblock copolymers as templates and phenolic resols as carbon precursors at 1001C.The monoliths show an ordered 2-D hexagonal mesostructure with a uniform pore size (B 3nm)and 3-D irregular macroporous scaffolds with a size of B 3m m.The high temperature synthesis approach is very efficient,with a high yield of 90wt%.This may be useful for the large-scale industrial production of ordered mesoporous carbonaceous materials.This approach allows the one-pot formation of both ordered mesostructures and irregular 3-D scaffolds,which will be of considerable importance for the facile production of hierarch-ical porous carbon materials.This work was funded by the NSFC (20721303and 20521140450),the State Key Basic Research Program of the PRC (2006CB202502,2006CB932302),the Shanghai Science &Technology Committee (06DJ14006),the Shanghai Nano-tech Promotion Center (0652nm024),the Shanghai leading academic discipline project (B108),the Australian Research Council (Discovery Project no.DP0773160)and Monash University.Y.Huang thanks the support from Fudan Graduate Innovation Funds.Notes and references1L.Z.Fan,Y.S.Hu,J.Maier,P.Adelhelm,B.Smarsly and M.Antonietti,Adv.Funct.Mater.,2007,17,3083–3087.2Y.Xia and R.Mokaya,J.Phys.Chem.C ,2007,111,10035–10039.3H.F.Yang,Q.H.Shi,X.Y.Liu,S.H.Xie,D.C.Jiang,F.Q.Zhang,C.Z.Yu,B.Tu and D.Y.Zhao,mun.,2002,2842–2843.4A.Taguchi,J.H.Smatt and M.Linden,Adv.Mater.,2003,15,1209–1211.5Y.S.Hu,P.Adelhelm,B.M.Smarsly,S.Hore,M.Antonietti and J.Maier,Adv.Funct.Mater.,2007,17,1873–1878.6A.H.Lu,J.H.Smatt and M.Linden,Adv.Funct.Mater.,2005,15,865–871.7X.Wang,K.N.Bozhilov and P.Feng,Chem.Mater.,2006,18,6373–6381.8Y.Deng,T.Yu,Y.Wan,Y.Shi,Y.Meng,D.Gu,L.Zhang,Y.Huang,C.Liu,X.Wu and D.Y.Zhao,J.Am.Chem.Soc.,2007,129,1690–1697.9Y.Huang,H.Q.Cai,T.Yu,F.Q.Zhang,F.Zhang,Y.Meng,D.Gu,Y.Wan,X.L.Sun,B.Tu and D.Y.Zhao,Angew.Chem.,Int.Ed.,2007,46,1089–1093.10(a )Y.Meng,D.Gu,F.Zhang,Y.F.Shi,L.Cheng,D.Feng,Z.X.Wu,Z.X.Chen,Y.Wan,A.Stein and D.Y.Zhao,Chem.Mater.,2006,18,4447–4464;(b )Y.Meng,D.Gu,F.Q.Zhang,Y.F.Shi,H.F.Yang,Z.Li,C.Z.Yu,B.Tu and D.Y.Zhao,Angew.Chem.,Int.Ed.,2005,44,7053–7059.11F.Q.Zhang,Y.Meng,D.Gu,Y.Yan,C.Z.Yu,B.Tu and D.Y.Zhao,J.Am.Chem.Soc.,2005,127,13508–13509.12F.Q.Zhang,Y.Meng,D.Gu,Y.Yan,Z.X.Chen,B.Tu and D.Y.Zhao,Chem.Mater.,2005,18,11652–11662.13C.Vercaemst,M.Ide,B.Allaert,N.Ledoux,F.Verpoort and P.Van Der Voort,mun.,2007,2261–2263.14B.L.Cushing,V.L.Kolesnichenko and C.J.O’Connor,Chem.Rev.,2004,104,3893–3946.15C.S.Cundy and P.A.Cox,Chem.Rev.,2003,103,663–701.16D.Brandhuber,H.Peterlik and N.Huesing,Small ,2006,2,503–506.17Y.Wan,Y.F.Shi and D.Y.Zhao,mun.,2007,897–926.18Y.Wan and D.Y.Zhao,Chem.Rev.,2007,2821–2860.Fig.5(a)SAXS patterns of as-synthesized and calcined carbonac-eous monoliths and (b)N 2sorption isotherms with the pore size distribution (inset)of the mesoporous carbon monolith after calcina-tion at 6001C.This journal iscThe Royal Society of Chemistry mun.,2008,2641–2643|2643D o w n l o a d e d b yE a s t C h i n a U n i v e r s i t y o f S c i e n c e & T e c h n o l o g y o n 15 M a r c h 2012P u b l i s h e d o n 09 M a y 2008 o n h t t p ://p u b s .r s c .o r g | d o i :10.1039/B 804716B。
Analytica Chimica Acta 691 (2011) 43–47Contents lists available at ScienceDirectAnalytica ChimicaActaj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /a caHierarchical CuO nanochains:Synthesis and their electrocatalytic determination of nitriteCao Xia a ,b ,Cai Xiaolan a ,b ,Wang Ning a ,∗,Guo Lin aa School of Chemistry and Environment,Beijing University of Aeronautics and Astronautics,Beijing,100083,China bSchool of Material Science and Engineering,Jiangsu University of Science and Technology,Jiangsu,212003,Chinaa r t i c l e i n f o Article history:Received 11November 2010Received in revised form 13February 2011Accepted 15February 2011Available online 23 February 2011Keywords:CuOHierarchical nanostructure Electrocatalysis Nitritea b s t r a c tMicrospheres with multilevel structure are widely used as removable catalysts,adsorbents,drug carri-ers and imaging contrast agents due to their hierarchical porosity.In this paper a facile wet chemical method for the synthesis of hierarchically nanostructured CuO chains assembled with small nanorods is reported.The as prepared nanomaterial is characterized using X-ray diffraction (XRD),field-emission scanning electron microscopy (FE-SEM)and high-resolution transmission electron microscopy (HRTEM).The autocatalytic response to nitrite ions is also carried out,which exhibits a wide linear range (0.004–3.7mM),sound sensitivity (177.9A mg −1dL −1cm −2),highly reproducible response (R.S.D.of 2.0%),and an excellent long-term stability.The good analytical performance,low cost and straightforward preparation method make this novel electrode material promising for the development of effective nitrite sensors.© 2011 Elsevier B.V. All rights reserved.1.IntroductionAdverse effects due to the build-up of high nitrite concentra-tions are major environmental concerns at the present stage [1].Consequently,researches on the development of electrochemical nitrite sensors with high sensitivity,fast response,good stability and low cost are receiving considerable attention [2,3].Platinum,palladium,rhodium,iridium and gold are important electro-oxidation catalysts.They are widely used for electrochemi-cal sensors due to their excellent chemical stability and catalytic activity [4–6].However,considering the high cost and limited reserves,now transition metal or their compounds are choosen as sound alternatives for scaled up commercial applications [7–22].For example,copper based materials,such as copper bulk metal [12],copper nanoparticles [13,14],copper alloys [15]and copper complexes [16],have been proved highly active for electro-chemical oxidation/reduction of carbohydrates [13],alcohols [15],amino acids/peptides [17],cyanide and nitrite [18].Hereinto,copper oxides catalysts show advantages such as low cost,sta-bility,as well as the absence of any oxygen limitation,which are helpful for the development of next generation electrochemical biosensors.However,in contrast with the synthesis and catalytic applica-tion of Cu [12–14,17]and Cu 2O [18,19],reports on the preparation∗Corresponding author.Tel.:+861082338212;fax:+861082338212.E-mail address:wangning@ (W.Ning).of hierarchically nanostructured CuO are rather rare,and little attention has been paid to their sensing performance related the mode of assembly.Meanwhile,inorganic catalysts often suffered from the very low surface area,structural collapse,and the as resulted poor catalytic efficiency [20].These problems can be gen-erally solved via two ways in the past.One is loading the oxides onto suitable oxides supports leading to highly dispersed surface,where time and energy-consuming calcination step is usually included [19–21].Another way is to use the rationally designed multilevel nanocatalyst,which can both offer high specific surface and avoid any possible complex loading process [3,13,14,22].For this rea-son,hierarchically nanostructured CuO particles may be an ideal candidate as electrocatalysts due to their extremely small size,anisotropy and the high specific surface areas [23].Furthermore,size and conformation-dependent electronic properties such as excellent sensitivity and rapid response may also be expected in contrast to the 0-D nanoparticles.Hence in this paper,the synthesis of hierarchical CuO chains assembled with small nanorods via a mild wet chemical method is reported.With the expectation that this CuO sens-ing material on the electrode surface may mediate the targeted heterogeneous chemical redox process while the converted iron oxides can be continuously recovered by the subsequent elec-tron transfer with the electrode substrate,an electrochemical nitrite electrode is fabricated and electrochemically character-ized.The high catalytic activity and excellent selectivity toward the oxidation of nitrite may trigger the possibilities of these nanomaterials towards diverse applications in biosensors and electroanalysis.0003-2670/$–see front matter © 2011 Elsevier B.V. All rights reserved.doi:10.1016/j.aca.2011.02.03744 C.Xia et al./Analytica Chimica Acta691 (2011) 43–472.Experimental2.1.MaterialsAll chemicals were purchased from Sinopharm Chemical Reagent,Beijing Co.,Ltd.and used without further purification. 2.2.InstrumentationScanning electron microscopy images were recorded from a Hitachi S4800coldfield emission scanning electron microscope (CFE-SEM).X-ray powder diffraction(XRD)pattern of the corrosion products was collected by a Rigaku X-ray diffractometer(Rigaku Goniometer PMG-A2,CN2155D2,wavelength=0.15147nm)with Cu K␣radiation.UV–vis absorption spectra were recorded with a Perkin-Elmer-950spectrometer.All the electrochemical tests were carried out at room temperatures under nitrogen atmosphere on a CHI660C electrochemical station,using a single compart-ment,three-electrode cell.Electrode potentials were measured with respect to an aqueous saturated calomel electrode(SCE).A Pt wire was used as the counter electrode.2.3.ProceduresHierarchically nanostructured CuO chains were synthe-sized through the following process. 1.2g of copper chloride (CuCl2·2H2O),6g of Poly(vinyl pyrrolidone)(PVP,Mw30,000), 10g of(NH4)2SO4and2g of NaOH were dissolved150mL of pure water to form a green solution by ultrasonication for30min.The mixture was stirred vigorously and refluxed for12h,the black powder was taken out and washed with ethanol and water several times and then dried in air for characterization.Spherical CuO nanoparticles were prepared as that reported previously[24].The hierarchically nanostructured CuO chains modified elec-trode(nc-CuOE)was prepared as follows[25]:First the surface of the glassy carbon electrode(GCE,3mm in diameter)for each exper-iment was mechanically polished with600grit sand-paper and then with0.050m␣-alumina powder.The polished surface rinsed with acetone and then with doubly distilled water.A chitosan(CH) (0.50%)solution was prepared by dissolving CH(50mg)in100mL of acetate buffer(0.05M,pH6.2)solution.5mg of nanostructured CuO chains were dispersed into transparent CH solution and mag-netic stirring for about30min at room temperature followed by ultrasonication for about1h.Then10l of nano CuO–CH solution was spread uniformly on to the surface of the pretreated GCE elec-trode.After dried at atmosphere,a nc-CuOE electrode was obtained. In addition,the nanoparticle CuO modified(np-CuOE)and bulk CuO modified electrode(b-CuOE)were also prepared forcomparison.Fig.1.The XRD pattern of the as prepared samples.3.Results and discussionThe XRD pattern of the as prepared samples is shown in Fig.1.The as-synthesized material can be identified as pure phase monoclinic CuO(JCPDS45-0937,space group C2/c(15), cell=4.685×3.426×5.13)and no other minor phase peaks were detected.SEM analysis of the samples(Fig.2)demonstrates the forma-tion of chain-like hierarchically nanostructured CuO nanomaterials with high uniformity on a large scale.Average size of the chains is measured to be about1m in diameter and several micrometers in length.From SEM image of higher magnification,it can be observed that the building blocks of the chain areflower like CuO spheres. All the petals grow from the center,with length of about500nm and diameter of about30–40nm.Detailed information on the assembled CuO chains was obtained from TEM and HRTEM(Fig.3A–C).It can be seen that the bundled nanorods are composed of one-dimensionally assembled nanopar-ticles with diameters of less than40nm(Fig.3B).The HRTEM image clearly exhibits the(002)lattice planes of hexagonal CuO,with a lattice spacing of0.26nm(Fig.3C).The synthetic method reported here is highly relevant because the hierarchically nanostructured chain-like CuO powders composed of orderly assembled particles is rather uncommon.The peculiar structure of the nanomaterials can be expected to achieve the merits of high specific area and structural stability.In this sense,the hierarchically nanostructured chain-like CuO crystals are expected to be good candidates for the catalysis based applications.The assembled crystals can providea Fig.2.SEM images of the as-prepared CuO samples.C.Xia et al./Analytica Chimica Acta691 (2011) 43–4745Fig.3.Typical TEM and HRTEM images of the as-prepared CuO samples.(A)and(B)TEM images,(C)and(D)HRTEM images. significant enhancement in effective electrode surface comparedwith its bulk solid counterpart.Fig.4shows the UV–vis spectra of CuO in the(a)absence and(b)presence of NaNO2recorded in dimethyl sulfoxide(DMSO).Itcan be seen that there is a distinct monomeric form Q-band with amaximum at825nm for CuO.However,on addition of NO2−,the Q-band of CuO splits into two peaks with maxima at648and818nm,which indicates the coordination of nitrite to the CuO and can beobserved even at very low CuO concentrations.Cyclic voltammograms(CVs)were recorded to understand theelectrocatalytic behaviors of nc-CuOE electrode in0.1M PBS solu-tions towards the oxidation of1mM NaNO2at a scanning rate of0.1V s−1.As witnessed from Fig.5,the nc-CuOE electrode showspronounced electrocatalytic effect for the oxidation of nitrite incomparison to the np-CuOE electrode and the b-CuOEelectrode.Fig.4.UV–vis absorption spectra of CuO recorded in DMSO,in the absence(a)and presence(b)of NaNO2.The enhanced peak currents for oxidation of NaNO2with oxida-tion potential of ca.+0.8382V is far more nagative than that with np-CuOE electrode(+0.875V)and b-CuOE electrode(+1.068V). However,the CVs of the nc-CuOE electrode run in a blank PBS solu-tion(without NaNO2)show no obvious peak,while no visible peak current decay is observed on the nc-CuOE electrode after repeti-tive potential scan for20cycles at the same solutions.Thus it can be deduced that little nano CuO powder has not been peeled off from the electrode surface.In the potential range within which Cu(II)/Cu(I)centers can be involved in a redox process,the material displays good con-ductivity due to the electronic and ionic transport.The oxidation process can be elucidated through an electrocatalytic mechanism involving the Cu(II)/Cu(I)centers,and the catalytic mechanismofFig.5.Cyclic voltammograms of the nc-CuOE electrode in0.1M phosphate buffer solutions(PBS,pH7.5)containing1mM NaNO2at scanning rate of0.1V s−1.(a)nc-CuOE electrode;(b)np-CuOE electrode(c)b-CuOE electrode;(d)n-CuOE electrode at the absence of NaNO2;Inset is the magnification of curve c and d.46 C.Xia et al./Analytica Chimica Acta691 (2011) 43–47Fig.6.Cyclic voltammograms of 1mM NaNO 2in different pH (a)5.1,(b)6.2,(c)7.0,(d)8.1(e)and 9.2solutions at the nc-CuOE electrode (from left to right)(A),and the dependences of the NaNO 2peak current (red line)and redox potential (black line)on the PBS solution pH (B)with a scan rate of 100mV s −1.(For interpretation of the references to color in this figure legend,the reader is referred to the web version of the article.)the CuO towards nitrite oxidation can be explained by the following scheme:Cu(II)+NO 2−+H 2O →Cu(I)+2H ++NO 3−(1)Cu(I)→Cu(II)+e −(2)The pH of the supporting electrolyte has a significant influence on the NaNO 2electro-oxidation at the nc-CuOE electrode by affect-ing both the peak current and peak potential.Fig.6A shows CVs of nc-CuOE electrode in 1mM NaNO 2with different pH values and Fig.6B illustrates the dependences of the NaNO 2anodic peak cur-rent and redox potential on the PBS solution pH.The anodic peak current increases with increasing solution pH,while the NaNO 2current peaks at pH 8.1.In addition,the anodic peak potentials for the oxidation of NaNO 2shifted negatively with increasing pH,showing that protons have taken part in the rate-determining step of the electrode processes.Plots of redox potential versus pH for the nc-CuOE electrode is linear in the pH range from 5.1to 9.2,as shown in Fig.6B.The slope in this pH range is −111mV/pH,which is close to the theoretical value of −112mV/pH for a 1e −/2H +reac-tion process [26].For the ternary mixture containing NaNO 2,PBS was used to control the pH of the mixture and the pH value of 7.5was chosen for the following experiment,which is much closer to physical conditions.In order to investigate electron transfer kinetics of CuO,cyclic voltammograms of electrodes were recorded at different scan rates.Fig.7shows the recorded cyclic voltammograms of the nc-CuOE electrode in pH 7.5PBS solutions containing 1mM NaNO 2at dif-ferent scan rates (10–100mV s −1).It is interesting to note thattheFig.7.Cyclic voltammograms of nc-CuOE electrode in 0.1M PBS solution (pH 7.5)containing 1mM NaNO 2at different scan rates (10–100mV s −1,from inner to out-side).Inset is a linear relationship between the anodic current and the square root of scan rate.oxidation peak potential (Eap)for NaNO 2shifts positively when the scan rate increases,indicating a kinetic control over the oxidation of NaNO 2at the redox sites of the nc-CuOE electrode.A Tafel slope of 110mV was obtained from the linear plot (Eap.vs.log V )for nc-CuOE electrode.This slope indicates the fact that a one-electron transfer process should be the rate determining step,assuming a transfer coefficient of 0.3<␣<0.7,and there is a greater probabil-ity that the reaction activated transition state forms the product.The influence of scan rate on the oxidation current of NaNO 2was also made (inset of Fig.7).There is a linear relation between the anodic peak currents and the square root of scan rate in the range of 10–100mV s −1with a correlation coefficient of 0.9948.These results suggest that the kinetics of the overall process is controlled by the diffusion process which is assumed to be related to the relatively slow diffusion of protons [20].CVs obtained at the nc-CuOE electrode in 0.1M PBS solutions containing different concentrations of NaNO 2are shown in Fig.8.The oxidation peak currents increase gradually with increasing the concentration of NaNO 2.Inset of Fig.8represents the calibration curve for the determination of NaNO 2at the nc-CuOE electrode.Lin-ear dependency could be observed between the current response and concentration of nitrite in the range of 0.004–3.7mM with a sensitivity and correlation coefficient of 12.69A mM −1and 0.9991.Furthermore,a low detection limit of 3×10−4mM nitrite is determined at the signal-to-noise ratio of 3.These results are comparable or better than previously reported [27–32](Table 1).In particular,the proposed method avoids using diffusion electron transfer mediators for the determination of nitrite,which is nec-essary for traditional non-enzyme-based biosensors and realizes a reagent-less nitrite biosensing.The hierarchicallynanostructuredFig.8.Cyclic voltammograms of nc-CuOE electrode in 0.1M PBS solutions at a scan rate of 0.1V s −1using different concentrations of NaNO 2(0.2–1.0mM,step is 0.2mM;1.5–3.5mM,step is 0.5mM).Inset is the relationships between the oxi-dation peak current and the NaNO 2concentrations.C.Xia et al./Analytica Chimica Acta 691 (2011) 43–4747Table 1Comparison of different modified electrodes for nitrite determination.ElectrodeLCR (M)LoD(M)Ref.Nano-MQPOE electrode5.0–3000 3.6[3]Cytc/l-Cys/P3MT/MWCNT/GCE 10–1000.5[27]nano-Pt/P3MT/GCE 8.0–1700 1.5[28]CNTs/PAA/GCE 3–4500 1.0[29]Thionine/ACNTs3.0–500 1.12[30]Ce(III)–EDTA complex 10–100004.8[32]Pt/Ch/GCE1.2–9000.4[5]Nc-CuOE electrode4.0–37000.3This workMQPOE:MnO 2/QPVP composite electrodes;Cyt c:Cytochrome c;l-Cys:l-Cysteine;P3MT:Poly-(3-methylthiophene);GCE:Glassy carbon electrode;MWCNT:Mul-tiwalled carbon nanotubes;CNTs:Carbon nanotubes;ACNTs:Aligned carbon nanotubes PAA:Poly(acrylic acid);Ch:Choline;LCR:Linear concentration range;LoD:Limit of detection.Table 2Determination of nitrite in picked vegetables samples (n =10).SamplesThe proposed method UV–vis detected (mg kg −1)Detected (mg kg −1)Added (mg kg −1)Found (mg kg −1)Recovery (%)R.S.D.(%)117.631027.0998.0 2.9017.26218.951028.1897.3 1.0018.58318.111028.45101.21.9718.69chain-like CuO powders provide larger contact area and well dis-tributed porosity between sensing materials and sensed species than the dispersed nanoparticle or bulk CuO powders.In addi-tion,due to small size of nanoparticles,the charge distribution on the surface may lead to less resistance to the diffusion of probe ions onto electrode surface than that of the bulk one.Thus,the CuO nanostructures coating can promote the transportation and accessibility of target molecules more effectively and facilitate the transfer of electrons or signals,which is the key to enhanced sen-sitivity and lowered nonfaradic behavior.Possible interference for the detection of nitrite on nc-CuOE electrode was investigated by addition of various interferents into the PBS solution (pH 7.0)containing 0.1mM nitrite.The results show that most of the interferents,such as NO 3−,HCO 3−,Ca 2+,Mg 2+donot interfere with the determination in a 100-fold concen-tration,while 50-fold amount of Cd 2+and Pd 2+,and 20-fold amount of SO 32−exhibit serious interference.Vegetables represent a natural source of exogenous nitrite,which may brings harm to human body at excessive levels of intake.The practical applicability of the as fabricated sensor was tested by analysis of nitrite in picked vegetables samples.The picked vegetables was firstly crushed adequately in the mortar then mixed with 200mL deionized water.Afterward,the solid of the mixture was filtered and separated.The residual solution was added into PBS (pH 7.5)for detection.As can be seen from Table 2,the recovery and R.S.D.are in the ranges of 97.3–101.2%and 1.0–2.9%.Meanwhile,the UV–visible experiment shows no sig-nificant statistical difference between the results obtained by the proposed method and standard methods at 95%confidence level (Table 2).The operational stability of the modified electrode was inves-tigated in continuous operation mode.The repeated experiments reveal good reproducibility with 3.2%deviation.The storage stabil-ity was examined at the same modified electrode in consecutive 30days.The response to the reduction of the same concentrationof nitrite at the optimum potential remains at 98.5%of the initial values.The good long-term stability could be attributed to both the structure stability and the preparation method of the thin films.4.ConclusionsIn summary,hierarchically nanostructured CuO chains were synthesized and employed for the investigation of the electrocat-alytic sensing of nitrite.Due to the intrinsic structural stability,ordered porosity and high specific surface area,the as modified nc-CuOE electrode showed both high selectivity and sensitivity in determination of nitrite.The good analytical performance,low cost,renewable feature and simple preparation procedures indicate their wide potential towards catalytic applications for the next gen-eration of low-cost,highly effective and sensitive nitrite sensors.AcknowledgmentThis project was financially supported by National Natural Science Foundation of China (NSFC No.50902007,973Pro-gram (2010CB934701),the Natural Science Foundation of Jiangsu Province (BK2010341),the Natural Science Foundation of Jiangsu Universities (No.09KJB430005)and Specialized Research Fund for the Qinglan Engineering Program of Jiangsu province.Supports by the Fundamental Research Funds for the Central Universities are also acknowledged (YMF1002016).References[1]I.K.Konstantinou,D.G.Hela,T.A.Albanis,Environ.Pollut.141(2006)555–570.[2]P.Tau,T.Nyokong,J.Electroanal.Chem.611(2007)10–18.[3]X.Cao,N.Wang,L.Guo,Sens.Actuators B Chem.137(2009)710–714.[4]A.De la Escosura-Muniz,M.B.Gonzalez-Garcia,A.Costa-Garcia,Electroanalysis 16(2004)1561–1568.[5]P.Wang,F.Li,X.Huang,Y.X.Li,L.Wang,mun.10(2008)195–199.[6]S.D.Yang,X.G.Zhang,H.Y.Mi,X.G.Ye,J.Power Sources 175(2008)26–32.[7]J.Davis,M.J.Moorcroft,S.J.Wilkins,pton,M.F.Cardosi,Analyst 125(2000)737–741.[8]J.Davis,pton,Anal.Chim.Acta 404(2000)241–247.[9]B.Sljukic,C.E.Banks,pton,Nano Lett.6(2006)1556–1558.[10]P.J.Hill,S.Alam,A.Katsuhiko,mun.2(2008)383–385.[11]L.Ozcan,Y.Sahin,H.Türk,Biosens.Bioelectron.24(2008)512–517.[12]H.Heli,M.Jafarian,M.G.Mahjani, F.Gobal,Electrochim.Acta 49(2004)4999–5006.[13]K.B.Male,S.Hrapovic,Y.Liu,D.Wang,J.H.T.Luong,Anal.Chim.Acta 516(2004)35–41.[14]H.Y.Liu,X.D.Su,X.F.Tian,Z.Huang,W.B.Song,J.Z.Zhao,Electroanalysis 18(2006)2055–2060.[15]I.H.Yeo,D.C.Johnson,J.Electroanal.Chem.495(2001)110–119.[16]Y.Hasebe,T.T.Gu,J.Electroanal.Chem.576(2005)177–181.[17]I.G.Casella,T.R.I.Cataldi,A.Guerrieri,E.Desimoni,Anal.Chem.Acta 335(1996)217–225.[18]B.Sljukic,C.E.Banks,A.Crossley,pton,Electroanalysis 19(2007)79–84.[19]Z.H.Ai,H.Y.Xiao,T.Mei,J.Liu,L.Z.Zhang,K.J.Deng,J.R.Qiu,J.Phys.Chem.C 112(2008)11929–11935.[20]ngley,C.Biljana,C.E.Banks,pton,Jpn.Soc.Anal.Chem.23(2007)165–170.[21]S.B.Khoo,F.Chen,Anal.Chem.74(2002)5734–5741.[22]N.Wang,X.Cao,X.Cai,Y.Xu,L.Guo,Analyst 135(2010)2106–2110.[23]X.Cao,N.Wang,X.Q.Lu,L.Guo,J.Electrochem.Soc.157(2010)K76–K79.[24]S.C.Lee,S.H.Park,S.M.Lee,J.B.Lee,H.J.Kim,Catal.Today 120(2007)358–362.[25]A.Kaushika,R.J.Khana,P.R.Solanki,P.Pandeya,J.Alam,S.Ahmad,B.D.Malhotra,Biosens.Bioelectron.24(2008)676–683.[26]P.Wang,X.Wang,X.Y.Jing,G.Zhu,Anal.Chim.Acta 424(2000)51–56.[27]S.C.William,G.Yoshitaka,J.Electroanal.Chem.583(2005)300–306.[28]O.A.Bolade,I.O.Kenneth,N.Tebello,Anal.Chim.Acta 51(2006)6470–6478.[29]W.Sun,X.Q.Li,Y.Wang,X.Li,Bioelectrochemistry 75(2009)170–175.[30]Y.Song,Y.T.Ma,Y.Wang,J.W.Di,Electrochim.Acta 55(2010)4909–4914.[31]Y.Zhou,H.Y.Xian,F.Li,S.N.Wu,Y.X.Li,Electrochim.Acta 55(2010)5905–5910.[32]A.Abbaspour,M.A.Mehrgardi,Talanta 67(2005)579–584.。
碱式钼酸铜的制备及其催化性质的研究王红红;牛和林【摘要】Cu3(MoO4)2(OH)2 nanosheets were synthesized by a fast and facile sonochemical method.The structure and morphology of as-synthesized products were characterized by X-ray diffraction (XRD),transmission electron microscopy (TEM) and fourier transform infrared spectroscopy (FT-IR).The removal efficiency of theCu3(MoO4)2(OH)2 was evaluated by removal of Congo red combining adsorption and photocatalysis technology.The result indicated thatCu3(MoO4)2(OH)2 nanosheets exhibited higher removal efficiency.%采用快速高效的超声化学法合成一种纳米片状的多金属氧酸盐(碱式钼酸铜).利用X-射线衍射仪(XRD)、透射电子显微镜(TEM)、傅里叶红外光谱仪(FT-IR)等分析测试手段对所得产物的结构和形貌进行表征.并通过吸附-光催化联合作用研究了碱式钼酸铜对刚果红(CR)的降解效果.结果表明纳米片状的碱式钼酸铜对CR有很好的降解效率.【期刊名称】《安徽大学学报(自然科学版)》【年(卷),期】2017(041)001【总页数】5页(P95-99)【关键词】碱式钼酸铜;吸附;光催化;刚果红【作者】王红红;牛和林【作者单位】安徽大学化学化工学院,安徽合肥 230601;安徽大学化学化工学院,安徽合肥 230601【正文语种】中文【中图分类】TQ426.83随着工业产业的日益崛起,水污染问题也逐渐加重,这对人类的健康生活产生了严重的影响,因此如何能够高效快速地治理水污染已经成为人们日益关注的焦点.除去废水中的染料是治理水污染问题的一个重要方面,染料的治理有很多种方法,例如臭氧化[1]、氧化[2]、化学混凝/絮凝[3]、浊点萃取[4]、离子交换[5]、正向渗透[6]、光催化[7]和吸附[8]等.其中吸附技术因其具有操作简单、成本低、效率高等特点,引起了研究者的广泛关注[9-10].但吸附技术也存在一定的缺陷,它只是使溶液中染料分子转移到吸附剂的表面而不能降解.同时,光催化技术在治理水污染中是研究的最为成熟的方法,但多数催化剂只有在紫外光的条件下(约占太阳光2%)才能达到较高的催化效率[11].而单一的吸附和光催化技术不能很好地有效降解有机污染物.因此,为了能够有效地利用太阳光(可见光约占太阳光的40%)进行催化降解有机污染物,制备同时具有吸附性能和光催化性质的新型材料已经成为了一种新的趋势[12-13].多金属氧酸盐(polyoxometalate),简称为多酸(POM), 它是由两个或多个过渡金属氧酸阴离子组成,并通过氧离子连接形成封闭的三维框架,是无机化学的一个重要研究领域.POMs由于具有新奇的结构,被广泛应用在催化、医学、磁化学和电化学等领域[14-17],已经成为当代研究中一类新型功能类型的分子材料.其中含铜钼酸的多金属氧酸盐[18-19]被广泛研究并用于催化降解有机物,但是对于碱式钼酸铜的研究却很少.因此,作者以乙酸铜、七钼酸铵为原料,首次采用一种操作简单、快速高效的超声方法,成功合成一种多金属氧酸盐碱式钼酸盐,并且研究该材料对水体中刚果红的降解效果.1.1 试剂与仪器一水合乙酸铜(Cu(CH3COO)2·H2O),七钼酸铵((NH4)6Mo7O24·4H2O),刚果红(Congo red,简称CR),所有使用的药品均为分析纯,使用时没有进一步提纯. 超声波细胞粉碎机(JY92-2D),宁波新芝生物科技有限股份公司;X射线粉末衍射仪(XRD, XD-3),北京普析通用仪器有限公司;透射电子显微镜(TEM, JEM-2100),日本电子株式会社;傅里叶红外光谱仪(NEXUS-870),美国尼高力仪器公司;紫外可见分光光度计(UV-3600),日本津岛公司;电子分析天平(AE240S),上海梅特勒-托利多仪器有限公司;氙灯光源(PLE-SXE300/300UV型),北京泊菲莱科技有限公司.1.2 产物的制备称取0.299 g乙酸铜和0.265 g钼酸铵分别溶解在25 mL蒸馏水中,混合上述两种溶液,磁力搅拌10 min,再将该悬浊液放入超声波细胞粉碎机中,设定条件为超声时间45 min,功率400 W,其中超声脉冲周期为9 s开,1 s关,超声采用高强度超声探头.当反应结束后,自然冷却到室温,样品分别用蒸馏水和无水乙醇高速离心洗涤各3次,最后将所制备的样品放入60 ℃真空干燥箱中干燥12 h,以备用.1.3 催化性能测试称取30 mg样品,超声分散于50 mL 2×10-5 M的CR溶液中,在光催化照射之前,将含有样品的悬浮液在黑暗条件下磁力搅拌1 h,使样品与CR完全达到吸附、脱附平衡.然后再将悬浮液在氙灯下照射(λ≥420 nm),每隔10 min取样5 mL,离心、分离,然后取上清液用紫外可见分光光度计测量在λ = 497 nm处吸光度的变化,以检测CR的降解程度,从而确定样品的催化活性.2.1 物相分析图1为所制备产物的X射线衍射图.从图1可知,产物在2θ值为12.6,21.3,25.3,33.4°处的衍射峰分别对应碱式钼酸铜的(0 2 0),(-1 0 1),(0 3 1),(-1 4 1)衍射晶面,与碱式钼酸铜的标准JCPD卡号(No. 36-0405)相吻合.在谱图中没有发现其他物质的衍射峰,表明合成的物质是碱式钼酸铜,并且纯度很高.2.2 形貌分析图2为碱式钼酸铜的透射电子显微镜(TEM)图和高分辨率透射电子显微镜图(HRTEM).由图2a 展示的产物的TEM图可以看到,它是片状的纳米材料,长约为200 nm,宽约为100 nm,厚约为20 nm.从图2b中清晰的晶格条纹可以看出产物具有很好的结晶性,同时所测量的晶格间距为0.298 nm,这与碱式钼酸铜的(2 1 1)晶面相一致.2.3 结构分析图3为碱式钼酸铜的傅里叶红外谱图.从图3可以看出,在827,917,963 cm-1处有吸收峰,是的特征吸收峰[21].3 346 cm-1处的吸收峰归结于O—H的伸缩振动,451 cm-1处的吸收峰是由于Cu—O的弯曲振动引起.另外在1 634 cm-1处的吸收峰归因于水分子的伸缩振动.这进一步辅证了制备的样品为碱式钼酸铜.2.4 催化性能通过催化降解CR研究碱式钼酸铜的催化活性,结果如图4所示.图4a为碱式钼酸铜对CR降解的紫外光谱图,可以看到悬浮液在黑暗条件下搅拌1 h, 碱式钼酸铜对CR吸附了57%,然后在可见光照射50 min后,CR降解了90%.在相同的条件下,不加入碱式钼酸铜,则CR的浓度基本没有变化(图4b).这表明碱式钼酸铜对CR有很好的降解效果.在实际生产和应用中,样品的重复利用性也是一个非常重要的性质.因此,每次实验后,离心收集样品,用水和乙醇各洗涤3次,60 ℃真空干燥箱烘干后,重新分散在CR溶液中.从图4c可知,碱式钼酸铜在重复利用5次后,其降解效率依然高达88%,显然,碱式钼酸铜具有优良的稳定性.2.5 吸附-光催化机制图5所示为碱式钼酸铜对CR的吸附-光催化机制.材料对染料的吸附作用,只是纯粹地使溶液中的染料分子吸附在吸附剂的外表面,而不能够进行降解,这使得吸附剂回收时,染料还需进一步处理.对于单一的光催化作用,在进行光降解时,只是催化与光催化剂接触的染料分子.而联合吸附与光催化作用会很大程度上提高对染料的降解.如图5所示,在黑暗条件下,碱式钼酸铜首先扮演吸附剂的角色,吸附大量的CR在吸附剂的表面上.然后在可见光照之后,碱式钼酸铜又充当光催化剂的作用,进一步催化降解吸附在光催化剂表面的CR,使CR氧化分解成CO2和H2O.因此,吸附和光催化的协同作用使得样品对CR有更好的降解效果.作者首次以乙酸铜、七钼酸铵、蒸馏水为原料,成功地合成纳米片状的碱式钼酸铜,借助XRD、TEM、FTIR等对其进行表征.并通过吸附和光催化的协同作用研究碱式钼酸铜对CR降解效果.结果表明碱式钼酸铜对CR的催化效率可达到90%,具有较好的降解效率,同时,5次循环催化后,碱式钼酸铜的降解效率依然很高.因此碱式钼酸铜在废水处理方面有很好的应用前景.【相关文献】[1] TIYA-DJOWE A, LAMINSI S, NOUPEYI G L, et al. Non-thermal plasma synthesis of sea-urchin like α-FeOOH for the catalytic oxidation of Orange II in aqueous solution[J]. Applied Catalysis B: Environmental, 2015, 176: 99-106.[2] MOGHADDAM S S, MOGHADDAM M R A, ARAMI M. Coagulation/flocculation process for dye removal using sludge from water treatment plant: optimization through responsesurface methodology[J]. Journal of Hazardous Materials, 2010, 175 (1/2/3): 651-657. [3] MELO R P F, NETO E L B, MOURA M C P A, et al. Removal of reactive blue 19 using nonionic surfactant in cloud point extraction[J]. Separation and Purification Technology, 2014, 138: 71-76.[4] LI X X, FANG S M, GE L, et al. Synthesis of flower-like Ag/AgCl-Bi2MoO6 plasmonic photocatalysts with enhanced visible-light photocatalytic performance[J]. Applied Catalysis B: Environmental, 2015, 176: 62-69.[5] YAO Y J, LU F, ZHU Y P, et al. Magnetic core-shell CuFe2O4@C3N4 hybrids for visible light photocatalysis of Orange II[J]. Journal of Hazardous Materials, 2015, 297: 224-233.[6] JIANG M, YANG W B, ZHANG Z W, et al. Adsorption of three pharmaceuticals on two magnetic ion-exchange resins[J]. Journal of Environmental Sciences-China, 2015, 31: 226-234.[7] ZHAO P, GAO B Y, YUE Q Y, et al. Explore the forward osmosis performance using hydrolyzed polyacrylamide as draw solute for dye wastewater reclamation in the long-term process[J]. Chemical Engineering Journal, 2015, 273: 316-324.[8] GUO H Y, GUO T F, ZHANG Q R, et al. Preparation of graphene oxide-based hydrogels as efficient dye adsorbents for wastewater treatment[J]. Nanoscale Research Letters, 2015, 10 (1): 931.[9] ZHU S Y, JIAO S H, LIU Z W, et al. High adsorption capacity for dye removal by CuZn hydroxyl double salts[J]. Environmental Science: Nano, 2014, 1 (2): 172-180.[10] CHENG B, LE Y, CAI W Q, et al. Synthesis of hierarchical Ni (OH)2 and NiO nanosheets and their adsorption kinetics and isotherms to Congo red in water[J]. Journal of Hazardous Materials, 2011, 185 (2/3): 889-897.[11] WANG X L, CHANG Z H, LIN H Y, et al. Two novel Anderson-type polyoxometalate-based metal-organic complexes with high-efficiency photocatalysis towards degradation of organic dyes under UV and visible light irradiation[J]. Rsc Advances, 2015, 5 (18): 14020-14026.[12] YUAN W W, YUAN P, LIU D, et al. In situ hydrothermal synthesis of a novel hierarchically porous TS-1/modified-diatomite composite for methylene blue (MB) removal by the synergistic effect of adsorption and photocatalysis[J]. Journal of Colloid and Interface Science, 2016, 462: 191-199.[13] XIONG D N, HUANG G F, ZHOU B X, et al. Facile ion-exchange synthesis of mesoporous Bi2S3/ZnS nanoplate with high adsorption capability and photocatalytic activity[J]. Journal of Colloid and Interface Science, 2016, 464: 103-109.[14] LIU L, HU L, LIU Q, et al. Dawson-type polyoxometalate-based oligomeric Hg (II)-acetylide hybrid: synthesis, characterization and materials properties[J]. Journal of Organometallic Chemistry, 2015, 792: 102-108.[15] SONG C Y, CHAI D F, ZHANG R R, et al. A silver-alkynyl cluster encapsulating a fluorescent polyoxometalate core: enhanced emission and fluorescence modulation[J]. Dalton Transactions, 2015, 44 (9): 3997-4002.[16] GAO W X, SUN X Y, NIU H L, et al. Phosphomolybdic acid functionalized covalent organic frameworks: structure characterization and catalytic properties in olefin epoxidation[J]. Microporous and Mesoporous Materials, 2015, 213: 59-67.[17] MENON D, THOMAS R T, NARAYANAN S, et al. A novel chitosan/polyoxometalate nano-complex for anti-cancer applications[J]. Carbohydrate Polymers, 2011, 84 (3): 887-893.[18] HUO Z H, ZANG D J, YANG S, et al. Synthesis and characterization of Lindqvist-type polyoxometalate-porphyrin copolymers[J]. Electrochim Acta, 2015, 179: 326-335.[19] LI N, MU B, LUE L, et al. Assembly of new polyoxometalate-templated metal-organic frameworks based on flexible ligands[J]. Journal of Solid State Chemistry, 2015, 226: 88-93.[20] PAL J, GANGULY M, MONDAL C, et al. Precursor salt assisted syntheses of high-index faceted concave hexagon and nanorod-like polyoxometalates[J]. Nanoscale, 2015, 7 (2): 708-719.[21] JIANG W J, FANG J, FAN Z Y, et al. Synthesis of nano-sized tabular lindgrenite (Cu3 (MoO4)2 (OH)2) by aqueous precipitation[J]. Journal of Inorganic Materials, 2011, 26 (4): 438-442.。
Society Chem.Mater.2010,22,3433–34403433DOI:10.1021/cm1002274Facile Synthesis of Hierarchically Ordered Porous Carbon via in Situ Self-Assembly of Colloidal Polymer and Silica Spheres and Its Use as aCatalyst SupportShiling Zhang,†Ling Chen,†Shuxue Zhou,†Dongyuan Zhao,‡,§and Limin Wu*,†,§†Department of Material Science,‡Department of Chemistry,and§Advanced Materials Laboratory,Fudan University,Shanghai200433,ChinaReceived January25,2010.Revised Manuscript Received April29,2010This article presents a one-pot method to synthesize hierarchically ordered porous carbons with interconnected macropores and mespores,via in situ self-assembly of colloidal polymer and silica spheres with sucrose as the carbon pared with other techniques,this procedure is veritably simple;neither presynthesis of the macropore/mesopore or crystal templates nor additional infiltration is needed,and the self-assembly of polymer spheres into the crystal template and the infiltration are finished in situ in the same system.The sizes of macropores and mesopores can be independently tuned by the sizes of polymer and silica spheres,respectively.The obtained bimodal porous carbons have large BET surface areas,large pore volumes,and partially graphi-tized frameworks.They show very good support of the Pt-Ru alloy catalyst in a direct methanol fuel cell.IntroductionOrdered porous carbon materials with tunable pore geo-metry and functionality have received a great deal of atten-tion due to their many applications,such as electrode materials for batteries,fuel cells,and supercapacitors,1-3 sorbents,4supports for many important catalytic processes,5 templates for the preparation of other nanostructured inor-ganic materials,6,7photonic waveguides,8,9and so on.10,11 Such diverse applications are directly related not only to their superior physical and chemical properties,such as good electric conductivity,high thermal conductivity,chemical stability,high surface area,tailorable surface properties,and low density,but also to their wide availability.Up to now,a variety of approaches have been devel-oped for the preparation of ordered micropore,meso-pore,or macropore carbons as reviewed by Hyeon,12 Ryoo,13and Stein.14For example,by using zeolites as templates,researchers successfully synthesized three-dimensional(3D)ordered microporous carbon mate-rials with uniform pores using various carbon precur-sors via infiltration and chemical vapor deposition (CVD).15,16Through a nanocasting approach combined with various ordered mesopore silica as templates17,18or a direct templating preparation using superamolecu-lar aggregates as templates,ordered mesopore carbons with different pore sizes and structures have been suc-cessfully synthesized.3,13,19-21By using inverse silica opal or colloidal crystals of polystyrene microshperes as templates,3D ordered macropore carbons could*To whom correspondence should be addressed.E-mail:lmw@fudan. .(1)Zhou,H.;Zhu,S.;Hibino,M.;Honma,I.;Ichihara,M.Adv.Mater.2003,15,2107–2111.(2)Chang,H.;S.Joo,H.;Pak,C.J.Mater.Chem.2007,17,3078–3088.(3)Liu,H.J.;Cui,W.J.;Jin,L.H.;Wang,C.X.;Xia,Y.Y.J.Mater.Chem.2009,19,3661–3667.(4)Fang,B.;Zhou,H.;Honma,I.J.Phys.Chem.B2006,110,4875–4880.(5)Yu,J.S.;Kang,S.;Yoon,S.B.;Chai,G.J.Am.Chem.Soc.2002,124,9382–9383.(6)Kim,J.Y.;Yoon,S.B.;Yu,J.S.Chem.Mater.2003,15,1932–1934.(7)Dibandjo,P.;Bois,L.;Chassagneux,F.;Cornu,D.;Letoffe,J.;Toury,B.;Babonneau,F.;Miele,P.Adv.Mater.2005,17,571–574.(8)Zakhidov,A.A.;Baughman,R.H.;Iqbal,Z.;Cui,C.X.;Khayr-ullin,I.;Dantas,S.O.;Marti,J.;Ralchenko,V.G.Science1998, 282,897–901.(9)Li,H.;Chang,L.;Wang,J.;Yang,L.;Song,Y.J.Mater.Chem.2008,18,5098–5103.(10)Yoo,W.C.;Kumar,S.;Wang,Z.;Ergang,N.S.;Fan,W.;Karanikolos,G.N.;McCormick,A.V.;Penn,R.L.;Tsapatsis, M.;Stein,A.Angew.Chem.,Int.Ed.2008,47,9096–9099. (11)Kyotani,T.Carbon2000,38,269–286.(12)Lee,J.;Kim,J.;Hyeon,T.Adv.Mater.2006,18,2073–2094.(13)Ryoo,R.;Joo,S.H.;Kruk,M.;Jaroniec,M.Adv.Mater.2001,13,677–681.(14)Stein,A.;Wang,Z.;Fierke,M.A.Adv.Mater.2009,21,265–293.(15)Hou,P.;Orikasa,H.;Yamazaki,T.;Matsuoka,K.;Tomita,A.;Setoyama,N.;Fukushima,Y.;Kyotani,T.Chem.Mater.2005,17, 5187–5193.(16)Ma,Z.;Kyotani,T.;Tomita,mun.2000,2365–2366.(17)Lu,A.;Kiefer,A.;Schmidt,W.;Sch€u th,F.Chem.Mater.2004,16,100–103.(18)Joo,S.H.;Choi,S.J.;Oh,I.;Kwak,J.;Liu,Z.;Terasaki,O.;Ryoo,R.Nature2001,412,169–172.(19)Deng,Y.;Yu,T.;Wan,Y.;Shi,Y.;Meng,Y.;Gu,D.;Zhang,L.;Huang,Y.;Liu,C.;Wu,X.;Zhao,D.J.Am.Chem.Soc.2007,129, 1690–1697.(20)Kosonen,H.;Valkama,S.;Nyk€a nen, A.;Toivanen,M.;Brinke,G.T.;Ruokolainen,J.;Ikkala,O.Adv.Mater.2006, 18,201–205.(21)Liang,C.;Hong,K.;Guiochon,G.A.;Mays,J.W.;Dai,S.Angew.Chem.,Int.Ed.2004,43,5785–5789./cmPublished on Web05/12/2010 r2010American Chemical3434Chem.Mater.,Vol.22,No.11,2010Zhang et al.also be successfully synthesized through infiltration or CVD.5,8,9,22-28Recently,much attention has been paid to prepare hierarchically ordered porous carbon materials,especially those possessing well-defined macropores and intercon-nected micropores and/or mesopores,because this bimodal porous structure can provide highly efficient mass transport through the macropores and high surface areas from micro/ mesopores,as well as selective accessibility of various sizes of species.For example,Stein et al.29and Yu et al.30,31 successfully prepared bimodal porous carbons by a multi-step templating method.First,3D ordered bimodal porous silica was prepared when polymer crystals were used as templates and then calcinated;then,the ordered bimodal porous silica was infiltrated with carbon precursors;and the composites were finally calcinated and subsequently etched to achieve hierarchically ordered porous carbons.Kana-mura et al.32reported the synthesis of bimodal carbons using polystyrene(PS)spheres and silica particles as tem-plates,and PS also served as the carbon source.Zhao et al.33successfully prepared ordered bimodal porous car-bon structures by the dual colloidal crystal/block copolymer template approach.First,the large and monodisperse silica microspheres(typically several hundred nanometers)self-assembled into3D crystal array by sedimentation,followed by solvent evaporation and thermal treatment.Second,the silica colloidal crystal was used as a template and impreg-nated with an amphiphilic triblock copolymer and phenolic resol precursors solution,which self-organized during the evaporation of the solvent and thermosetting of phenolic resol.The composite was finally carbonized and treated with HF solution to achieve hierarchically ordered porous carbons.The large silica hard template and triblock copo-lymer soft template were responsibile for the formation of macropores and mesopores,respectively. Unfortunately,although the above pioneering works are very interesting,the preparation procedure,which includes presynthesis of ordered porous silica or crystals as templates,infiltration,carbonization and dissolution, seem to be time-consuming and arduous,and the meso-pore size of the obtained carbon materials is either too small or untunable.These disadvantages severely limittheir practical applications.Herein,we report,for the firsttime,a novel and simple method we call an in situ seif-assembly strategy of colloidal polymer and silica spheres toprepare hierarchically ordered porous carbons with inter-connected macropores and mespores.In this appraoch,colloidal polymer spheres are first blended with colloidalsilica and then with sucrose and sulfuric acid to obtainstable dispersion.When this dispersion is cast on a glasssubstrate and treated at different temperatures,followedby etching in HF solution to remove silica,a hierarchicallyordered porous carbon can be directly yielded.In compar-ison with previous techniques,our in situ self-assemblyapproach at least has the following merits:(i)the syntheticprocedure is veritably quite simple,neither presynthesis ofthe macropore/mesopore or crystal templates nor addi-tional infiltration is needed,and both the self-assembly ofpolymer spheres into the crystal template and the infiltra-tion are finished in situ in the same system;(ii)the macro-pore and mespore sizes can be easily tuned by changingpolymer and silica particles in diameter,respectively,andthe mesopore size can be modulated in the whole mesoporerange(2-50nm);(iii)soft polymer spheres(T g=21°C)rather than hard PS spheres are used,this can prepareintermediate composite films(not powder),and sulfuricacid is added into the stable dispersion as the carbonizationcatalyst,which makes carbonization occur at a relativelylower temperature;(iv)this approach is mild and can beused for the mass of production of hierarchically orderedporous carbon.Preliminary research shows that this bi-modal porous carbon as supports can significantly increasethe catalytic activity of the Pt-Ru catalyst.Experimental SectionMaterials.Methyl methacrylate(MMA),butyl acrylate(BA),acrylic acid(AA),and sucrose(T m=185°C)were purchasedfrom Sinopharm Chemical Reagent Corp.Auxiliary monomers:allyloxy hydroxypropyl sodium sulfonate(HAPS,40wt%ofsolid content in aqueous solution)was kindly donated by Shuang-jian Trading Corp.Ltd.(China).The initiator,ammoniumpersulfate(APS),was obtained from Shanghai Guanghua Che-mical Reagent Corp.(China).Colloidal silica:R1050(50nm,pH9,solid content of50wt%,and zeta potential of-43.5mV)was provided by Eka Chemicals Corp.(Sweden),Snowtex50(20nm,pH10,solid content of50wt%,and zeta potential of -48.0mV)was obtained from Nissan Chemicals(Japan),and Nexsil8(9nm,pH10,solid content of30wt%,and zeta potentialof-47.1mV)was supplied by Nyacol Nanotechnologies,Inc.(USA).H2PtCl636H2O and RuCl3were purchased from Aldrich. Nafion(10wt%)was provided by DuPont -mercially available30wt%Pt-Ru/C(weight ratio Pt/Ru=2:1)catalyst was purchased from E-TEK Inc.Synthesis of Colloidal Polymer Spheres.P(MMA-BA-AA)coplolymer latex was synthesized by semibatch surfactant-freeemulsion polymerization.34The size of polymer spheres could beadjusted by the amount of monomers.Three kinds of polymerlatex with sizes of475,370,and280nm were obtained.The glasstransition temperature of the polymer was about21°C.(22)Lee,K.T.;Lytle,J.C.;Ergang,N.S.;Oh,S.M.;Stein,A.Adv.Funct.Mater.2005,15,547–556.(23)Lei,Z.;Zhang,Y.;Wang,H.;Ke,Y.;Li,J.;Li,F.;Xing,J.J.Mater.Chem.2001,11,1975–1977.(24)Zhou,Z.;Yan,Q.;Su,F.;Zhao,X.J.Mater.Chem.2005,15,2569–2574.(25)Chai,G.S.;Yoon,S.B.;Yu,J.S.;Choi,J.H.;Sung,Y.E.J.Phys.Chem.B2004,108,7074–7079.(26)Su,F.;Zhao,X.;Wang,Y.;Zeng,J.;Zhou,Z.;Lee,J.Y.J.Phys.Chem.B2005,109,20200–20206.(27)Yoon,S.B.;Chai,G.S.;Kang,S.K.;Yu,J.S.;Gierszal,K.P.;Jaroniec,M.J.Am.Chem.Soc.2005,127,4188–4189.(28)Baumann,T.F.;Satcher,J.H.,Jr.Chem.Mater.2003,15,3745–3747.(29)Wang,Z.;Li,F.;Ergang,N.S.;Stein,A.Chem.Mater.2006,18,5543–5553.(30)Chai,G.S.;Shin,S.;Yu,J.S.Adv.Mater.2004,16,2057–2061.(31)Fang,B.;Kim,J.H.;Kim,M.;Yu,J.S.Chem.Mater.2009,21,789–796.(32)Woo,S.W.;Dokko,K.;Nakano,H.;Kanamura,K.J.Mater.Chem.2008,18,1674–1680.(33)Deng,Y.;Liu,C.;Yu,T.;Liu,F.;Zhang,F.;Wan,Y.;Zhang,L.;Wang,C.;Tu,B.;Webley,P.A.;Wang,H.;Zhao,D.Chem.Mater.2007,19,3271–3277.(34)You,B.;Wen,N.;Shi,L.;Wu,L.;Zi,J.J.Mater.Chem.2009,19,3594–3597.Article Chem.Mater.,Vol.22,No.11,20103435 Preparation of Hierarchically Ordered Porous Carbons.Thehierarchically ordered porous carbons were prepared through anin situ self-assembly approach of colloidal polymer spheres andsilica particles.For a typical procedure,20g of the polymer latexwas first blended with1.3g of silica sol under magnetic stirring for1h to obtain nanocomposite latex and then with0.45g of sucrosepowder at room temperature for10min,followed by the additionof0.45g of10wt%of sulfuric acid and0.1g of water understirring for another10min to obtain stable dispersion;the polymersolid content of dispersion was modulated with water to15wt%.The typical mass ratio of polymer,silica,sucrose,and sulfuric acidwas100:15:15:1.5.The as-prepared dispersion was cast on glasssubstrates by the pouring method and directly dried in an oven at110°C for2h,then at160°C for6h,followed by heating to700°Cwith a rate of5°C/min under highly pure nitrogen flow(1L/min),and kept at700°C for another2h to decompose polymer spheresand carbonize sucrose.The obtained carbon/silica composite wascooled in pure nitrogen and immersed in10wt%of HF aqueoussolution to remove silica(hazard:HF is highly toxic and corrosiveand must be handled in fume hoods with proper personal protec-tion equipment),followed by washing with deionized water anddrying at60°C to yield hierarchically ordered porous carbons.Carbon Supported Pt-Ru catalyst.The carbon supportedPt-Ru catalyst was prepared by a borohydride reductionmethod.5The metal loading was kept at30wt%(weight ratioPt/Ru=2:1)to allow a comparison with a commercial catalyst,which was also confirmed by TGA and EDX measurements.The thin film electrode used as the work electrode in theelectrochemical measurement was manufactured as follows:3550mg of porous carbon supported Pt-Ru catalyst powder and250μL of Nafion(10wt%)were ultrasonically dispersed in10mL of isopropanol.A100μL portion of the resultant ink wasdropped onto an electrode surface.Then,the electrode was driedat70°C.The cyclic voltammograms measurements in the0.5MH2SO4þ1.0M CH3OH solution was carried out in a potentio-stat of ECO CHEMIE B.V Model T30/FRA2,and a Pt sheetand saturated calomel electrodes were used as the counter andreference electrodes,respectively.The potential scan was carriedout between0.05and0.75V with a scan rate of10mV s-1undera temperature of40°C.Characterization.SEM was conducted with a Philips XL30field emission microscope at an accelerating voltage of10kV.High-resolution SEM(HR-SEM)was conducted on a HitachiS-4800microscope(Japan)operated at1.0kV.Transmissionelectron microscopy(TEM)images were taken with a JEOL2011microscope(Japan)operating at200kV.For the TEM observa-tions,the samples were dispersed in ethanol and then dried on aholey carbon film Cu grid.N2adsorption-desorption isothermswere measured at77K on a Micromeritics ASAP2020gassorptometer(USA)after the carbon and carbon/silica compositewas degassed at423K to20μTorr for12h.The specific surfaceareas were determined from nitrogen adsorption using the Bru-nauer-Emmett-Teller(BET)equation.By using the Barret-Joyner-Halenda(BJH)model,the pore volumes and pore sizedistributions were derived from the adsorption branches ofisotherms,and the total pore volumes were estimated from theadsorbed amount at a relative pressure P/P0of0.992.Ramanspectra were obtained with a Dilor LabRam-1B microscopicRaman spectrometer(France),using a He-Ne laser with anexcitation wavelength of632.8nm.XRD measurement wascarried out with a Rigaku D/max-rB X-ray diffractometer with Cu K R radiation(λ=1.54056A)at a scanning rate of0.02°per second at2θranging from5to60°.Results and discussionPreparation of the Hierarchically Ordered Porous Car-bon.The hierarchically ordered porous carbons were prepared via an in situ self-assembly of colloidal polymer and silica spheres with sucrose as the carbon source and sulfuric acid as the catalyst,as schematically described in Scheme1.First,colloidal poly(MMA-BA-AA)spheres were blended with colloidal silica,followed by sucrose and sulfuric acid to obtain stable dispersion.This disper-sion was then cast on a glass substrate and directly dried in an oven at110°C for2h to cause a transparent film, followed at160°C for6h for preliminary carbonization by sulfuric acid to obtain black composite films,and then fully carbonized at700°C in N2for another2h to yield iridescent black silica/carbon composite films after the decomposition of polymer spheres and sucrose.The obtained silica/carbon composite was finally etched by HF solution to remove silica to directly form iridescent black hierarchically ordered porous carbon.The typical cross-sectional SEM images of the treated composite by different temperatures are demonstrated in Figure1.After treatment at110and160°C,although an individual polymer sphere is not visible due to high drying temperatures,the periodic arrangement of these spheres can still be observed(Figure1a and b).Since soft polymer spheres(T g=21°C)rather than hard PS or PMMA spheres were used as the template,on the one hand,small silica particles occupied the interstitial voids between large polymer spheres;on the other hand,suc-rose molecules and some of the polymer chains could easily diffuse and reversely fill the remaining free volumes among the silica particles.This availed the connection of neighboring macropores,greatly shortening the prepara-tion time,and the robust intermediate composite films (not powder;Figure S1in Supporting Information)were Scheme1.Schematic Illustration for the Preparation of Hier-archically Ordered PorousCarbon(35)Chen,L.;Guo,M.;Zhang,H.;Wang,X.Electrochim.Acta2006,52,1191–1198.3436Chem.Mater.,Vol.22,No.11,2010Zhang et al.also in favor of the mass production of bimodal porous carbons.Furthermore,the surrounding nanosilica parti-cles and sucrose crystallites could restrain the deforma-tion and coalescence of these soft polymer spheres,which could guarantee these monodisperse and spherical poly-mer spheres to tightly pile into 3D ordered arrays.34The results are also consistent with previous works using viscous liquids.36,37After being completely carbonized,the obtained silica/carbon composite reveals a 3D or-dered porous structure (Figure 1c).The macropore size estimated from SEM is about 350nm,around 5%of shrinkage of the lattice structure compared to that of polymer spheres.These 3D ordered macropores are con-necting each other by small windows with a size of 120nm that are caused by the contacting area between neighbor-ing polymer spheres.After the removal of silica micro-spheres,the product also presents 3D interconnection of the ordered macropores with small windows,indicating that the macrostructure of the colloidal polymer crystals can be well retained (Figure 1d).Moreover,this structure appears to be typical face-centered cubic (fcc),which can be observed in the (100)plane in Figure 1d,with each pore surrounded by six equal pores in layers perpendicular to the (111)direction (Figure.1e).Effects of Silica and Polymer Sizes.Figure 2displays the cross-sectional SEM and HRTEM images of 3D ordered porous carbons prepared from different sizes of silica.The inserted HRSEM images in Figure 2a -c clearly reveal that the macropores are interconnected by windows and that the pore walls are composed of sphe-rical mesopores.The HRTEM images in Figure 2d and e also show mesopores between macropores,indicating ahierarchically ordered porous structure.Moreover,the mesopore size increases from about 9to 50nm with the increase of colloidal silica size from around 9to 50nm,indicating that the mesopore size can be easily and independently adjusted in the full mesopore range by the size of silica particles.This cannot be easily realized in other templating approaches.29-31,33When the polymer spheres with a size of 280or 475nm were used,this hierarchically ordered porous carbon could also be prepared,as indicated in Figure 3.These results show that the higher ratio of small nanoparticles to large sphere sizes than the theoretical 0.155can also form an organization arrangement,38which is consistent with a previous report.39This is probably because some polymer chains of the soft polymer spheres in our system could contrarily diffuse into the voids between silica particles as the dispersion was dried at 110°C,causing a ratio higher than the theoretical value.Effects of Silica and Sucrose Contents.When too low of an amount of silica,i.e.,5wt %,was used,a collapsing skeleton was observed (Figure 4a),while the windows were dramatically decreased in size,even closed completely,when too much silica,i.e.,35wt %,was embedded (Figure 4b).If the amount of silica further increased to 40wt %,a meso-pore domain formed due to the silica domain (Figure 4c).This suggests that the efficient infiltration of silica into the voids between polymer spheres is playing a critical role for the successful preparation of hierarchically ordered porous carbon structures in our in situ self-assembly approach:too low an amount of silica is not enough for the formation of an integrated silica framework,but excess silica would impede the contact of neighboring polymer spheres except for filling in the voids between polymer spheres andevenFigure 1.Typical cross-sectional SEM images of (a)composite films treated at 110°C,(b)composite films treated at 160°C,(c)porous silica/carbon composite film,(d)bimodal porous carbon,and (e)surface SEM image of bimodal porous carbon (polymer spheres,370nm;silica colloid,9nm;sucrose,15wt %;silica,15wt %).(36)Kim,S.H.;Kim,S.H.;Yang,S.M.Adv.Mater.2009,21,3771–3775.(37)Jiang,P.;McFarland,M.J.J.Am.Chem.Soc.2004,126,13778–13786.(38)Iskandar,F.;Mikrajuddin;Okuyama,K.Nano Lett.2002,2,389.(39)Wang,J.;Li,Q.;Knoll,W.;Jonas,U.J.Am.Chem.Soc.2006,128,15606–15607.Article Chem.Mater.,Vol.22,No.11,20103437form a silica domain.On the basis of the above results,15wt %of silica is appropriate for the formation of an ordered porous structure.When 5or 15wt %of sucrose was used,hierarchically ordered porous carbons with interconnected macropores and mesopores could also be obtained,as indicated by Figure 5.But excess sucrose crystallites would cumber the osculation of neighboring polymer spheres,leading to closed windows,just as excess silica does.Adsorption and XRD Analyses.Figure 6demonstrates the typical nitrogen adsorption -desorption isotherms at 77K and the pore size distributions of hierarchically ordered porous carbons.The corresponding pore sizes,BET surface areas,and pore volumes are summarized in Table 1.All these ordered porous carbons display type IV isotherms with a H2hysteresis,which is the characteric of mesopores.The carbons prepared from 280,370,and 475nm polymer spheres and 9nm silica have a calculated pore size of approximately 9nm from the adsorption branch by the BJH model (inset of Figure 6a),and almost equal BET surface areas and pore volumes,with as high as 781,818,and 765m 2/g,and 1.23,1.29,and 1.22cm 3STP/g,Figure 3.Cross-sectional SEM images of the porous carbon films prepared from (a)280nm polymer spheres,(b)475nm polymer spheres (inserts are corresponding amplified SEM image;,silica particle size is 9nm).Figure 4.Cross-sectional SEM images of the porous carbon films prepared from different amounts of silica:(a)5wt %,(b)35wt %,and (c)40wt %(the sizes of polymer and silica particles are 370and 9nm,respectively).Figure 2.Typical cross-sectional SEM and HRTEM images of the bimodal porous carbons prepared from different sizes of silica.(a,d,and e)9nm,(b)and f)20nm,and (c)50nm (bar in HRTEM images:d is 200nm;bars in e and f are 50nm;insets are corresponding HRSEM images for macro-pore wall,and all bars are 100nm;polymer sphere size is 370nm).3438Chem.Mater.,Vol.22,No.11,2010Zhang et al.respectively (Table 1),which are comparable with those from these approaches of presynthesizing templates.29-33These results indicate that the sizes of polymer spheres have little impact on the BET surface area and the pore volume.However,as sucrose content decreases from 15to 5wt %,the surface area and total pore volume increase from 781to 850.1m 2/g,and from 1.23to 1.61cm 3STP/g,respectively,indicating that both of them can be tuned by sucrose content.For the carbons prepared from silica with a size of 9,20,and 50nm,the capillary condensation steps shift to low relative pressure when the mesopore size decreases from 50to 9nm (Figure 6b).The samples from 9and 20nm silica have pore sizes of 9and 22nm (inset of Figure 6b),which are consistent with the sizes of silica beads used.However,the sample from 50nm silica possesses a wider mesoporedistribution in the whole mesopore region.This is probably attributed to the increasing interstices between hollow carbon spheres and the windows connecting mesosized carbon spheres besides mesopores.The specific surface area decreases from 781and 509to 527m 2/g,while pore volume is not varied when the size of silica increases from 9and 20to 50nm,indicating that the specific surface area can also be controlled by the size of the silica beads used.The Raman spectrum (Figure 7)shows two strong broad bands at 1320and 1585cm -1that can be assigned to the D and G bands of the disordered and graphitized carbons,respectively.Considering that the G band is associated with graphitic sp 2carbon structures,40the comparableintensityFigure 5.Cross-sectional SEM images of the porous carbons prepared from different sucrose content:(a)5wt %;(b)15wt %(polymer spheres,370nm;silica,15wt%).Figure 6.Typical N 2adsorption isotherms and pore size distribution (inset)of some ordered bimodal porous carbons (a)from different sizes of polymer spheres;(b)from different sizes of silica (P280,P370,and P475denote polymer spheres with sizes of 280,370,and 475nm,reslectively;S9,S20and S50denote silica beads with sizes of 9,20,and 50nm,respectively).Table 1.Structural Parameters of Porous Silica/Carbon Composite,Silica,and Carbon asampleamacropore size (nm)cmesopore diameter (nm)d BET surface area (m 2g -1)total pore volume (cm 3STP g -1)micorpore volume (cm 3STP g -1)P280-S9composite 260980.0840.034P280-S9silica 2602040.330.00P280-S9-0.05b 2609.0850 1.610.04P280-S92609.0781 1.230.11P280-S2026022.2509 1.120.09P280-S5026050.0c 527 1.120.12P370-S93509.0818 1.290.11P475-S94409.07651.220.10aBoth silica and sucrose contents:15wt %.b Silica content,15wt %;sucrose content,5wt %.c By SEM images.d From the PSD max.(40)Sadezky,A.;Muckenhuber,H.;Grothe,H.;Niessner,R.;Poschl,U.Carbon 2005,43,1731–1742.Article Chem.Mater.,Vol.22,No.11,20103439of D and G bands (the intensity ratio,∼1.22)suggests that the hierarchically ordered porous carbon materials are partially composed of graphites.The XRD pattern (inset of Figure 7)shows two broad reflections at 2θof ca.22and 44°,which can be attributed to turbostratic packing of graphene sheets in amorphous carbon.Formation Mechanism of Hierarchically Ordered Porous Carbons.On the basis of the above observations,the formation mechanism of hierarchically ordered porous carbons via the in situ self-assembly approach can be explained as follows:When the dispersion of colloidal polymer spheres,colloidal silica,sucrose,and sulfuric acid was cast on glass substrates and directly dried in an oven at 110°C,the charged colloidal polymer spheres came into close contact via capillaries as water evapo-rated.During this in situ self-assembly process,the de-formation and coalescence of these soft polymer spheres were restrained due to the surrounding nanosilica parti-cles and sucrose crystallites.34This could guarantee that these monodisperse and spherical polymer spheres tightly pile into 3D ordered arrays,in which the voids were filled in situ with nanosilica particles as well as sucrose crystal-lites in the same system.When this dried transparent film was treated at 160°C for preliminary carbonization,then at 700°C for full carbonization,polymer spheres were removed,leaving behind macropore silica/carbon com-posites,which reveals a characteristic of multilayer ad-sorption on macropore solids.The silica in the pore walls was acting as a rigid framework to prevent large shrink-age of the pores during the pyrolytic removal of organic components.This composite was finally etched by HF solution to remove silica to form hierarchically ordered porous carbons with interconnected macropores and mesopores.The whole process neither needs the presynth-esis of the macropore/mesopore or crystal templates nor additional infiltration;both the polymer crystal and the infiltration are finished in situ in the same system.As Support of the Pt -Ru Alloy Catalyst.The obtained hierarchically ordered porous carbons were further used as the support of the Pt -Ru alloy catalyst and compared with the commercially available E-TEK catalysts with Pt -Ru alloy supported on carbon.31,41The Pt -Ru alloy catalystswere prepared by the borohydride reduction method 5using our porous carbon with a 350nm macropore size and 50nm mesopore size as a catalyst support.The TEM image of the supported catalysts show a homogeneous dispersion of Pt -Ru alloy metal particles with around 2nm (Figure 8).The cyclic voltammograms of porous carbon supported Pt -Ru catalyst and the commercial E-TEK catalyst,as illustrated in Figure 9,display that the specific mass current density at the same potential for the porous carbon sup-ported catalyst is considerably higher than that for the commercial catalyst in the forward as well as the reverse scan.This indicates that the Pt -Ru catalyst supported on our bimodal porous carbon has higher catalytic activity than the commercial E-TEK catalyst.This should be attri-buted to the higher surface and efficient diffusion of metha-nol and oxidized product in the 3D interconnected macro-pores and mesopores.ConclusionsOn the basis of this study,a novel and feasible approach to prepare hierarchically ordered porous carbons with interconnected macropores and mespores has been pro-posed via an in situ self-assembly approach.This is the first example of the self-assembly of soft colloidal poly-mer spheres and silica particles in carbon sourcesolutionFigure 7.Typical Raman spectrum and XRD pattern (inset)of the bimodal porouscarbon.Figure 8.TEM image of the porous supported Pt -Rucatalyst.Figure 9.Cyclic voltamograms for a porous carbon supported Pt -Ru catalyst as compared with that of a commercial catalyst for methanol oxidation.(41)Wen,Z.;Liu,J.;Li,J.Adv.Mater.2008,20,743–747.。