GMP2010实施指南---取样方法
- 格式:docx
- 大小:1.31 MB
- 文档页数:6


2010版gmp实施指南(无菌)无菌制剂 GMP实施指南 14.6过滤除菌14.6过滤除菌本节旨在提供系统的方法对除菌过滤工艺进行选择和验证。
除菌过滤过程中的关键设备为除菌(级)过滤器,对其进行的具体确认和验证项目的相关原理和要求等,将在以下14.6.1~14.6.3中详细阐述。
按照不同药品分类,需要、推荐、可选和评估后确定是否进行的确认和验证项目,总结为下表。
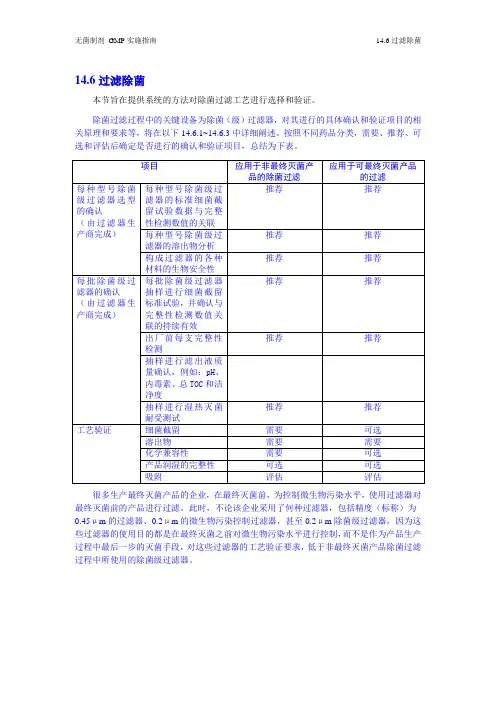
项目应用于非最终灭菌产应用于可最终灭菌产品品的除菌过滤的过滤每种型号除菌每种型号除菌级过推荐推荐级过滤器选型滤器的标准细菌截的确认留试验数据与完整(由过滤器生性检测数值的关联产商完成) 每种型号除菌级过推荐推荐滤器的溶出物分析构成过滤器的各种推荐推荐材料的生物安全性每批除菌级过每批除菌级过滤器推荐推荐滤器的确认抽样进行细菌截留(由过滤器生标准试验,并确认与产商完成) 完整性检测数值关联的持续有效出厂前每支完整性推荐推荐检测抽样进行滤出液质量确认,例如:pH、内毒素、总TOC和洁净度抽样进行湿热灭菌推荐推荐耐受测试工艺验证细菌截留需要可选溶出物需要需要化学兼容性需要可选产品润湿的完整性可选可选吸附评估评估很多生产最终灭菌产品的企业,在最终灭菌前,为控制微生物污染水平,使用过滤器对最终灭菌前的产品进行过滤。
此时,不论该企业采用了何种过滤器,包括精度(标称)为0.45μm的过滤器、0.2μm的微生物污染控制过滤器,甚至0.2μm 除菌级过滤器,因为这些过滤器的使用目的都是在最终灭菌之前对微生物污染水平进行控制,而不是作为产品生产过程中最后一步的灭菌手段,对这些过滤器的工艺验证要求,低于非最终灭菌产品除菌过滤过程中所使用的除菌级过滤器。
错误~文档中没有指定样式的文字。
无菌制剂GMP实施指南14.6.1除菌级过滤器的验证/细菌截留【法规要求】《药品生产质量管理规范》2010修订版:附录1 无菌药品第九十条对可最终灭菌的产品不得以除菌过滤工艺替代最终灭菌工艺。
14.6过滤除菌本节旨在提供系统的方法对除菌过滤工艺进行选择和验证。
除菌过滤过程中的关键设备为除菌(级)过滤器,对其进行的具体确认和验证项目的相关原理和要求等,将在以下14.6.1~14.6.3中详细阐述。
按照不同药品分类,需要、推荐、可选和评估后确定是否进行的确认和验证项目,总结为下表。
很多生产最终灭菌产品的企业,在最终灭菌前,为控制微生物污染水平,使用过滤器对最终灭菌前的产品进行过滤。
此时,不论该企业采用了何种过滤器,包括精度(标称)为0.45μm的过滤器、0.2μm的微生物污染控制过滤器,甚至0.2μm除菌级过滤器,因为这些过滤器的使用目的都是在最终灭菌之前对微生物污染水平进行控制,而不是作为产品生产过程中最后一步的灭菌手段,对这些过滤器的工艺验证要求,低于非最终灭菌产品除菌过滤过程中所使用的除菌级过滤器。
14.6.1除菌级过滤器的验证/细菌截留【法规要求】编者理解:在这里法规要求的第一个要点是在采用除菌过滤方法时,首先确认采用的过滤器为“除菌级”的,即“除菌过滤器”。
达到此要求后,除菌过滤法中的其它无菌保障措施才有意义。
定义过滤器是否为除菌级,需要依据过滤器的微生物截留能力,并完成相关的标准方法确认和工艺验证。
而过滤药液所用的时间、流速、温度、滤出总量、过滤器二侧压力(压差)、药液对微生物的生存性的影响和过滤器的重复使用等情况,都是可能影响过滤器细菌截留能力的重要因素,需要在验证过程中考虑并确证。
【背景介绍】除菌过滤是指除去流体中微生物的工艺过程,该过程不应对产品质量产生不良影响。
包括液体和气体除菌过滤。
药品生产中采用的除菌过滤膜的孔径一般不超过0.22um(或者0.2um,这两种标称没有区别)。
当膜过滤器在1960年代出现在市场上时,0.45um孔径的膜被认为是“除菌级”的液体过滤器,并被成功应用于注射剂的除菌过滤。
这些过滤器采用黏质沙雷氏菌(Serratia marcescens)进行挑战确认。


药品GMP 认证检查指南通则国家食品药品监督管理局药品安全监管司国家食品药品监督管理局药品认证管理中心一、机构与人员【检查核心】药品生产和质量管理的组织机构对保证药品生产全过程受控至关重要;适当的组织机构及人员配备是保证药品质量的关键因素;人员的职责必须以文件形式明确规定;培训是实施药品GMP的重要环节。
【检查条款及方法】*0301 企业是否建立药品生产和质量管理机构,明确各级机构和人员的职责。
1.看企业组织机构图,查生产质量管理组织机构及功能设置(图示),是否涵盖生产、质量、物料仓储、设备、销售及人员管理等内容,并有负责培训的职能部门/人员。
2.查企业分管生产及质量的负责人、生产及质量管理中层干部基本情况,内容包括:姓名、职务、职称、学历、毕业院校、所学专业、从药年限、所在岗位等。
3.生产管理部门和质量管理部门的负责人通常有一些共同的质量责任,如:3.1 制订书面规程和其他文件;3.2 对生产环境的监控;3.3 工厂卫生;3.4 工艺验证和分析仪器的校验;3.5 人员培训,包括质量保证系统及其实施;3.6 供应商的审计;3.7 被委托(加工或包装)方的批准和监督;3.8 物料和产品贮存条件的确定和监控;3.9 记录的归档;3.10 对GMP 实施情况加以监控等;3.11 因监控某些影响质量的因素而进行取样、试验或调查。
4. 质量管理部门的主要职责不得委派给他人,例如,仓库负责人不得决定某批产品能否放行出厂,分管厂长不得跳过质量管理部门对怀疑有质量问题的产品做出合格与否的决定。
质量管理部门的职责应以文件形式规定,通常包括以下各项:4.1 建立原材料、中间体、包装材料、标签和成品的放行或拒收系统;4.2 批准工艺规程、取样方法、质量标准、检验方法和其他质量控制规程;4.3 审查、批准原料、包装材料、中间产品、待包装产品和成品;4.4 确保物料、中间体、成品都经过适当的检测并有测试报告;4.5 审核评价批记录,在决定放行前,审核已完成关键步骤的批生产记录和实验室控制记录,确保各种重要偏差已进行调查并已有纠正措施;4.6 确保对质量相关的投诉进行调查并予以适当处理;4.7 批准和监督由被委托方承担的委托检验;4.8 检查本部门、厂房和设备的维护情况;4.9 确保所需的验证(包括检验方法的验证)以及控制设备的校准都已进行;4.10 确保有稳定性数据支持中间体或成品的复验期/有效期及储存条件;4.11 对产品质量情况定期进行回顾及审核;4.12 确保本部门人员都已经过必要的GMP 及岗位操作的基础培训和继续培训,并根据实际需要适当调整培训计划。