guide-annex_3_en
- 格式:pdf
- 大小:325.10 KB
- 文档页数:86

ANNEX 1附录ⅠMANUFACTURE OF STERILE MEDICINAL PRODUCTS无菌药品的生产Principle原则The manufacture of sterile products is subject to special requirements in order to minimize risks of microbiological contamination, and of particulate and pyrogen contamination. Much depends on the skill, training and attitudes of the personnel involved. Quality Assurance is particularly important, and this type of manufacture must strictly follow carefully established and validated methods of preparation and procedure. Sole reliance for sterility or other quality aspects must not be placed on any terminal process or finished product test.为将微生物、微粒和热原污染的风险降低至最小限度,无菌药品的生产应有各种特殊要求。
这在很大程度上取决于生产人员的技能、所接受的培训及其工作态度。
质量保证极为重要,无菌药品的生产必须严格按照精心制订并经验证的方法及规程进行。
产品的无菌或其它质量特性绝不能仅依赖于任何形式的最终操作或成品检验。
Note:注:This guidance does not lay down detailed methods for determining the microbiological and particulate cleanliness of air, surfaces etc. Reference should be made to other documents such as the EN/ISO Standards.本指南没有制定详细的空气和物体表面等的微生物和微粒的测定方法,相关事项可以参照其他文件,诸如欧洲/国际标准。

CAPE PARROTIDENTIFICATION GUIDECompiled on behalf of the Scientific Authority of SouthAfricaMay 2015(English only / Únicamente en inglés / Seulement en anglais)AC28 Doc. 21.1Annex 5A guide to distinguish the Cape Parrot (Poicephalusrobustus) and the Grey-headed Parrot (Poicephalusfuscicollis suahelicus)BackgroundUntil recently the Cape parrot (Poicephalus robustus robustus) was not recognised as a separate species from the grey-headed parrot (Poicephalus robustus fuscicollis) and both these taxa were therefore included together in Appendix II to CITES and traded under the name Poicephalus robustus. The Cape parrot, a South African endemic, has a restricted and fragmented distribution with an estimated global population size of 1000 – 1500 individuals representing considerably less than 500 breeding pairs. The species is listed as Endangered in the South African Red Data Book for Birds, as critically endangered on the current Threatened or Protected species list (section 56 of the National Environmental Management: Biodiversity Act No. 10 (NEMBA) of 2004), as specially protected in the province of KwaZulu-Natal and is a protected species in the Eastern Cape province of South Africa. However, due to the grey-headed parrot and Cape parrot not being recognised as two separate species, both taxa are together considered as Least Concern at a global level.However, genetic work recently conducted by the University of KwaZulu-Natal (paper submitted and accepted by PLOSONE – revision in progress) indicates clear genetic differentiation between P.r. robustus and the P.r. suahelicus - P.r. fuscicollis cluster, supporting previous recommendations that the Cape parrot should be viewed as a separate species, namely P. robustus, and that the grey-headed parrot and brown-necked parrot should be grouped under the P. fuscicollis species complex as P.f. suahelicus and P.f. fuscicollis respectively.The Cape parrot (Poicephalus robustus) has been the focus of much debate over the past few years, from its taxonomic position, to habitat use and population declines. Various factors have contributed to population declines in the species such as the illegal harvest of individuals from the wild for the pet trade, habitat destruction and diseases, specifically psittacine beak and feather virus (Wirminghaus et al. 1999).In preparation for the Cape parrot being treated as a separate species under CITES, the South African National Biodiversity Institute developed an identification guide to assist law enforcers with identifying the Cape parrot and distinguishing it from the grey-headed parrot. The guide highlights the main morphological and colour differences between the two species. It consists of1. A brief description of the two species,2. Tables listing the main ecological, morphological and biological differencesbetween the two species (tables 1 & 2 and fig. 1),3. Photographs of both sexes annotated to show the colour differences betweenthe two species (fig. 2, 3, & 4), and4. A colour palette (table 3) to assist in recognising the various coloursdescribed.Brief description of the Cape parrotThe Cape parrot has small body dimensions, a narrow bill; the head colour is olive, yellow to golden.The head, throat and neck are olive yellow to golden brown. The body and the wings are dark green. Thighs and outer edges of the wings orange-red and the tail and flight feathers are bottle-green to black. Females typically have an orange-red forehead. Males have a dark-earth fore crown and no colouration on the forehead. Juveniles resemble the female but without red on the tibia and wing edges.Brief description of the grey-headed parrotThe grey-headed parrot has large body dimensions; the bill is heavier basally with the apex tapered and longer. Head colour is silver grey.The grey-headed parrot is generally larger than the Cape parrot and has a short tail. The body is green. Adults normally have orange-red patches on shoulders and thighs. Just like the Cape parrot, females have a well-defined orange-red forehead. Similar to the Cape parrot, juveniles resemble females with the orange-red colour on the forehead.Table 1: Ecological, morphological and biological differences between the Cape and the grey-headed parrot.5Table 2: Morphometric comparison of the Cape and grey-headed parrot.678Figure 1:Different body parts of a bird (bird anatomy).9Figure 2: Male Cape parrot (P. robustus) (left) and male grey-headed parrot (P. fuscicollis suahelicus ) (right)Fore crown colour is rusty grey-brownFore crown is dark earth-brownNape and Collar colours are warm-rusty brown Nape and Collar yellow green Upper chest is yellowgreenUpper chest colour isrusty grey brown10 Figure 3:Male grey-headed parrot.11Figure 4:Female grey-headed parrot (left) and female Cape parrot (right).Table 3: Colour palette showing the different colours occurring on the head and body of the Cape and the grey-headed parrotOlive green Olive green Rusty grey-brown Silvery grey-brown12Orange brown Orange brown Warm grey-brown Yellow green Yellow green Rusty grey-brown Grey-green13Dark oily-green Dark green Olive-greenBlue-green Blue-green Leaf-green Blue-green1415ReferencesDOWNS, C. T. 2005. Abundance of the endangered Cape parrot, Poicephalus robustus, in South Africa: implications for its survival. African Zoology 4(1): 15-24.PERRIN, M. 2012. Parrots of Africa, Madagascar and the Mascarene Islands. Wits University Press. 1 Jan Smuts Avenue. Johannesburg.WIRMINGHAUS, J.O., DOWNS, C.T., SYMES, C.T. & PERRIN, M.R. 1999. Conservation of the Cape parrot in southern Africa. South African Journal of Wildlife Research 29: 118–129.WIRMINGHAUS. J. O., DOWNS. C. T., PERRIN. M. R. and SYMES. C. T. 2002. Taxonomic relationships of the subspecies of the Cape Parrot Poicephalus robustus (Gmelin).Journal of Natural History. 36: 361-378.16。

NuMicro®FamilyArm® ARM926EJ-S BasedNuMaker-HMI-N9H30User ManualEvaluation Board for NuMicro® N9H30 SeriesNUMAKER-HMI-N9H30 USER MANUALThe information described in this document is the exclusive intellectual property ofNuvoton Technology Corporation and shall not be reproduced without permission from Nuvoton.Nuvoton is providing this document only for reference purposes of NuMicro microcontroller andmicroprocessor based system design. Nuvoton assumes no responsibility for errors or omissions.All data and specifications are subject to change without notice.For additional information or questions, please contact: Nuvoton Technology Corporation.Table of Contents1OVERVIEW (5)1.1Features (7)1.1.1NuMaker-N9H30 Main Board Features (7)1.1.2NuDesign-TFT-LCD7 Extension Board Features (7)1.2Supporting Resources (8)2NUMAKER-HMI-N9H30 HARDWARE CONFIGURATION (9)2.1NuMaker-N9H30 Board - Front View (9)2.2NuMaker-N9H30 Board - Rear View (14)2.3NuDesign-TFT-LCD7 - Front View (20)2.4NuDesign-TFT-LCD7 - Rear View (21)2.5NuMaker-N9H30 and NuDesign-TFT-LCD7 PCB Placement (22)3NUMAKER-N9H30 AND NUDESIGN-TFT-LCD7 SCHEMATICS (24)3.1NuMaker-N9H30 - GPIO List Circuit (24)3.2NuMaker-N9H30 - System Block Circuit (25)3.3NuMaker-N9H30 - Power Circuit (26)3.4NuMaker-N9H30 - N9H30F61IEC Circuit (27)3.5NuMaker-N9H30 - Setting, ICE, RS-232_0, Key Circuit (28)NUMAKER-HMI-N9H30 USER MANUAL3.6NuMaker-N9H30 - Memory Circuit (29)3.7NuMaker-N9H30 - I2S, I2C_0, RS-485_6 Circuit (30)3.8NuMaker-N9H30 - RS-232_2 Circuit (31)3.9NuMaker-N9H30 - LCD Circuit (32)3.10NuMaker-N9H30 - CMOS Sensor, I2C_1, CAN_0 Circuit (33)3.11NuMaker-N9H30 - RMII_0_PF Circuit (34)3.12NuMaker-N9H30 - RMII_1_PE Circuit (35)3.13NuMaker-N9H30 - USB Circuit (36)3.14NuDesign-TFT-LCD7 - TFT-LCD7 Circuit (37)4REVISION HISTORY (38)List of FiguresFigure 1-1 Front View of NuMaker-HMI-N9H30 Evaluation Board (5)Figure 1-2 Rear View of NuMaker-HMI-N9H30 Evaluation Board (6)Figure 2-1 Front View of NuMaker-N9H30 Board (9)Figure 2-2 Rear View of NuMaker-N9H30 Board (14)Figure 2-3 Front View of NuDesign-TFT-LCD7 Board (20)Figure 2-4 Rear View of NuDesign-TFT-LCD7 Board (21)Figure 2-5 Front View of NuMaker-N9H30 PCB Placement (22)Figure 2-6 Rear View of NuMaker-N9H30 PCB Placement (22)Figure 2-7 Front View of NuDesign-TFT-LCD7 PCB Placement (23)Figure 2-8 Rear View of NuDesign-TFT-LCD7 PCB Placement (23)Figure 3-1 GPIO List Circuit (24)Figure 3-2 System Block Circuit (25)Figure 3-3 Power Circuit (26)Figure 3-4 N9H30F61IEC Circuit (27)Figure 3-5 Setting, ICE, RS-232_0, Key Circuit (28)Figure 3-6 Memory Circuit (29)Figure 3-7 I2S, I2C_0, RS-486_6 Circuit (30)Figure 3-8 RS-232_2 Circuit (31)Figure 3-9 LCD Circuit (32)NUMAKER-HMI-N9H30 USER MANUAL Figure 3-10 CMOS Sensor, I2C_1, CAN_0 Circuit (33)Figure 3-11 RMII_0_PF Circuit (34)Figure 3-12 RMII_1_PE Circuit (35)Figure 3-13 USB Circuit (36)Figure 3-14 TFT-LCD7 Circuit (37)List of TablesTable 2-1 LCD Panel Combination Connector (CON8) Pin Function (11)Table 2-2 Three Sets of Indication LED Functions (12)Table 2-3 Six Sets of User SW, Key Matrix Functions (12)Table 2-4 CMOS Sensor Connector (CON10) Function (13)Table 2-5 JTAG ICE Interface (J2) Function (14)Table 2-6 Expand Port (CON7) Function (16)Table 2-7 UART0 (J3) Function (16)Table 2-8 UART2 (J6) Function (16)Table 2-9 RS-485_6 (SW6~8) Function (17)Table 2-10 Power on Setting (SW4) Function (17)Table 2-11 Power on Setting (S2) Function (17)Table 2-12 Power on Setting (S3) Function (17)Table 2-13 Power on Setting (S4) Function (17)Table 2-14 Power on Setting (S5) Function (17)Table 2-15 Power on Setting (S7/S6) Function (18)Table 2-16 Power on Setting (S9/S8) Function (18)Table 2-17 CMOS Sensor Connector (CON9) Function (19)Table 2-18 CAN_0 (SW9~10) Function (19)NUMAKER-HMI-N9H30 USER MANUAL1 OVERVIEWThe NuMaker-HMI-N9H30 is an evaluation board for GUI application development. The NuMaker-HMI-N9H30 consists of two parts: a NuMaker-N9H30 main board and a NuDesign-TFT-LCD7 extensionboard. The NuMaker-HMI-N9H30 is designed for project evaluation, prototype development andvalidation with HMI (Human Machine Interface) function.The NuMaker-HMI-N9H30 integrates touchscreen display, voice input/output, rich serial port serviceand I/O interface, providing multiple external storage methods.The NuDesign-TFT-LCD7 can be plugged into the main board via the DIN_32x2 extension connector.The NuDesign-TFT-LCD7 includes one 7” LCD which the resolution is 800x480 with RGB-24bits andembedded the 4-wires resistive type touch panel.Figure 1-1 Front View of NuMaker-HMI-N9H30 Evaluation BoardNUMAKER-HMI-N9H30 USER MANUAL Figure 1-2 Rear View of NuMaker-HMI-N9H30 Evaluation Board1.1 Features1.1.1 NuMaker-N9H30 Main Board Features●N9H30F61IEC chip: LQFP216 pin MCP package with DDR (64 MB)●SPI Flash using W25Q256JVEQ (32 MB) booting with quad mode or storage memory●NAND Flash using W29N01HVSINA (128 MB) booting or storage memory●One Micro-SD/TF card slot served either as a SD memory card for data storage or SDIO(Wi-Fi) device●Two sets of COM ports:–One DB9 RS-232 port with UART_0 used 75C3232E transceiver chip can be servedfor function debug and system development.–One DB9 RS-232 port with UART_2 used 75C3232E transceiver chip for userapplication●22 GPIO expansion ports, including seven sets of UART functions●JTAG interface provided for software development●Microphone input and Earphone/Speaker output with 24-bit stereo audio codec(NAU88C22) for I2S interfaces●Six sets of user-configurable push button keys●Three sets of LEDs for status indication●Provides SN65HVD230 transceiver chip for CAN bus communication●Provides MAX3485 transceiver chip for RS-485 device connection●One buzzer device for program applicationNUMAKER-HMI-N9H30 USER MANUAL●Two sets of RJ45 ports with Ethernet 10/100 Mbps MAC used IP101GR PHY chip●USB_0 that can be used as Device/HOST and USB_1 that can be used as HOSTsupports pen drives, keyboards, mouse and printers●Provides over-voltage and over current protection used APL3211A chip●Retain RTC battery socket for CR2032 type and ADC0 detect battery voltage●System power could be supplied by DC-5V adaptor or USB VBUS1.1.2 NuDesign-TFT-LCD7 Extension Board Features●7” resolution 800x480 4-wire resistive touch panel for 24-bits RGB888 interface●DIN_32x2 extension connector1.2 Supporting ResourcesFor sample codes and introduction about NuMaker-N9H30, please refer to N9H30 BSP:https:///products/gui-solution/gui-platform/numaker-hmi-n9h30/?group=Software&tab=2Visit NuForum for further discussion about the NuMaker-HMI-N9H30:/viewforum.php?f=31 NUMAKER-HMI-N9H30 USER MANUALNUMAKER-HMI-N9H30 USER MANUAL2 NUMAKER-HMI-N9H30 HARDWARE CONFIGURATION2.1 NuMaker-N9H30 Board - Front View Combination Connector (CON8)6 set User SWs (K1~6)3set Indication LEDs (LED1~3)Power Supply Switch (SW_POWER1)Audio Codec(U10)Microphone(M1)NAND Flash(U9)RS-232 Transceiver(U6, U12)RS-485 Transceiver(U11)CAN Transceiver (U13)Figure 2-1 Front View of NuMaker-N9H30 BoardFigure 2-1 shows the main components and connectors from the front side of NuMaker-N9H30 board. The following lists components and connectors from the front view:NuMaker-N9H30 board and NuDesign-TFT-LCD7 board combination connector (CON8). This panel connector supports 4-/5-wire resistive touch or capacitance touch panel for 24-bits RGB888 interface.Connector GPIO pin of N9H30 FunctionCON8.1 - Power 3.3VCON8.2 - Power 3.3VCON8.3 GPD7 LCD_CSCON8.4 GPH3 LCD_BLENCON8.5 GPG9 LCD_DENCON8.7 GPG7 LCD_HSYNCCON8.8 GPG6 LCD_CLKCON8.9 GPD15 LCD_D23(R7)CON8.10 GPD14 LCD_D22(R6)CON8.11 GPD13 LCD_D21(R5)CON8.12 GPD12 LCD_D20(R4)CON8.13 GPD11 LCD_D19(R3)CON8.14 GPD10 LCD_D18(R2)CON8.15 GPD9 LCD_D17(R1)CON8.16 GPD8 LCD_D16(R0)CON8.17 GPA15 LCD_D15(G7)CON8.18 GPA14 LCD_D14(G6)CON8.19 GPA13 LCD_D13(G5)CON8.20 GPA12 LCD_D12(G4)CON8.21 GPA11 LCD_D11(G3)CON8.22 GPA10 LCD_D10(G2)CON8.23 GPA9 LCD_D9(G1) NUMAKER-HMI-N9H30 USER MANUALCON8.24 GPA8 LCD_D8(G0)CON8.25 GPA7 LCD_D7(B7)CON8.26 GPA6 LCD_D6(B6)CON8.27 GPA5 LCD_D5(B5)CON8.28 GPA4 LCD_D4(B4)CON8.29 GPA3 LCD_D3(B3)CON8.30 GPA2 LCD_D2(B2)CON8.31 GPA1 LCD_D1(B1)CON8.32 GPA0 LCD_D0(B0)CON8.33 - -CON8.34 - -CON8.35 - -CON8.36 - -CON8.37 GPB2 LCD_PWMCON8.39 - VSSCON8.40 - VSSCON8.41 ADC7 XPCON8.42 ADC3 VsenCON8.43 ADC6 XMCON8.44 ADC4 YMCON8.45 - -CON8.46 ADC5 YPCON8.47 - VSSCON8.48 - VSSCON8.49 GPG0 I2C0_CCON8.50 GPG1 I2C0_DCON8.51 GPG5 TOUCH_INTCON8.52 - -CON8.53 - -CON8.54 - -CON8.55 - -NUMAKER-HMI-N9H30 USER MANUAL CON8.56 - -CON8.57 - -CON8.58 - -CON8.59 - VSSCON8.60 - VSSCON8.61 - -CON8.62 - -CON8.63 - Power 5VCON8.64 - Power 5VTable 2-1 LCD Panel Combination Connector (CON8) Pin Function●Power supply switch (SW_POWER1): System will be powered on if the SW_POWER1button is pressed●Three sets of indication LEDs:LED Color DescriptionsLED1 Red The system power will beterminated and LED1 lightingwhen the input voltage exceeds5.7V or the current exceeds 2A.LED2 Green Power normal state.LED3 Green Controlled by GPH2 pin Table 2-2 Three Sets of Indication LED Functions●Six sets of user SW, Key Matrix for user definitionKey GPIO pin of N9H30 FunctionK1 GPF10 Row0 GPB4 Col0K2 GPF10 Row0 GPB5 Col1K3 GPE15 Row1 GPB4 Col0K4 GPE15 Row1 GPB5 Col1K5 GPE14 Row2 GPB4 Col0K6GPE14 Row2GPB5 Col1 Table 2-3 Six Sets of User SW, Key Matrix Functions●NAND Flash (128 MB) with Winbond W29N01HVS1NA (U9)●Microphone (M1): Through Nuvoton NAU88C22 chip sound input●Audio CODEC chip (U10): Nuvoton NAU88C22 chip connected to N9H30 using I2Sinterface–SW6/SW7/SW8: 1-2 short for RS-485_6 function and connected to 2P terminal (CON5and J5)–SW6/SW7/SW8: 2-3 short for I2S function and connected to NAU88C22 (U10).●CMOS Sensor connector (CON10, SW9~10)–SW9~10: 1-2 short for CAN_0 function and connected to 2P terminal (CON11)–SW9~10: 2-3 short for CMOS sensor function and connected to CMOS sensorconnector (CON10)Connector GPIO pin of N9H30 FunctionCON10.1 - VSSCON10.2 - VSSNUMAKER-HMI-N9H30 USER MANUALCON10.3 - Power 3.3VCON10.4 - Power 3.3VCON10.5 - -CON10.6 - -CON10.7 GPI4 S_PCLKCON10.8 GPI3 S_CLKCON10.9 GPI8 S_D0CON10.10 GPI9 S_D1CON10.11 GPI10 S_D2CON10.12 GPI11 S_D3CON10.13 GPI12 S_D4CON10.14 GPI13 S_D5CON10.15 GPI14 S_D6CON10.16 GPI15 S_D7CON10.17 GPI6 S_VSYNCCON10.18 GPI5 S_HSYNCCON10.19 GPI0 S_PWDNNUMAKER-HMI-N9H30 USER MANUAL CON10.20 GPI7 S_nRSTCON10.21 GPG2 I2C1_CCON10.22 GPG3 I2C1_DCON10.23 - VSSCON10.24 - VSSTable 2-4 CMOS Sensor Connector (CON10) FunctionNUMAKER-HMI-N9H30 USER MANUAL2.2NuMaker-N9H30 Board - Rear View5V In (CON1)RS-232 DB9 (CON2,CON6)Expand Port (CON7)Speaker Output (J4)Earphone Output (CON4)Buzzer (BZ1)System ResetSW (SW5)SPI Flash (U7,U8)JTAG ICE (J2)Power ProtectionIC (U1)N9H30F61IEC (U5)Micro SD Slot (CON3)RJ45 (CON12, CON13)USB1 HOST (CON15)USB0 Device/Host (CON14)CAN_0 Terminal (CON11)CMOS Sensor Connector (CON9)Power On Setting(SW4, S2~S9)RS-485_6 Terminal (CON5)RTC Battery(BT1)RMII PHY (U14,U16)Figure 2-2 Rear View of NuMaker-N9H30 BoardFigure 2-2 shows the main components and connectors from the rear side of NuMaker-N9H30 board. The following lists components and connectors from the rear view:● +5V In (CON1): Power adaptor 5V input ●JTAG ICE interface (J2) ConnectorGPIO pin of N9H30Function J2.1 - Power 3.3V J2.2 GPJ4 nTRST J2.3 GPJ2 TDI J2.4 GPJ1 TMS J2.5 GPJ0 TCK J2.6 - VSS J2.7 GPJ3 TD0 J2.8-RESETTable 2-5 JTAG ICE Interface (J2) Function●SPI Flash (32 MB) with Winbond W25Q256JVEQ (U7); only one (U7 or U8) SPI Flashcan be used●System Reset (SW5): System will be reset if the SW5 button is pressed●Buzzer (BZ1): Control by GPB3 pin of N9H30●Speaker output (J4): Through the NAU88C22 chip sound output●Earphone output (CON4): Through the NAU88C22 chip sound output●Expand port for user use (CON7):Connector GPIO pin of N9H30 FunctionCON7.1 - Power 3.3VCON7.2 - Power 3.3VCON7.3 GPE12 UART3_TXDCON7.4 GPH4 UART1_TXDCON7.5 GPE13 UART3_RXDCON7.6 GPH5 UART1_RXDCON7.7 GPB0 UART5_TXDCON7.8 GPH6 UART1_RTSCON7.9 GPB1 UART5_RXDCON7.10 GPH7 UART1_CTSCON7.11 GPI1 UART7_TXDNUMAKER-HMI-N9H30 USER MANUAL CON7.12 GPH8 UART4_TXDCON7.13 GPI2 UART7_RXDCON7.14 GPH9 UART4_RXDCON7.15 - -CON7.16 GPH10 UART4_RTSCON7.17 - -CON7.18 GPH11 UART4_CTSCON7.19 - VSSCON7.20 - VSSCON7.21 GPB12 UART10_TXDCON7.22 GPH12 UART8_TXDCON7.23 GPB13 UART10_RXDCON7.24 GPH13 UART8_RXDCON7.25 GPB14 UART10_RTSCON7.26 GPH14 UART8_RTSCON7.27 GPB15 UART10_CTSCON7.28 GPH15 UART8_CTSCON7.29 - Power 5VCON7.30 - Power 5VTable 2-6 Expand Port (CON7) Function●UART0 selection (CON2, J3):–RS-232_0 function and connected to DB9 female (CON2) for debug message output.–GPE0/GPE1 connected to 2P terminal (J3).Connector GPIO pin of N9H30 Function J3.1 GPE1 UART0_RXDJ3.2 GPE0 UART0_TXDTable 2-7 UART0 (J3) Function●UART2 selection (CON6, J6):–RS-232_2 function and connected to DB9 female (CON6) for debug message output –GPF11~14 connected to 4P terminal (J6)Connector GPIO pin of N9H30 Function J6.1 GPF11 UART2_TXDJ6.2 GPF12 UART2_RXDJ6.3 GPF13 UART2_RTSJ6.4 GPF14 UART2_CTSTable 2-8 UART2 (J6) Function●RS-485_6 selection (CON5, J5, SW6~8):–SW6~8: 1-2 short for RS-485_6 function and connected to 2P terminal (CON5 and J5) –SW6~8: 2-3 short for I2S function and connected to NAU88C22 (U10)Connector GPIO pin of N9H30 FunctionSW6:1-2 shortGPG11 RS-485_6_DISW6:2-3 short I2S_DOSW7:1-2 shortGPG12 RS-485_6_ROSW7:2-3 short I2S_DISW8:1-2 shortGPG13 RS-485_6_ENBSW8:2-3 short I2S_BCLKNUMAKER-HMI-N9H30 USER MANUALTable 2-9 RS-485_6 (SW6~8) FunctionPower on setting (SW4, S2~9).SW State FunctionSW4.2/SW4.1 ON/ON Boot from USB SW4.2/SW4.1 ON/OFF Boot from eMMC SW4.2/SW4.1 OFF/ON Boot from NAND Flash SW4.2/SW4.1 OFF/OFF Boot from SPI Flash Table 2-10 Power on Setting (SW4) FunctionSW State FunctionS2 Short System clock from 12MHzcrystalS2 Open System clock from UPLL output Table 2-11 Power on Setting (S2) FunctionSW State FunctionS3 Short Watchdog Timer OFFS3 Open Watchdog Timer ON Table 2-12 Power on Setting (S3) FunctionSW State FunctionS4 Short GPJ[4:0] used as GPIO pinS4Open GPJ[4:0] used as JTAG ICEinterfaceTable 2-13 Power on Setting (S4) FunctionSW State FunctionS5 Short UART0 debug message ONS5 Open UART0 debug message OFFTable 2-14 Power on Setting (S5) FunctionSW State FunctionS7/S6 Short/Short NAND Flash page size 2KBS7/S6 Short/Open NAND Flash page size 4KBS7/S6 Open/Short NAND Flash page size 8KBNUMAKER-HMI-N9H30 USER MANUALS7/S6 Open/Open IgnoreTable 2-15 Power on Setting (S7/S6) FunctionSW State FunctionS9/S8 Short/Short NAND Flash ECC type BCH T12S9/S8 Short/Open NAND Flash ECC type BCH T15S9/S8 Open/Short NAND Flash ECC type BCH T24S9/S8 Open/Open IgnoreTable 2-16 Power on Setting (S9/S8) FunctionCMOS Sensor connector (CON9, SW9~10)–SW9~10: 1-2 short for CAN_0 function and connected to 2P terminal (CON11).–SW9~10: 2-3 short for CMOS sensor function and connected to CMOS sensorconnector (CON9).Connector GPIO pin of N9H30 FunctionCON9.1 - VSSCON9.2 - VSSCON9.3 - Power 3.3VCON9.4 - Power 3.3V NUMAKER-HMI-N9H30 USER MANUALCON9.5 - -CON9.6 - -CON9.7 GPI4 S_PCLKCON9.8 GPI3 S_CLKCON9.9 GPI8 S_D0CON9.10 GPI9 S_D1CON9.11 GPI10 S_D2CON9.12 GPI11 S_D3CON9.13 GPI12 S_D4CON9.14 GPI13 S_D5CON9.15 GPI14 S_D6CON9.16 GPI15 S_D7CON9.17 GPI6 S_VSYNCCON9.18 GPI5 S_HSYNCCON9.19 GPI0 S_PWDNCON9.20 GPI7 S_nRSTCON9.21 GPG2 I2C1_CCON9.22 GPG3 I2C1_DCON9.23 - VSSCON9.24 - VSSTable 2-17 CMOS Sensor Connector (CON9) Function●CAN_0 Selection (CON11, SW9~10):–SW9~10: 1-2 short for CAN_0 function and connected to 2P terminal (CON11) –SW9~10: 2-3 short for CMOS sensor function and connected to CMOS sensor connector (CON9, CON10)SW GPIO pin of N9H30 FunctionSW9:1-2 shortGPI3 CAN_0_RXDSW9:2-3 short S_CLKSW10:1-2 shortGPI4 CAN_0_TXDSW10:2-3 short S_PCLKTable 2-18 CAN_0 (SW9~10) Function●USB0 Device/HOST Micro-AB connector (CON14), where CON14 pin4 ID=1 is Device,ID=0 is HOST●USB1 for USB HOST with Type-A connector (CON15)●RJ45_0 connector with LED indicator (CON12), RMII PHY with IP101GR (U14)●RJ45_1 connector with LED indicator (CON13), RMII PHY with IP101GR (U16)●Micro-SD/TF card slot (CON3)●SOC CPU: Nuvoton N9H30F61IEC (U5)●Battery power for RTC 3.3V powered (BT1, J1), can detect voltage by ADC0●RTC power has 3 sources:–Share with 3.3V I/O power–Battery socket for CR2032 (BT1)–External connector (J1)●Board version 2.1NUMAKER-HMI-N9H30 USER MANUAL2.3 NuDesign-TFT-LCD7 -Front ViewFigure 2-3 Front View of NuDesign-TFT-LCD7 BoardFigure 2-3 shows the main components and connectors from the Front side of NuDesign-TFT-LCD7board.7” resolution 800x480 4-W resistive touch panel for 24-bits RGB888 interface2.4 NuDesign-TFT-LCD7 -Rear ViewFigure 2-4 Rear View of NuDesign-TFT-LCD7 BoardFigure 2-4 shows the main components and connectors from the rear side of NuDesign-TFT-LCD7board.NuMaker-N9H30 and NuDesign-TFT-LCD7 combination connector (CON1).NUMAKER-HMI-N9H30 USER MANUAL 2.5 NuMaker-N9H30 and NuDesign-TFT-LCD7 PCB PlacementFigure 2-5 Front View of NuMaker-N9H30 PCB PlacementFigure 2-6 Rear View of NuMaker-N9H30 PCB PlacementNUMAKER-HMI-N9H30 USER MANUALFigure 2-7 Front View of NuDesign-TFT-LCD7 PCB PlacementFigure 2-8 Rear View of NuDesign-TFT-LCD7 PCB Placement3 NUMAKER-N9H30 AND NUDESIGN-TFT-LCD7 SCHEMATICS3.1 NuMaker-N9H30 - GPIO List CircuitFigure 3-1 shows the N9H30F61IEC GPIO list circuit.Figure 3-1 GPIO List Circuit NUMAKER-HMI-N9H30 USER MANUAL3.2 NuMaker-N9H30 - System Block CircuitFigure 3-2 shows the System Block Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-2 System Block Circuit3.3 NuMaker-N9H30 - Power CircuitFigure 3-3 shows the Power Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-3 Power Circuit3.4 NuMaker-N9H30 - N9H30F61IEC CircuitFigure 3-4 shows the N9H30F61IEC Circuit.Figure 3-4 N9H30F61IEC CircuitNUMAKER-HMI-N9H30 USER MANUAL3.5 NuMaker-N9H30 - Setting, ICE, RS-232_0, Key CircuitFigure 3-5 shows the Setting, ICE, RS-232_0, Key Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-5 Setting, ICE, RS-232_0, Key Circuit3.6 NuMaker-N9H30 - Memory CircuitFigure 3-6 shows the Memory Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-6 Memory Circuit3.7 NuMaker-N9H30 - I2S, I2C_0, RS-485_6 CircuitFigure 3-7 shows the I2S, I2C_0, RS-486_6 Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-7 I2S, I2C_0, RS-486_6 Circuit3.8 NuMaker-N9H30 - RS-232_2 CircuitFigure 3-8 shows the RS-232_2 Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-8 RS-232_2 Circuit3.9 NuMaker-N9H30 - LCD CircuitFigure 3-9 shows the LCD Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-9 LCD Circuit3.10 NuMaker-N9H30 - CMOS Sensor, I2C_1, CAN_0 CircuitFigure 3-10 shows the CMOS Sensor,I2C_1, CAN_0 Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-10 CMOS Sensor, I2C_1, CAN_0 Circuit3.11 NuMaker-N9H30 - RMII_0_PF CircuitFigure 3-11 shows the RMII_0_RF Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-11 RMII_0_PF Circuit3.12 NuMaker-N9H30 - RMII_1_PE CircuitFigure 3-12 shows the RMII_1_PE Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-12 RMII_1_PE Circuit3.13 NuMaker-N9H30 - USB CircuitFigure 3-13 shows the USB Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-13 USB Circuit3.14 NuDesign-TFT-LCD7 - TFT-LCD7 CircuitFigure 3-14 shows the TFT-LCD7 Circuit.Figure 3-14 TFT-LCD7 CircuitNUMAKER-HMI-N9H30 USER MANUAL4 REVISION HISTORYDate Revision Description2022.03.24 1.00 Initial version NUMAKER-HMI-N9H30 USER MANUALNUMAKER-HMI-N9H30 USER MANUALImportant NoticeNuvoton Products are neither intended nor warranted for usage in systems or equipment, anymalfunction or failure of which may cause loss of human life, bodily injury or severe propertydamage. Such applications are deemed, “Insecure Usage”.Insecure usage includes, but is not limited to: equipment for surgical implementation, atomicenergy control instruments, airplane or spaceship instruments, the control or operation ofdynamic, brake or safety systems designed for vehicular use, traffic signal instruments, all typesof safety devices, and other applications intended to support or sustain life.All Insecure Usage shall be made at customer’s risk, and in the event that third parties lay claimsto Nuvoton as a result of customer’s Insecure Usage, custome r shall indemnify the damagesand liabilities thus incurred by Nuvoton.。

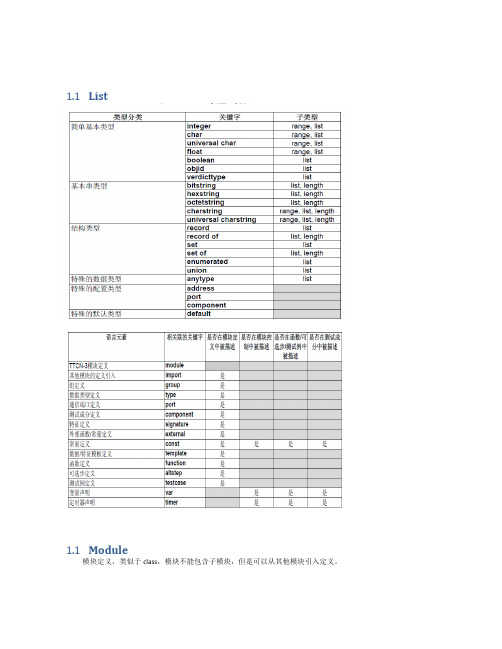
1.1List1.1Module模块定义,类似于class,模块不能包含子模块,但是可以从其他模块引入定义。
定义部分:定义测试成分、通信端口、数据类型、常数、测试数据模板、函数、测试端口上调用的过程特征(signatures)、测试例等等。
控制部分:分调用测试例并控制它们的执行。
控制部分也可以声明(局部)变量等,程序语句(如ifelse和do-while)可以用于各个测试例的选择和执行顺序。
TTCN-3不支持全局变量的概念。
1.2Type用户自定义数据类型type float pi (3.1415926);type set MySetType{integer field1,charstring field2}1.3Template模板(Templates)用于传送一个特定值的集合或是测试接收的值的集合是否与模板说明匹配。
a) 模板提供了一种组织和重复使用测试数据的方法b) 模板能够被参数化;c) 模板允许匹配机制;d) 模板既能够用于基于消息的通信,也能用于基于过程的通信1.4ComponentThe component type defines which ports are associated with a component.•It is also possible to declare constants, variables and timers local to a particular component type.•These declarations are visible to all testcases, functions and altsteps that run on an instance of the given component type. This shall be explicitly stated using the runs on keyword (see clause 16) in the testcase, function or altstep header.1.5Port && messagePort:端口可以基于消息(message标识),和基于过程(procedure)的,甚至是二者混合(mixed)的。

COMMISSION IMPLEMENTING DECISIONof 25 November 2013on Guidelines on Annex I to Regulation (EC) No 1223/2009 of the European Parliament and of the Council on cosmetic products(Text with EEA relevance)(2013/674/EU)THE EUROPEAN COMMISSION,Having regard to the Treaty on the Functioning of the European Union,Having regard to Regulation (EC) No 1223/2009 of the European Parliament and of the Council of 30 November 2009 on cosmetic products ( 1 ), and in particular the third subparagraph of Article 10(1) thereof,Whereas:(1) It is essential that cosmetic products made available on the Union market be safe for human health when used under normal and reasonably foreseeable conditions of use. To that end, Regulation (EC) No 1223/2009 requires that, in order to establish that a cosmetic product is safe under those conditions, cosmetic products undergo a safety assessment. (2) The operator designated as the responsible person in accordance with Regulation (EC) No 1223/2009 is to ensure that, for each cosmetic product which is to be placed on the Union market, a cosmetic product safety report is drawn up on the basis of the relevant information and in accordance with the requirements laid down in Annex I to Regulation (EC) No 1223/2009. (3) In order to facilitate the understanding of the requirements of Annex I to Regulation (EC) No 1223/2009 by all undertakings, and especially small and medium-size enterprises, the Regulation requires that the Commission adopts appropriate guidelines. (4) This Decision sets out appropriate guidelines on Annex I to Regulation (EC) No 1223/2009. They were developedwith the contribution of the relevant stakeholders, including representatives of small and medium-sized enterprises. (5) The guidelines should assist responsible persons in complying with their regulatory obligations. However, they are not meant to replace the knowledge and expertise of the qualified safety assessor, as required by Article 10(2) of Regulation (EC) No 1223/2009, who should remain the only professional allowed to carry out the cosmetic product safety assessment as described in Part B of Annex I. (6) The measures provided for in this Decision are in accordance with the opinion of the Standing Committee on Cosmetic Products,HAS ADOPTED THIS DECISION: Article 1 The guidelines to enable undertakings to comply with the requirements laid down in Annex I to Regulation (EC) No 1223/2009 on cosmetic products are set out in the Annex to this Decision. Article 2 This Decision shall enter into force on the twentieth day following that of its publication in the Official Journal of the European Union .Done at Brussels, 25 November 2013. For the CommissionThe President José Manuel BARROSO ( 1 ) OJ L 342, 22.12.2009, p. 59.ANNEXGUIDELINES ON ANNEX I TO REGULATION (EC) No 1223/2009 ON THE COSMETIC PRODUCT SAFETYREPORT1. INTRODUCTIONArticle 11 of Regulation (EC) No 1223/2009 requires that a product information file is drawn up for each product before it is placed on the market. The product information file should be updated when necessary and kept readily accessible, in electronic or other format, at the address of the responsible person given on the label, to the competent authorities for market surveillance purposes for a period of 10 years following the placing on the market of the last batch of the product.The most important element of the product information file, from a safety point of view, is the cosmetic product safety report referred to in Article 10(1). The other elements are a clear description of the cosmetic product, a description of the method of manufacturing and a statement on compliance with good manufacturing practice, the proof of the effects claimed, and data on animal testing (1).Where the responsible person drawing up the cosmetic product safety report is not the manufacturer of the product, they should ensure they have access to all the technical and scientific skills necessary to obtain reliable cosmetic product safety information and an appropriate safety assessment to demonstrate that the product they are responsible for is safe, in accordance with Article 3 of Regulation (EC) No 1223/2009. They may therefore need to involve not only the safety assessor, but also the manufacturer, the suppliers of raw materials, and other technical experts.In any case, the responsible person is to ensure that the intended use of the cosmetic product and the anticipated systemic exposure to individual ingredients in a final formulation are taken into account in the safety assessment;an appropriate weight-of-evidence approach is used in the safety assessment for reviewing data from all existing sources; the cosmetic product safety report is kept up to date in view of additional relevant information generated subsequent to placing the product on the market (2).The cosmetic product safety assessment, as set out in Part B of Annex I to Regulation (EC) No 1223/2009, is to be carried out by a qualified safety assessor. The responsible person and the safety assessor should work closely together to ensure that the safety of the product is properly assessed and documented and that the assessment is kept up to date. The responsible person and the safety assessor should gather all the necessary information as required by Part A of Annex I to Regulation (EC) No 1223/2009.The cosmetic product safety report should be drawn up in a transparent way and should be well-argued and easily understood.The Cosmetic Product Safety Report is an expert piece of work made up of different modules and where the information required under Part A may be stored in different databases. The report, which should contain, as a minimum, all the information indicated in Annex I to Regulation (EC) No 1223/2009, should appear under the same or similar headings for ease of reference of the competent authorities. However, it may be sufficient to provide under each heading a clear reference to a document containing the information and readily available in electronic or printed format.1223/2009—COSMETICPRODUCTREPORTSAFETY(EC)2. ANNEXITOREGULATIONNoIn accordance with Annex I to Regulation (EC) No 1223/2009, the Cosmetic Product Safety Report is to contain, ‘as a minimum’, the information required under each of the headings of Part A and Part B.Part A aims to gather all the data necessary for the safety assessment of the product, while Part B sets out the reasoning, starting from the data, for drawing conclusions as to the safety of the product.(1) Article 11(2) of Regulation (EC) No 1223/2009.(2) Article 10(1) of Regulation (EC) No 1223/2009.The structure and content of the safety report should reflect the requirements of Annex I to Regulation (EC) No 1223/2009. However, if the report does not directly contain the required information, it should provide a reference to another readily available source.The responsible person is to ensure that the Cosmetic Product Safety Report is kept up to date in the light of additional relevant information emerging after the product has been placed on the market (1).PRODUCTCOSMETICSAFETYINFORMATION—3. PARTAPart A of the cosmetic product safety report is intended to gather the data necessary to prove that the cosmetic product is safe. The information should enable the safety assessor to clearly identify and quantify, based on the identified hazards, the risks a cosmetic product may present to human health.A hazard may arise, for example, from the raw materials, the manufacturing process, the packaging, theconditions of use of the product, the microbiological specifications, the quantities used, the toxicological profile of the substances, etc.As Part A of Annex I to Regulation (EC) No 1223/2009 requires that the data listed under its headings are provided as a minimum, any discrepancy with regards to the requirements of Part A should be justified.Part A of Annex I to Regulation (EC) No 1223/2009 lists the data that is to be available, ‘as a minimum’, for the safety assessor to be able to carry out the safety assessment.In addition to the minimum data listed in Part A of Annex I to Regulation (EC) No 1223/2009, the safety assessor can use any additional data, where relevant. On the other hand, they, or the responsible person, may consider that, depending on the type of product, some of the required data are not relevant or necessary to assess the safety of the product (e.g. preservation challenge test). In this case, the absence of specific data is to be clearly justified in Part A and the justification is to be repeated and validated by the safety assessor in their reasoning in Part B. The responsible person should check the presence of the required data or the justification for their absence.The data required by Part A can be drawn from any reliable source. Examples include: data from suppliers, scientific literature, experience gained with similar or other product categories, results of studies on the product itself or on the substances it contains, available data on similar formulations, or computer models. The safety report should highlight the relevance of the data in relation to the product.The guidance published by the EU scientific committees concerned with risk assessment (2), as well as the recommendations of national competent authorities or professional organisations, may provide further helpful support.3.1. Quantitative and qualitative composition of the cosmetic productThe aim of that section of the cosmetic product safety report is to provide the exact quantitative and qualitative composition of the finished product, starting from the raw materials. Raw materials are substances or mixtures used in the manufacturing of the cosmetic product. The intended function of each substance is to be indicated.The complete product composition is to be specified, stating the name and identity (qualitative) of each raw material (including chemical name, INCI, CAS, Einecs/ELINCS, where possible), and the amount of each raw material, stating the weight percentage (quantitative). Ranges should not be used, unless this can be justified(e.g. viscosity or pH adjusters). If concentration ranges are unavoidable, toxicological considerations and calculations should be based on the highest concentration figure. It may also be useful to indicate the supplier(s) of the raw materials.(1) Article 10(1)(c) of Regulation (EC) No 1223/2009.(2) The SCCS’s Notes of Guidance for the Testing of Cosmetic Ingredients and their Safety Evaluation, 8th Revision, SCCS/1501/12, and itssubsequent updates.All substances entering into the composition of commercial mixtures supplied as raw materials (including directly added preservatives, antioxidants, chelators, buffering agents, solvents, other additives, etc.) are to be identified and quantified in the formula of the finished product. This also applies to all substances indirectly added to the product, such as preservatives used for preserving raw materials. The intended function of each substance is to be indicated.When chemically well-defined substances are present, their quantity and molecular formula should be given together with their analytical specifications (degree of purity, identification of major impurities, criteria and test methods used).When complex ingredients are present, their nature and quantity together with a clear definition of the mixture and the material(s) used should be given in order to identify the substances with regard to their composition and effects (manufacturing and purification processes, including physical, chemical, enzymatic, biotechnological and microbiological steps). The purity criteria and test methods used should be provided. Examples of complex ingredients include those of mineral, botanical, animal or biotechnological origin. The scope of the information needed on complex ingredients, depending on their nature and origin, is explicitly listed in the Scientific Committee for Consumer Safety (SCCS) Note of Guidance (1).When a mixture of both chemically well-defined substances and complex ingredients is present, the above guidance also applies.Where any fragrance (or flavour) compound comprising a mixture of fragrance (or flavour) ingredients and functional components with olfactory, odour-enhancing, odour-protecting or blending properties is formulated and intentionally added to a cosmetic product to impart a scent (or flavour) or to cover a malodour, its identification is to include the name and code number as well as the identity of the supplier. Qualitative and quantitative information about regulated substances in the fragrance (or flavour) compound and information relevant for a safety assessment should be disclosed to the responsible person and the safety assessor, and should be included in the safety report.3.2. Physical/chemical characteristics and stability of the cosmetic productThe aim of that section of the cosmetic product safety report is to describe the relevant physical and chemical specifications of the substances or mixtures used and the cosmetic product itself. These specifications are crucial for an appropriate safety assessment, as they may influence the safety of a cosmetic product. For example, physico-chemical properties, in combination with other information, can help the safety assessor determine the need to investigate relevant toxicological parameters.In addition, the physico-chemical characteristics of the substances or mixtures and finished products set the benchmark against which the products and the raw materials can be considered acceptable from a quality point of view (2).That section of the cosmetic product safety report also requires an assessment of the stability of the cosmetic product, under reasonably foreseeable storage conditions. The aim is to evaluate if the stability of the cosmetic product affects the safety and quality of the product, and to use the information to determine its minimum durability and period-after-opening (PAO).3.2.1. Physical/chemical characteristics of substances or mixturesThis description is to include the most relevant physico-chemical properties of each substance and mixture contained in the product, for example: chemical identification, physical form, molecular weight, solubility, partition coefficient, substance purity, other parameters relevant for the characterisation of specific substances and mixtures, and, for polymers, the average molecular weight and range.(1) SCCS Notes of Guidance, para. 3-6.2, pp. 35-36.(2) This point is relevant in the context of Good Manufacturing Practices, and is explicitly addressed by the relevant standard EN ISO22716:2007. More specifically, it matches the requirements for the release of raw materials and the finished product.Where relevant, the particle-size distribution curve of substances should be included in the physico-chemical characteristics, especially for nanomaterials.Cosmetics manufacturers should ensure that the specifications of raw materials are properly documented by their suppliers. Specifications should be available for each raw material actually used in the product. Based on function, additional specifications may be needed. For UV absorbers, for instance, the absorption spectra should be provided.For each description of physico-chemical properties and specifications (for each substance and mixture contained in the product), the reference methods should be stated in the safety report.3.2.2. Physical/chemical characteristics of the finished cosmetic productThis description is to contain the specifications of the finished product. Each specification should be given with relevant limits, e.g. pH between 5.5 and 6.5.For each description of physico-chemical properties and specifications of the finished product, the reference methods should be stated in the cosmetic product safety report.3.2.3. Stability of the cosmetic productAs the requirement is to assess the stability of the cosmetic product under reasonably foreseeable storage conditions, if stability is dependent on storage conditions, information about these conditions should be passed on throughout the supply chain, and, if relevant for the end user, it should be indicated on the labelling of the product.The methodology used to determine the product’s minimum durability should be described. Any specific preservation precautions should be mentioned.All available data used to justify the indicated minimum durability should be listed in the safety report. In order to determine the coherence of the stability study conducted, and to check the relevance of the date of minimum durability chosen for the product, the description of the tests specific to the stability study and the results of those tests should be included in the cosmetic product safety report. In addition, the following should also be provided:(1) evidence that the composition of the product used for stability testing corresponds to the product actuallyplaced on the market;(2) the results of the preservative efficacy study, e.g. challenge test, if applicable (1);(3) when applicable, the period-after-opening (PAO) (2) and its justification.The SCCS has recommended that ‘relevant stability tests, adapted to the type of cosmetic product and its intended use, should be carried out. To make sure that no stability problems are induced by the type of container and packaging used, physical stability tests are currently carried out with inert containers and those intended to be used on the market.’ (3)(1) See section 3.3 on Microbiological quality.(2) See ‘Practical implementation of Article 6(1)(c) of the Cosmetics Directive (76/768/EEC): Labelling of product durability: “period of timeafter opening” ’ (Council Directive 76/768/EEC, OJ L 262, 27.9.1976, p. 169) http://ec.europa.eu/consumers/sectors/cosmetics/files/doc/ wd-04-entr-cos_28_rev_version_adoptee20040419_en.pdf(3) SCCS Notes of Guidance, para. 4-3.3, p. 74.3.3. Microbiological qualityThe aim of that section of the cosmetic product safety report is to determine the acceptable microbiological specifications of the raw materials (substances or mixtures) and finished product from a microbiological point of view. In accordance with Annex I to Regulation (EC) No 1223/2009, particular attention is to be paid to the microbiological specifications of cosmetic products intended to be used on sensitive body parts and on specific populations. In addition, information regarding microbiological quality is essential in order to justify the effectiveness of the preservation system and justify the indicated minimum durability of the cosmetic product stored under appropriate conditions and period-after- opening (PAO) (1) of the finished product in terms of safety.The microbiological specifications of the raw materials (substances or mixtures) and cosmetic product are to form part of the safety assessment. Particular attention is to be paid to the microbiological specifications of cosmetic products intended to be used around the eyes, on mucous membranes in general, on damaged skin (e.g. skin care products suitable for atopic or irritated skin), on children under three years of age, on elderly people or on persons with compromised immune responses.3.3.1. Microbiological quality of substances and mixturesThe main parameters for microbiological quality are the original level of contamination and the possibility of microbial growth. Particular attention should be paid to the raw materials (substances and mixtures) most susceptible to microbial growth (e.g. water-based mixtures, protein-rich materials, plant or animal raw materials).On the other hand, there are raw materials which do not support microbial growth, e.g. organic solvents.3.3.2. Microbiological quality of the finished cosmetic productConcerning microbiological susceptibility, there is a difference between three product categories:(1) low microbiological risk products (e.g. products with an alcohol content > 20 %, products based on organicsolvents, high/low-pH products), for which neither a preservation challenge test nor microbiological quality tests on the finished product are necessary. A scientific justification is to be provided, however;(2) single-use products, and products which cannot be opened (e.g. for which the packaging allows dosing theproduct without it coming in contact with the air), for which only microbiological quality tests on the finished product are necessary. A scientific justification is to be provided, however;(3) all other products, for which both a preservation challenge test and microbiological quality tests on thefinished product are necessary.Specific ‘Guidelines on Microbiological Quality of the Finished Product’ are provided in the SCCS Notes of Guidance (2).3.4. Impurities, traces, information about the packaging materialThe aim of that section of the cosmetic product safety report is to assess whether the cosmetic product contains substances that have not been intentionally added to the formulation, and which may have an impact on its safety.Impurities are unintended substances in raw materials.(1) The ‘date of minimum durability’ is the date until which the cosmetic product, stored under appropriate conditions, will continue tofulfil its initial function and, in particular, will remain safe; the PAO is the period of time after opening for which the product can be used without any harm to the consumer. See ‘Practical implementation of Article 6(1)(c) of the Cosmetics Directive (76/768/EEC): Labelling of product durability: “period of time after opening” ’.(2) SCCS Notes of Guidance, para 4-4, pp. 75–76.A trace is a small quantity of an unintended substance in the finished product. Traces are to be evaluatedwith regard to safety of the finished product. When traces of prohibited substances are present, evidence of their technical unavoidability are also to be provided.Traces can originate from the following sources: impurities in the raw materials/substances; the manufacturing process; potential chemical evolution/interaction and/or migration of substances in the product that could occur under normal storage conditions and/or through contact with the packaging material.Because substances may migrate from the packaging to the formulation, the relevant characteristics of the packaging material are to be considered.In accordance with point 4 of Annex I to Regulation (EC) No 1223/2009, the section on ‘Impurities, traces, information about the packaging material’ is to address three specific issues:(a) the purity of substances and mixtures;(b) in case of traces of prohibited substances, evidence of their technical unavoidability;(c) the relevant characteristics of the packaging material, in particular purity and stability.In practical terms, those elements may be interpreted as follows:(a) precise definition of impurities and traces (see 3.4.1);(b) evidence of technical unavoidability of prohibited substances (see 3.4.2);(c) potential release of substances from the packaging or possible deterioration of the product in contact with thepackaging (see 3.4.3).For the analysis of impurities and packaging material, data from suppliers are of crucial importance and should be preferred.3.4.1. Purity of substances and mixturesThe presence of unintended substances, such as impurities and traces, can have an impact on the safety of the finished product. The cosmetic product safety report is to include data on the purity of raw materials (substances and mixtures) and the identification of the toxicologically relevant unintended substances. These substances should be taken into account in the safety assessment of the product.Impurities are unintended substances in raw materials.A trace is a small quantity of an unintended substance in the finished product.The presence of traces in the finished product can be evaluated in two ways:(a) through the specifications/technical data for each raw material, based on knowledge of the process for manufacturing the raw material (origin of substance, production process, synthesis route, extraction process, solvent used, etc.);(b) through a physico-chemical analysis of possible impurities in raw materials and, if necessary, in the finalproduct (e.g. nitrosamines which are potentially generated during or after the manufacturing process).Traces of prohibited substances are dealt with in paragraph 3.4.2 of these Guidelines.Some traces have regulatory concentration limits. For the presence of traces of substances that are not prohibited, and for which there are no regulatory concentration limits, but which could be expected to impact consumer safety, the safety assessment needs to be carried out by the safety assessor.3.4.2. Evidence of the technical unavoidability of traces of prohibited substancesWhile the procedure outlined in paragraph 3.4.1 should be followed for all known impurities and traces to evaluate their toxicological impact, further investigation is required for prohibited substances present as traces in the finished product (1).When such presence is technically unavoidable, the cosmetics manufacturers are required to provide evidence of the technical unavoidability. That means they have to justify the presence of those traces by all necessary means.The presence of traces of prohibited substances should be kept as low as is reasonably achievable under good manufacturing practices. In addition, the safety assessor has to decide whether their levels are toxicologically acceptable and whether the product is still safe.Especially in the case of non-threshold genotoxic and carcinogenic substances (2), the cosmetic industry should keep improving its best practices in order to eliminate these substances (ALARA principle (3)) in the finished cosmetic product. The main concern is to ensure the protection of human health, as required by Article 3 of Regulation (EC) No 1223/2009.Traces generated by the degradation of substances within the final product (stability issues), by preservation or transport problems, or by the interaction of raw materials should be avoided through good manufacturing practices, or possibly through re-formulation of the product.3.4.3. The relevant characteristics of packaging materialPackaging material means the container (or primary packaging) that is in direct contact with the formulation. The relevant characteristics of packaging materials in direct contact with the final product are important for the safety of the cosmetic product. Reference to Regulation (EC) No 1935/2004 of the European Parliament and of the Council (4) could be useful.Experience with similar formulation/packaging combinations already on the market provides useful indications.Materials that have been developed for food packaging have often already been tested, so relevant information on stability and migration may be available. Additional testing may not be required. However, more evaluation may be needed for new or novel packaging.The combination of packaging material, formulation of the cosmetic product and contact with the external environment may have an impact on the safety of the finished product, due to the following factors:(a) interaction between the product and the packaging material;(b) barrier properties of the packaging material;(c) substance migration from/to the packaging material.The information on relevant characteristics of the packaging materials in direct contact with the product should allow an estimation of potential risks. Relevant characteristics could include, for example, the following:(a) composition of the packaging material, including technical substances such as additives;(b) technically unavoidable impurities;(c) possible migration from the packaging.(1) Article 17 of Regulation (EC) No 1223/2009 establishes that traces of prohibited substances are only permitted if they are technicallyunavoidable and if they have no impact on the safety of the cosmetic products.(2) The ‘non-threshold genotoxic and carcinogenic substances’ are the genotoxic and carcinogenic substances without a threshold for thecarcinogenic-genotoxic effects.(3) Opinion of the Scientific Committee on a request from EFSA related to A Harmonised Approach for Risk Assessment of SubstancesWhich are both Genotoxic and Carcinogenic, the EFSA Journal (2005) 282, pp. 1-31.(4) OJ L 338, 13.11.2004, p. 4.。
欧盟:临床阶段的GMP要求和QP放行欧盟临床阶段的D Si f i d S h itt PAREXELDr Siegfried Schmitt, PAREXELEU GMP REQUIREMENTS AT THE CLINICAL DEVELOPMENT STAGE & QP RELEASEAGENDAGMPs for Investigational Medicinal Products (IMP) and their Qualified Person (QP) releasetheir Qualified Person(QP)releaseNew developments and requirements in the EUN d l t d i t i th EUINVESTIGATIONAL MEDICINAL PRODUCTS (IMP)()Definition of Investigational Medicinal Products(IMPs)“a pharmaceutical form of an active substance or placebo beingftested or used as a reference in a clinical trial,including products already with a marketing authorization but used or assembled (formulated or packaged)in a way different from the authorised form,or when used for an unauthorised indication,or when used to gain further information about the authorised form.”An IMP must be registered in the EudraCT database/3rya6w7NON INVESTIGATIONAL MEDICINAL PRODUCTS (NIMP)Definition of Non Investigational Medicinal Products(NIMPs) Products which are not the object of investigation(i.e.other than the tested product,placebo or active comparator)may be supplied)to subjects participating in a trial and used in accordance with the protocol.For instance,some clinical trial protocols require the use of medicinal products such as support or rescue/escape medication for preventive,diagnostic or therapeutic reasons and/or to ensure q p j y y that adequate medical care is provided for the subject.They may also be used in accordance with the protocol to induce a physiological response./3rya6w7()THE QUALIFIED PERSON (QP)The person defined in Article 48 of Directive 2001/83/EC and Article 52 of Directive 2001/82/ECThe QP is the only person that can release product to be placed on the market in the EUplaced on the market in the EUThe QP is personally liable for the release decisionThe regulations apply also to IMPsTHE EUROPEAN UNIONEU -LEGAL INSTRUMENTSPrimary EU Law:Treaty of the Function of the European Union(TFEU)Treaty of the Function of the European Union (TFEU) Secondary EU Law:Medicinal(Drug) Secondary EU Law:a) Legislative Acts:Regulation, Directive, Decisionb)Non Legislati e ActsMedicinal (Drug)Products:E d L V l1b) Non-Legislative Acts:Recommendation, Opinionc) New Legal Acts (created byEudraLex Vol 1(human) &EudraLex Vol 5the Treaty of Lisbon):Delegated Act, ImplementingAct(veterinary)http://europa.eu/legislation_summaries/institutional_affairs/treaties/lisbon_treaty/ai0032_en.htm_y_EU REGULATIONSA regulation is a legal instrument that is immediately enforceableExample:Council Regulation(EEC)No2309/93on Community procedures for the authorization and supervision of medicinal products for human and veterinary use and establishing a European Agency for the Evaluation of Medicinal ProductsEU DIRECTIVES•Require Member States to achieve a certain result•Do not dictate the means of achieving the result•The contents of the Directive must be transposed into national law by Member States-and carried out by Member State National Competent Authorities“National Authorities”Example:Commission Directive2003/94/EC on the principles E l C i i Di ti th i i l and guidelines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human useEU GUIDELINESDo not have the force of law,but represent the agreed views of regulators on a certain topic.It is possible not to follow them if scientifically justifiedAre complemented by Questions and Answers to clarify specific points in guidelines•EMA scientific guidelines-/d2qvbjx•The GMP guide-/crj4qb3g p y j q•The guideline publication process-/c6dpjrxEU LEGISLATION ON THE WEBEU Legislationhttp://eur-lex.europa.eu/en/index.htmLegislation for Medicinal Products in the European Union http://ec.europa.eu/health/index_en.htmGXP FOR IMPS -APPLICABLE REGULATIONSThe regulations cover the lifecycle from development to discontinuationEU GXP FOR IMPS -APPLICABLE REGULATIONS EudraLex Volume4Annex13Manufacture of InvestigationalMedicinal ProductsEudraLex Volume4Parts I,II and IIIEudraLex Volume4Annex16Certification by a Qualified person and Batch Release July2001Draft revision of Annex162013http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm htt///h lth/d t/d l/l4/i d htEU GXP FOR IMPS -PHASE APPROPRIATEPDA Technical Report 56 GXP FOR IMPS VERSUS GXP FOR COMMERCIAL PRODUCTyParticularly for Clinical Phases I and II•Changes need to be documented,but do not need to go through a formal change approval process•Deviations will be the exceptiony y •Analytical methods need to be documented and suitability established,but do not need to be validated •Documentation has to be controlled,but laboratory journals may be used instead of batch recordsb d i t d f b t h d•Operational and analytical personnel may be the same •Critical parameters and quality attributes not fully established(e.g.yield not a critical parameter)GXP FOR IMPS VERSUS GXP FOR COMMERCIAL PRODUCTWhat must be in place for IMPs and Commercial product:•A formal quality system•Job descriptions and training•Calibration program•Appropriate environmental conditions(minar Air Fumehood(LAF)or HVAC)•Process controls•Rationales,e.g.for changes,analytical method selection, process and process controlsGXP FOR IMPS -SOME SPECIFIC ISSUESSterilisation steps in IMP manufacture•Must be fully validated•Associated analytical methods must be fully validatedBiotech Products•Production scale for Clinical Phase III material must be equivalent to commercial scale(preferably the same equipment)GXP FOR IMPS -SOME SPECIFIC ISSUESClinical Trial LogisticsPharmaceutical Engineering Nov/Dec 2014 GXP FOR IMPS -SOME SPECIFIC ISSUESClinical Trial LogisticsPAREXEL InternationalGXP FOR IMPS -SOME SPECIFIC ISSUESOptimising IMP UtilisationPAREXEL InternationalTHE ROLE OF THE QP IN IMP RELEASEThe QP is responsible for certifying that each batch of IMP has been produced and tested/checked in accordance with:•EU GMPG•The Product Specification File•The IMPD(Investigational Medicinal Product Dossier)or the CTA(Clinical Trial Authorisation)Th P d t S ifi ti Fil(di A t th EU •The Product Specification File(according Annex13to the EU-GMP Guide)THE ROLE OF THE QP IN IMP RELEASEThe IMP QP is accountable from manufacture all the way to the patient by:•assessing GMP issuesG•participating in inspections and audits at sites involved in the manufacturing and distribution of IMPs•being a reliable contact for the health authorities•knowing applicable legislation and processes including k i li bl l i l ti d i l di exceptions which may impact the quality and safety of the IMP •being involved in complaint handling and recall processes 2014QP RELEASE -ABOUT THE QPThere is very little harmonisation in Europe regarding the prerequisites for becoming a QPE.g.in the UK a biologist,a chemist or a pharmacist will be eligible to become a QP if they have chartered status with the respective Royal Societyg y Q p y yE.g.in Italy the QP must be employed by the MAIHE.g.in Germany the QP must have police clearance certificationQP RELEASE -THERE IS A PRICE TO PAYWentworth Pharmaceutical QP Survey2015THE BASIS OF THE QP DECLARATIONFor human and veterinary medicinal products,the QP declaration should be based upon an audit of the active substance manufacturers.It is established good practice that the audit should be conducted at the manufacturing site i.e.an on-site auditAudits should be by or on behalf of the Manufacturing Importation Authorisation Holder(MIAH),by suitably trained p p(),y p y and experienced person(s),who may be a third party contractorThe audit cannot be replaced by GMP certificates from a relevant competent authorityQP RELEASE -TEMPLATES FOR API AND IMPEMA Guidance for the template for the qualified person’s declaration concerning GMP compliance of active substance manufacture The QP declaration template2014“The template”-/pmnkfggThe template-/o63p7ogp q p g Template for the qualified person’s declaration concerning GMP compliance of investigational medicinal products manufactured in non-EU countries-/pnh4m68QP RELEASE -TEMPLATES FOR BATCH RELEASE Internationally harmonised requirements for batch certification -/posc88wThis certificate may also be used for active pharmaceutical ingredients and investigational medicinal products used in clinical trial authorisations.t i l th i tiIMP RELEASE -TWO-STEP PROCESSStep1Certification by the QPStep2Release following fulfilment of the requirements of Article9(Commencement of a clinical trial)of Directive(C f)f2001/20/EC-i.e.the clinical trial must be authorisedQP RELEASE -SOME SPECIFIC ISSUESQ:How do you perform batch record review of batches produced in China,when they are not bilingual or translated? How about the following options:f•Not required•Only in audits•Translation of one example•Full translation of every batch•Only summaryQP RELEASE -SOME SPECIFIC ISSUESQ:How do you perform batch record review of batches produced in China,when they are not bilingual or translated?fA:A translation of one batch record as an example and a summary of each batch would be required together with a CoA.It is important that the QP can understand the process and whether there were any excursions,CAPAs,changes,etc., and what they were and how they were concludedPlease note:The QP has to release each individual batchQP RELEASE -SOME SPECIFIC ISSUESQ:Can a member state can invalidate a procedure because a GMP Certificate was not provided for an API for which the QP declaration was provided?A:No.A satisfactory QP Declaration is always necessary and is normally sufficient to confirm that the manufacture of active pharmaceutical ingredients(APIs)comply with Good Manufacturing Practice(GMP),as required by Article8g p()Paragraph3(ha)of Directive2001/83/EC-http://tinyurl com/p7uecnqCMDh/268/2012/p7uecnqGXP FOR IMPS -INDUSTRY EXPERIENCEThere will be a shortage of QPs as many QPs are due to retire in the coming yearsf Q For logistical reasons it is often practical to use the QP services of a Contract Research Organisation(CRO)often rely on third party audit reports,provided they are QPs reportsprepared by suitably qualified auditorsy Q p It may be advisable to have a second QP named as a back-up optionSUMMARY AND OUTLOOK¾The level of GMP applied to IMP manufacture has to increase depending on the clinical phase.Rationales for (necessary)changes need to be documented¾IMPs require a QP release in Europe¾specific requirements for QPs differ in each EU Themember state¾IMP manufacture,IMP supply and clinical trial management,pp y g are intricately linked-working with a suitably qualified and experienced third party may be advisableYOUR PRESENTERSiegfried Schmitt, PhD FRSC CChem CSciPrincipal ConsultantPAREXEL International+44 7824 592401siegfried.schmitt@。
Guidelines related to the Pressure Equipment Directive2014/68/EU (PED)In order to ensure a coherent application of the Pressure Equipment Directive 2014/68/EU (replacing the Directive 97/23/EC (PED) as of 19 July 2016), Guidelines are developed and agreed by the Commission's Working Group "Pressure" (WGP).This working group is composed of representatives of Member States, European federations, the Notified Bodies Forum and CEN and chaired by a representative of the Commission services. The PED Guidelines developed for Directive 97/23/EC will systematically be reviewed and possibly issued as a PED Guideline under the new Directive 2014/68/EU. Also new Guidelines may be issued to support the implementation of the Directive. This work is in progress and the new or updated Guidelines will be made available as soon as they are endorsed by the Working Group "Pressure" (WGP).Remarks or questions concerning this document should be addressed via the email to the unit in the European Commission dealing with the Pressure Equipment Directive:GROW-PRESSURE-EQUIPMENT@ec.europa.euStatus of the guidelinesThe PED Guidelines are not a legally binding interpretation of the Directive. The legally binding text remains that of the Directive. However, the PED Guidelines represent a reference for ensuring consistent application of the Directive. They represent, unless indicated differently in the respective guideline text, the unanimous opinion of the Member States.Classification of the guidelinesThe guidelines carry a x/yy type identification- (x) relates to the subject (A, B, C etc…),- the second (yy) is a sequential numbering.Remark: To facilitate the transition to the new Guidelines the sequential number is maintained as far as possible (e.g. Guideline A-24 under the new PED 2014/68/EU corresponds to Guideline 1-24 under PED 97/23/EC)The letter x refers to one of the following subjects:A.S COPE AND EXCLUSIONS OF THE DIRECTIVEB.C LASSIFICATION AND CATEGORIESC.A SSEMBLIESD.E VALUATION ASSESSMENT PROCEDURESE.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ON DESIGNF.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ONMANUFACTURINGG.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ON MATERIALSH.I NTERPRETATION OF OTHER ESSENTIAL SAFETY REQUIREMENTSI.M ISCELLANEOUSJ.G ENERAL-HORIZONTAL QUESTIONSDocument historyVersion Date Comment1.0 31/3/2015 Includes PED 2014-68-EU Guidelines from the WGP meeting of 11/03/2015A.S COPE AND EXCLUSIONS OF THE DIRECTIVEGuideline A-24Guideline related to: Article 2, point (12)Question According to the definition of Article 2 point 12 fluids may contain a suspension of solids.Is a system of solid pieces or liquid drops distributed in a gas still a fluidin the sense of the PED?Answer YesReasonNote 1 A gas containing pieces of solids or drops of liquid is also to be considered a fluid.suspensionKeywords fluid,Accepted by Working Party Guidelines (WPG) on: 28/11/2014Accepted by Working Group Pressure (WGP) on: 11/03/2015Guideline A–54Guideline related to: Article 1 paragraph 2(s)Question How to understand the exclusion in PED Article 1 paragraph 2 (s) related to equipment covered by the regulations on transport of dangerousgoods?Answer This exclusion shall be read in the context of the scope of PED which applies to the design, manufacture and conformity assessment of pressure equipment.The exclusion only applies when the listed regulations on transport ofdangerous goods includes construction and conformity assessmentrequirements for the equipment concerned.ReasonNote In the context of the listed regulations on transport of dangerous goods, the term construction traditionally refers to design and manufacture.KeywordsAccepted by Working Party Guidelines (WPG) on: 28/11/2014Accepted by Working Group Pressure (WGP) on: 11/03/2015B.C LASSIFICATION AND CATEGORIESGuideline B-21Guideline related to: Annex I sections 2.2.1 and 2.3,Annex II Table 1 Annex IITable 6Question How does one define an unstable gas as referred to in Tables 1 & 6 of Annex II of PED?Answer An unstable gas in this context is a gas liable to transform itselfspontaneously, producing a sudden pressure increase.Such transformation as an example can result from a relatively small variationof an operating parameter (e.g. pressure, temperature, presence of catalysingmaterial) in a confined volume.This includes gases that are classified as chemically unstable gases accordingto CLP Regulation (EC) No 1272/2008 as amended.Typical examples of unstable gases: acetylene (UN 1001), methyl acetylene(UN 1060), vinylfluoride (UN 1860), ozone and dinitrogen oxide (UN 1067).For further examples, see Table 35.1 of the UN Manual of Tests and Criteria. NotegasKeywords UnstableAccepted by Working Party Guidelines (WPG) on: 12/02/2015Accepted by Working Group Pressure (WGP) on: 11/03/2015Guideline B-41Guideline related to: Article 13 QuestionWhere to find additional information on classification of fluids based on PED Article 13 as of 1 June 2015?AnswerAs of 1 June 2015 classification of fluids is based on article 13 of PED 2014/68/EU. Article 13 paragraph 1 (a) lists the physical and health hazard classes and categories for substances and mixtures included in Group 1. The classification is based on the CLP Regulation (EC) No 1272/2008. The table below provides an overview of the hazard classes and categories and the corresponding hazard statements according the CLP Regulation including references to the criteria and label elements in the CLP Regulation.CLP hazard classes and categories (as listed in article 13 of PED) Criteria according to Annex I to CLP Hazard statementaccording toCLP Label elementsaccording to Annex I to CLP(i) unstable explosives or explosives of Divisions 1.1, 1.2, 1.3, 1.4 and 1.5; Section 2.1.2 H200, H201, H202, H203, H204, H205 Table 2.1.2 (ii) flammable gases, category 1 and 2; Section 2.2.2 H220, H221 Table 2.2.3 (iii) oxidising gases, category 1;Section 2.4.2H270 Table 2.4.2 (iv) flammable liquids, category 1 and 2;Section 2.6.2 H224, H225 Table 2.6.2 (v) flammable liquids, category 3 where the maximum allowable temperature is above the flashpoint;Section 2.6.2 H226Table 2.6.2(vi) flammable solids, category 1 and 2; Section 2.7.2 H228 Table 2.7.2(vii) self-reactive substances and mixtures, type A to F;Section 2.8.2 H240, H241, H242 Table 2.8.1 (viii) pyrophoric liquids, category 1; Section 2.9.2 H250 Table 2.9.2 (ix) pyrophoric solids, category 1;Section 2.10.2H250 Table 2.10.2 (x) substances and mixtures which in contact with water emit flammable gases, category 1,2 and 3;Section 2.12.2 H260, H261Table 2.12.2(xi) oxidising liquids, category 1, 2 andSection 2.13.2 H271, H272 Table 2.13.2 3;(xii) oxidising solids, category 1, 2 andSection 2.14.2 H271, H272 Table 2.14.2 3;(xiii) organic peroxides types A to F; Section 2.15.2 H240, H241,Table 2.15.1H242Table 3.1.1 H300 Table 3.1.3 (xiv) acute oral toxicity, category 1 and2;(xv) acute dermal toxicity, category 1Table 3.1.1 H310 Table 3.1.3 and 2;Table 3.1.1 H330, H331 Table 3.1.3 (xvi) acute inhalation toxicity, category1, 2 and 3;(xvii) specific target organ toxicity –Table 3.8.1 H370 Table 3.8.4 single exposure, category 1.Note 1 Article 13 paragraph 1 (a) also states that "Group 1 comprises alsosubstances and mixtures contained in pressure equipment with amaximum allowable temperature which exceeds the flash point of thefluid". The purpose of this provision is to ensure that the flammabilityhazard is properly addressed for those substances and mixtures which arenot classified as flammable under the CLP Regulation (based on thetemperature criteria of the CLP Regulation) but which are presenting thishazard due to the maximum allowable temperature (TS).For example, Heat transfer oils are not classified as flammable liquidsaccording to the CLP Regulation because their flashpoint is above 60 °C(see CLP Regulation Annex I, Table 2.6.1 in Section 2.6 FlammableLiquids, 2.6.2 Classification criteria). However, if the maximumallowable temperature (TS) is above the flashpoint, the hazard of heattransfer oil corresponds to a Group 1 fluid.Note 2 Please note that the CLP Regulation is subject to adaptations to technical progress and therefore the information in the table above should bechecked with the version of the CLP Regulation in force at the time theequipment is placed on the market.Note 3 For questions related to the CLP Regulation please consult your national CLP-helpdesks. Further information on the CLP Regulation can be foundon the European Chemicals Agency (ECHA) website:www.echa.europa.eu. On the ECHA website there is also a list with thecontact details of all national CLP-helpdesks.fluids, classificationKeywords CLPRegulation,Accepted by Working Party Guidelines (WPG) on: 12/02/2015Accepted by Working Group Pressure (WGP) on: 11/03/2015C.A SSEMBLIESD.E VALUATION ASSESSMENT PROCEDURESE.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ON DESIGNF.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ONMANUFACTURINGG.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ON MATERIALSH.I NTERPRETATION OF OTHER ESSENTIAL SAFETY REQUIREMENTSI.M ISCELLANEOUSJ.G ENERAL-HORIZONTAL QUESTIONS。
EUROPEAN COMMISSIONHEALTH AND CONSUMERS DIRECTORATE-GENERALPublic Health and Risk AssessmentPharmaceuticalsBrussels,SANCO/C8/AM/sl/ares(2010)1064599EudraLexThe Rules Governing Medicinal Products in the European UnionVolume 4Good Manufacturing PracticeMedicinal Products for Human and Veterinary UseAnnex 11: Computerised SystemsLegal basis for publishing the detailed guidelines: Article 47 of Directive 2001/83/EC on the Community code relating to medicinal products for human use and Article 51 of Directive 2001/82/EC on the Community code relating to veterinary medicinal products. This document provides guidance for the interpretation of the principles and guidelines of good manufacturing practice (GMP) for medicinal products as laid down in Directive 2003/94/EC for medicinal products for human use and Directive 91/412/EEC for veterinary use.Status of the document: revision 1Reasons for changes: t he Annex has been revised in response to the increased use of computerised systems and the increased complexity of these systems. Consequential amendments are also proposed for Chapter 4 of the GMP Guide.Deadline for coming into operation: 30 June 2011Commission Européenne, B-1049 Bruxelles / Europese Commissie,B-1049 Brussel - BelgiumTelephone: (32-2) 299 11 11PrincipleThis annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill certain functionalities.The application should be validated; IT infrastructure should be qualified.Where a computerised system replaces a manual operation, there should be no resultant decrease in product quality, process control or quality assurance. There should be no increase in the overall risk of the process.General1. Risk ManagementRisk management should be applied throughout the lifecycle of the computerised system taking into account patient safety, data integrity and product quality. As part of a risk management system, decisions on the extent of validation and data integrity controls should be based on a justified and documented risk assessment of the computerised system.2. PersonnelThere should be close cooperation between all relevant personnel such as Process Owner, System Owner, Qualified Persons and IT. All personnel should have appropriate qualifications, level of access and defined responsibilities to carry out their assigned duties. 3. Suppliers and Service Providers3.1 When third parties (e.g. suppliers, service providers) are used e.g. to provide, install, configure, integrate, validate, maintain (e.g. via remote access), modify or retain a computerised system or related service or for data processing, formal agreements must exist between the manufacturer and any third parties, and these agreements should include clear statements of the responsibilities of the third party. IT-departments should be considered analogous.3.2 The competence and reliability of a supplier are key factors when selecting a product or service provider. The need for an audit should be based on a risk assessment.3.3 Documentation supplied with commercial off-the-shelf products should be reviewed by regulated users to check that user requirements are fulfilled.3.4 Quality system and audit information relating to suppliers or developers of software and implemented systems should be made available to inspectors on request.Project Phase4. Validation4.1 The validation documentation and reports should cover the relevant steps of the life cycle. Manufacturers should be able to justify their standards, protocols, acceptance criteria, procedures and records based on their risk assessment.4.2 Validation documentation should include change control records (if applicable) and reports on any deviations observed during the validation process.4.3 An up to date listing of all relevant systems and their GMP functionality (inventory) should be available.For critical systems an up to date system description detailing the physical and logical arrangements, data flows and interfaces with other systems or processes, any hardware and software pre-requisites, and security measures should be available.4.4 User Requirements Specifications should describe the required functions of the computerised system and be based on documented risk assessment and GMP impact. User requirements should be traceable throughout the life-cycle.4.5 The regulated user should take all reasonable steps, to ensure that the system has been developed in accordance with an appropriate quality management system. The supplier should be assessed appropriately.4.6 For the validation of bespoke or customised computerised systems there should be a process in place that ensures the formal assessment and reporting of quality and performance measures for all the life-cycle stages of the system.4.7 Evidence of appropriate test methods and test scenarios should be demonstrated. Particularly, system (process) parameter limits, data limits and error handling should be considered. Automated testing tools and test environments should have documented assessments for their adequacy.4.8 If data are transferred to another data format or system, validation should include checks that data are not altered in value and/or meaning during this migration process.Operational Phase5. DataComputerised systems exchanging data electronically with other systems should include appropriate built-in checks for the correct and secure entry and processing of data, in order to minimize the risks.6. Accuracy ChecksFor critical data entered manually, there should be an additional check on the accuracy of the data. This check may be done by a second operator or by validated electronic means. The criticality and the potential consequences of erroneous or incorrectly entered data to a system should be covered by risk management.7. Data Storage7.1 Data should be secured by both physical and electronic means against damage. Stored data should be checked for accessibility, readability and accuracy. Access to data should be ensured throughout the retention period.7.2 Regular back-ups of all relevant data should be done. Integrity and accuracy of back-up data and the ability to restore the data should be checked during validation and monitored periodically.8. Printouts8.1 It should be possible to obtain clear printed copies of electronically stored data.8.2 For records supporting batch release it should be possible to generate printouts indicating if any of the data has been changed since the original entry.9. Audit TrailsConsideration should be given, based on a risk assessment, to building into the system the creation of a record of all GMP-relevant changes and deletions (a system generated "audit trail"). For change or deletion of GMP-relevant data the reason should be documented. Audit trails need to be available and convertible to a generally intelligible form and regularly reviewed.10. Change and Configuration ManagementAny changes to a computerised system including system configurations should only be made in a controlled manner in accordance with a defined procedure.11. Periodic evaluationComputerised systems should be periodically evaluated to confirm that they remain in a valid state and are compliant with GMP. Such evaluations should include, where appropriate, the current range of functionality, deviation records, incidents, problems, upgrade history, performance, reliability, security and validation status reports.12. Security12.1 Physical and/or logical controls should be in place to restrict access to computerised system to authorised persons. Suitable methods of preventing unauthorised entry to the system may include the use of keys, pass cards, personal codes with passwords, biometrics, restricted access to computer equipment and data storage areas.12.2 The extent of security controls depends on the criticality of the computerised system. 12.3 Creation, change, and cancellation of access authorisations should be recorded.12.4 Management systems for data and for documents should be designed to record the identity of operators entering, changing, confirming or deleting data including date and time.13. Incident ManagementAll incidents, not only system failures and data errors, should be reported and assessed. The root cause of a critical incident should be identified and should form the basis of corrective and preventive actions.14. Electronic SignatureElectronic records may be signed electronically. Electronic signatures are expected to:a. have the same impact as hand-written signatures within the boundaries of thecompany,b. be permanently linked to their respective record,c. include the time and date that they were applied.15. Batch releaseWhen a computerised system is used for recording certification and batch release, the system should allow only Qualified Persons to certify the release of the batches and it should clearly identify and record the person releasing or certifying the batches. This should be performed using an electronic signature.16. Business ContinuityFor the availability of computerised systems supporting critical processes, provisions should be made to ensure continuity of support for those processes in the event of a system breakdown (e.g. a manual or alternative system). The time required to bring the alternative arrangements into use should be based on risk and appropriate for a particular system and the business process it supports. These arrangements should be adequately documented and tested.17. ArchivingData may be archived. This data should be checked for accessibility, readability and integrity. If relevant changes are to be made to the system (e.g. computer equipment or programs), then the ability to retrieve the data should be ensured and tested.GlossaryApplication: Software installed on a defined platform/hardware providing specific functionalityBespoke/Customized computerised system: A computerised system individually designed to suit a specific business processCommercial of the shelf software: Software commercially available, whose fitness for use is demonstrated by a broad spectrum of users.IT Infrastructure: The hardware and software such as networking software and operation systems, which makes it possible for the application to function.Life cycle: All phases in the life of the system from initial requirements until retirement including design, specification, programming, testing, installation, operation, and maintenance.Process owner: The person responsible for the business process.System owner: The person responsible for the availability, and maintenance of a computerised system and for the security of the data residing on that system.Third Party: Parties not directly managed by the holder of the manufacturing and/or import authorisation.。
最新IEC标准--EN IEC62776双端LED灯管安全要求.pdf的翻译®注册商标国际电工委员会34A/1501/NP新工作项目提案投保人IEC SC 34A日期的建议2011-07-05TC / SC34A秘书处BSI英国流通日期2011-07-22截止日期为投票2011-10-28一个新的工作项目范围内现有的技术委员会或小组委员会的建议应提交中央办公室。
建议将被分配到P-成员的技术委员会或小组委员会的表决它变成了工作方案,并介绍O-成员的信息。
投保人可能是一个全国委员会IEC,秘书处本身,另一个技术委员会或小组委员会会议上,联络,组织,标准化管理委员会或咨询委员会,或总书记。
为提出和证明一个新的准则工作项目在ISO / IEC导则第1部分,附录C(见提取物背页),这种形式是不被用于修订或修改现有的出版物。
“建议(投保人可完成)标题的建议普通照明用双端LED灯-安全规范。
标准技术规格范围(ISO / IEC导则第2部分,第6.2.1节中定义的)本标准规定了的安全性和互换性的要求,交换操作的测试方法和必要条件,以表明符合双端帽G5和G13的LED灯,用于更换荧光灯同样的帽子,其额定功率高达60 W,额定电压高达250 V。
目的和理由,包括市场的相关性,它是否是一个建议的水平标准(指南108)1)和关系到安全(指南104),EMC(指南107),环境因素(指南109)和质量保证(指南102)。
(作为附件附加一个单独的页面,如果有必要)1)其他TC / SC的要求表明他们的兴趣,如果有的话,这NP TC / SC书记的双端荧光灯在办公照明,路灯照明,安装在大容量工业照明等等。
双端LED灯的目的是作为一个可能的G5-G13-皑皑的荧光灯的替代品。
本标准的保障措施的变化从荧光灯,LED灯和落后的变化,从LED灯,荧光灯进行了安全LED灯,并在指定的交换条件。
目标日期第一张CD-IS / TS2012年10月会议的估计数2会议次数:2每年第一次会议的日期和地点:2011年10月,代尔夫特/ NL建议工作方法电子邮件协作工具要考虑的相关文件IEC 62560,IEC 61199其他国际机构的活动项目之间的关系联络组织需要在ISO或IEC的协调®版权所有©2011,IEC国际电工委员会。
ANNEX I‘A NNEX14INTRODUCTORY NOTES TO THE LIST IN ANNEX15Note1:The list sets out the conditions required for all products to be considered as sufficiently worked or processed within the meaning of Articles69and100.Note2:2.1.The first two columns in the list describe the product obtained.The first column gives the heading number orchapter number used in the Harmonised System and the second column gives the description of goods used in that system for that heading or chapter.For each entry in the first two columns,a rule is specified in column3 or4.Where,in some cases,the entry in the first column is preceded by an“ex”,this signifies that the rules in column3or4apply only to the part of that heading as described in column2.2.2.Where several heading numbers are grouped together in column1or a chapter number is given and thedescription of products in column2is therefore given in general terms,the adjacent rules in column3or4 apply to all products which,under the Harmonised System,are classified in headings of the chapter or in any of the headings grouped together in column1.2.3.Where there are different rules in the list applying to different products within a heading,each indent containsthe description of that part of the heading covered by the adjacent rules in column3or4.2.4.Where,for an entry in the first two columns,a rule is specified in both columns3and4,the exporter may opt,as an alternative,to apply either the rule set out in column3or that set out in column4.If no origin rule is given in column4,the rule set out in column3is to be applied.Note3:3.1.The provisions of Articles69and100,concerning products having acquired originating status which are used inthe manufacture of other products,shall apply,regardless of whether this status has been acquired inside the factory where these products are used or in another factory in the beneficiary country or republic or in the Community.Example:An engine of heading8407,for which the rule states that the value of the non originating materials which may be incorporated may not exceed40%of the ex works price,is made from“other alloy steel roughly shaped by forging”of heading ex7224.If this forging has been forged in the beneficiary country or republic from a non originating ingot,it has already acquired originating status by virtue of the rule for heading ex7224in the list.The forging can then count as originating in the value-calculation for the engine,regardless of whether it was produced in the same factory or in another factory in the beneficiary country or republic.The value of the non-originating ingot is thus not taken into account when adding up the value of the non-originating materials used.3.2.The rule in the list represents the minimum amount of working or processing required,and the carrying-out ofmore working or processing also confers originating status;conversely,the carrying-out of less working or processing cannot confer originating status.Thus,if a rule provides that non-originating material,at a certain level of manufacture,may be used,the use of such material at an earlier stage of manufacture is allowed,and the3.3.Without prejudice to Note3.2,where a rule uses the expression“Manufacture from materials of any heading”,then materials of any heading(s)(even materials of the same description and heading as the product)may be used,subject,however,to any specific limitations which may also be contained in the rule.However,the expression“Manufacture from materials of any heading,including other materials of heading...”or “Manufacture from materials of any heading,including other materials of the same heading as the product”means that materials of any heading(s)may be used,except those of the same description as the product as given in column2of the list.3.4.When a rule in the list specifies that a product may be manufactured from more than one material,this meansthat one or more materials may be used.It does not require that all be used.Example:The rule for fabrics of headings5208to5212provides that natural fibres may be used and that chemical materials,among other materials,may also be used.This does not mean that both have to be used;it is possible to use one or the other,or both.3.5.Where a rule in the list specifies that a product must be manufactured from a particular material,the conditionobviously does not prevent the use of other materials which,because of their inherent nature,cannot satisfy the rule.(See also Note6.2below in relation to textiles.)Example:The rule for prepared foods of heading1904,which specifically excludes the use of cereals and their derivatives, does not prevent the use of mineral salts,chemicals and other additives which are not products from cereals.However,this does not apply to products which,although they cannot be manufactured from the particular materials specified in the list,can be produced from a material of the same nature at an earlier stage of manufacture.Example:In the case of an article of apparel of ex Chapter62made from non-woven materials,if the use of only non-originating yarn is allowed for this class of article,it is not possible to start from non-woven cloth–even if non-woven cloths cannot normally be made from yarn.In such cases,the starting material would normally be at the stage before yarn–that is,the fibre stage.3.6.Where,in a rule in the list,two percentages are given for the maximum value of non originating materials thatcan be used,then these percentages may not be added together.In other words,the maximum value of all the non-originating materials used may never exceed the higher of the percentages given.Furthermore,the individual percentages must not be exceeded,in relation to the particular materials to which they apply.Note4:4.1.The term“natural fibres”is used in the list to refer to fibres other than artificial or synthetic fibres.It is restrictedto the stages before spinning takes place,including waste,and,unless otherwise specified,includes fibres which have been carded,combed or otherwise processed,but not spun.4.2.The term“natural fibres”includes horsehair of heading0503,silk of headings5002and5003,as well aswool-fibres and fine or coarse animal hair of headings5101to5105,cotton fibres of headings5201to5203,4.3.The terms“textile pulp”,“chemical materials”and“paper-making materials”are used in the list to describe thematerials,not classified in Chapters50to63,which can be used to manufacture artificial,synthetic or paper fibres or yarns.4.4.The term“man-made staple fibres”is used in the list to refer to synthetic or artificial filament tow,staple fibresor waste,of headings5501to5507.Note5:5.1.Where,for a given product in the list,reference is made to this Note,the conditions set out in column3shallnot be applied to any basic textile materials used in the manufacture of this product and which,taken together, represent10%or less of the total weight of all the basic textile materials used.(See also Notes5.3and5.4below.)5.2.However,the tolerance mentioned in Note5.1may be applied only to mixed products which have been madefrom two or more basic textile materials.The following are the basic textile materials:—silk;—wool;—coarse animal hair;—fine animal hair;—horsehair;—cotton;—paper-making materials and paper;—flax;—true hemp;—jute and other textile bast fibres;—sisal and other textile fibres of the genus Agave;—coconut,abaca,ramie and other vegetable textile fibres;—synthetic man-made filaments;—artificial man-made filaments;—current-conducting filaments;—synthetic man-made staple fibres of polypropylene;—synthetic man-made staple fibres of polyester;—synthetic man-made staple fibres of polyacrylonitrile;—synthetic man-made staple fibres of polyimide;—synthetic man-made staple fibres of polytetrafluoroethylene;—synthetic man-made staple fibres of poly(phenylene sulphide);—synthetic man-made staple fibres of poly(vinyl chloride);—other synthetic man-made staple fibres;—artificial man-made staple fibres of viscose;—other artificial man-made staple fibres;—yarn made of polyurethane segmented with flexible segments of polyether,whether or not gimped;—yarn made of polyurethane segmented with flexible segments of polyester,whether or not gimped;—products of heading5605(metallised yarn)incorporating strip consisting of a core of aluminium foil or of a core of plastic film whether or not coated with aluminium powder,of a width not exceeding5mm, sandwiched by means of a transparent or coloured adhesive between two layers of plastic film;—other products of heading5605.Example:A yarn,of heading5205,made from cotton fibres of heading5203and synthetic staple fibres of heading5506,is a mixed yarn.Therefore,non-originating synthetic staple fibres which do not satisfy the origin-rules(which require manufacture from chemical materials or textile pulp)may be used,provided that their total weight does not exceed10%of the weight of the yarn.Example:A woollen fabric,of heading5112,made from woollen yarn of heading5107and synthetic yarn of staple fibresof heading5509,is a mixed fabric.Therefore,synthetic yarn which does not satisfy the origin-rules(which require manufacture from chemical materials or textile pulp),or woollen yarn which does not satisfy the origin-rules(which require manufacture from natural fibres,not carded or combed or otherwise prepared for spinning),or a combination of the two,may be used,provided that their total weight does not exceed10%of the weight of the fabric.Example:Tufted textile fabric,of heading5802,made from cotton yarn of heading5205and cotton fabric of heading5210,is a only mixed product if the cotton fabric is itself a mixed fabric made from yarns classified in two separate headings,or if the cotton yarns used are themselves mixtures.Example:If the tufted textile fabric concerned had been made from cotton yarn of heading5205and synthetic fabric of heading5407,then,obviously,the yarns used are two separate basic textile materials and the tufted textile fabric is,accordingly,a mixed product.5.3.In the case of products incorporating“yarn made of polyurethane segmented with flexible segments of polyether,5.4.In the case of products incorporating“strip consisting of a core of aluminium foil or of a core of plastic filmwhether or not coated with aluminium powder,of a width not exceeding5mm,sandwiched by means of a transparent or coloured adhesive between two layers of plastic film”,this tolerance is30%in respect of this strip. Note6:6.1.Where,in the list,reference is made to this Note,textile materials(with the exception of linings and interlinings),which do not satisfy the rule set out in the list in column3for the made-up product concerned,may be used, provided that they are classified in a heading other than that of the product and that their value does not exceed 8%of the ex-works price of the product.6.2.Without prejudice to Note6.3,materials,which are not classified within Chapters50to63,may be used freelyin the manufacture of textile products,whether or not they contain textiles.Example:If a rule in the list provides that,for a particular textile item(such as trousers),yarn must be used,this does not prevent the use of metal items,such as buttons,because buttons are not classified within Chapters50to63.For the same reason,it does not prevent the use of slide-fasteners,even though slide-fasteners normally contain textiles.6.3.Where a percentage-rule applies,the value of materials which are not classified within Chapters50to63must betaken into account when calculating the value of the non-originating materials incorporated.Note7:7.1.For the purposes of headings ex2707,2713to2715,ex2901,ex2902and ex3403,the“specific processes”arethe following:(a)vacuum-distillation;(b)redistillation by a very thorough fractionation-process(1);(c)cracking;(d)reforming;(e)extraction by means of selective solvents;(f)the process comprising all of the following operations:processing with concentrated sulphuric acid,oleumor sulphuric anhydride;neutralisation with alkaline agents;decolourisation and purification with naturally-active earth,activated earth,activated charcoal or bauxite;(g)polymerisation;(h)alkylation;(i)isomerisation.7.2.For the purposes of headings2710,2711and2712,the“specific processes”are the following:(a)vacuum-distillation;(b)redistillation by a very thorough fractionation-process(1);(c)cracking;(d)reforming;(e)extraction by means of selective solvents;(f)the process comprising all of the following operations:processing with concentrated sulphuric acid,oleumor sulphuric anhydride;neutralisation with alkaline agents;decolourisation and purification with naturally-active earth,activated earth,activated charcoal or bauxite;(g)polymerisation;(h)alkylation;(ij)isomerisation;(k)in respect of heavy oils of heading ex2710only,desulphurisation with hydrogen,resulting in a reduction of at least85%of the sulphur-content of the products processed(ASTM D1266-59T method);(l)in respect of products of heading2710only,deparaffining by a process other than filtering;(m)in respect of heavy oils of heading ex2710only,treatment with hydrogen,at a pressure of more than20bar and a temperature of more than250°C,with the use of a catalyst,other than to effect desulphurisation, when the hydrogen constitutes an active element in a chemical reaction.The further treatment,with hydrogen,of lubricating oils of heading ex2710(e.g.hydrofinishing or decolourisation),in order,more especially,to improve colour or stability shall not,however,be deemed to be a specific process;(n)in respect of fuel oils of heading ex2710only,atmospheric distillation,on condition that less than30%of these products distils,by volume,including losses,at300°C,by the ASTM D86method;(o)in respect of heavy oils other than gas oils and fuel oils of heading ex2710only,treatment by means of a high-frequency electrical brush-discharge.(p)in respect of crude products(other than petroleum jelly,ozokerite,lignite wax or peat wax,paraffin wax containing by weight less than0,75%of oil)of heading ex2712only,de-oiling by fractional crystallisation.7.3.For the purposes of headings ex2707,2713to2715,ex2901,ex2902and ex3403,simple operations,such ascleaning,decanting,desalting,water-separation,filtering,colouring,marking,obtaining a sulphur-content as a result of mixing products with different sulphur-contents,or any combination of these operations or like operations,do not confer origin.'ANNEX II‘A NNEX15LIST OF WORKING OR PROCESSING REQUIRED TO BE CARRIED OUT ON NON-ORIGINATING MATERIALS IN ORDER THAT THE PRODUCT MANUFACTURED CAN OBTAIN ORIGINATING STATUSHS heading Description of product Working or processing,carried out on non-originating materials,which confers originating status(1)(2)(3)or(4)Chapter1Live animals All the animals of Chapter1shall bewholly obtainedChapter2Meat and edible meat offal Manufacture in which all the materialsof Chapters1and2used are whollyobtainedChapter3Fish and crustaceans,molluscs and otheraquatic invertebrates Manufacture in which all the materials of Chapter3used are wholly obtainedex Chapter4Dairy produce;birds'eggs;naturalhoney;edible products of animal origin,not elsewhere specified or included;except for:Manufacture in which all the materials of Chapter4used are wholly obtained0403Buttermilk,curdled milk and cream,yoghurt,kephir and other fermented oracidified milk and cream,whether or notconcentrated or containing added sugaror other sweetening matter or flavouredor containing added fruit,nuts or cocoa Manufacture in which:–all the materials of Chapter4used are wholly obtained,–all the fruit juice(except that of pineapple,lime or grapefruit)of heading2009used is originating,and –the value of all the materials of Chapter17used does not exceed30% of the ex-works price of the productex Chapter5Products of animal origin,not elsewherespecified or included;except for:Manufacture in which all the materials of Chapter5used are wholly obtainedex0502Prepared pigs',hogs'or boars'bristlesand hair Cleaning,disinfecting,sorting and straightening of bristles and hairChapter6Live trees and other plants;bulbs,rootsand the like;cut flowers and ornamentalfoliage Manufacture in which:–all the materials of Chapter6used are wholly obtained,and–the value of all the materials used does not exceed50%of the ex-works price of the productChapter7Edible vegetables and certain roots andtubers Manufacture in which all the materials of Chapter7used are wholly obtainedChapter8Edible fruit and nuts;peel of citrus fruitsor melonsManufacture in which:–all the fruit and nuts used are whollyobtained,and–the value of all the materials ofChapter17used does not exceed30%of the value of the ex-works price ofthe productex Chapter9Coffee,tea,matéand spices;except for:Manufacture in which all the materialsof Chapter9used are wholly obtained0901Coffee,whether or not roasted ordecaffeinated;coffee husks and skins;coffee substitutes containing coffee inany proportion Manufacture from materials of any heading0902Tea,whether or not flavoured Manufacture from materials of anyheadingex0910Mixtures of spices Manufacture from materials of anyheadingChapter10Cereals Manufacture in which all the materialsof Chapter10used are wholly obtainedex Chapter11Products of the milling industry;malt;starches;inulin;wheat gluten;except for:Manufacture in which all the cereals, edible vegetables,roots and tubers of heading0714or fruit used are wholly obtainedex1106Flour,meal and powder of the dried,shelled leguminous vegetables ofheading0713Drying and milling of leguminous vegetables of heading0708Chapter12Oil seeds and oleaginous fruits;miscellaneous grains,seeds and fruit;industrial or medicinal plants;straw andfodder Manufacture in which all the materials of Chapter12used are wholly obtained1301Lac;natural gums,resins,gum-resins andoleoresins(for example;balsams)Manufacture in which the value of all the materials of heading1301used does not exceed50%of the ex-works price of the product1302Vegetable saps and extracts;pecticsubstances,pectinates and pectates;agar-agar and other mucilages andthickeners,whether or not modified,derived from vegetable products:–Mucilages and thickeners,modified, derived from vegetable products Manufacture from non-modified mucilages and thickeners–Other Manufacture in which the value of allthe materials used does not exceed50%of the ex-works price of the productChapter14Vegetable plaiting materials;vegetableproducts not elsewhere specified orincluded Manufacture in which all the materials of Chapter14used are wholly obtainedex Chapter15Animal or vegetable fats and oils andtheir cleavage products;prepared ediblefats;animal or vegetable waxes;exceptfor:Manufacture from materials of any heading,except that of the product1501Pig fat(including lard)and poultry fat,other than that of heading0209or1503:–Fats from bones or waste Manufacture from materials of anyheading,except those of heading0203,0206or0207or bones of heading0506–Other Manufacture from meat or edible offal ofswine of heading0203or0206or ofmeat and edible offal of poultry ofheading02071502Fats of bovine animals,sheep or goats,other than those of heading1503–Fats from bones or waste Manufacture from materials of anyheading,except those of heading0201,0202,0204or0206or bones ofheading0506–Other Manufacture in which all the materialsof Chapter2used are wholly obtained1504Fats and oils and their fractions,of fishor marine mammals,whether or notrefined,but not chemically modified:–Solid fractions Manufacture from materials of anyheading,including other materials ofheading1504–Other Manufacture in which all the materialsof Chapters2and3used are whollyobtainedex1505Refined lanolin Manufacture from crude wool grease ofheading15051506Other animal fats and oils and theirfractions,whether or not refined,butnot chemically modified:–Solid fractions Manufacture from materials of anyheading,including other materials ofheading1506–Other Manufacture in which all the materialsof Chapter2used are wholly obtained 1507to1515Vegetable oils and their fractions:–Soya,ground nut,palm,copra,palm kernel,babassu,tung and oiticica oil, myrtle wax and Japan wax,fractions of jojoba oil and oils for technical or industrial uses other than the manufacture of foodstuffs for human consumption Manufacture from materials of any heading,except that of the product–Solid fractions,except for that of jojoba oil Manufacture from other materials of headings1507to1515–Other Manufacture in which all the vegetablematerials used are wholly obtained1516Animal or vegetable fats and oils andtheir fractions,partly or whollyhydrogenated,inter-esterified,re-esterifiedor elaidinised,whether or not refined,but not further prepared Manufacture in which:–all the materials of Chapter2used are wholly obtained,and–all the vegetable materials used are wholly obtained.However,materials of headings1507,1508,1511 and1513may be used1517Margarine;edible mixtures orpreparations of animal or vegetable fatsor oils or of fractions of different fats oroils of this Chapter,other than ediblefats or oils or their fractions ofheading1516Manufacture in which:–all the materials of Chapters2and4 used are wholly obtained,and–all the vegetable materials used are wholly obtained.However,materials of headings1507,1508,1511 and1513may be usedChapter16Preparations of meat,of fish or ofcrustaceans,molluscs or other aquaticinvertebrates Manufacture:–from animals of Chapter1,and/or–in which all the materials of Chapter3 used are wholly obtainedex Chapter17Sugars and sugar confectionery;exceptfor:Manufacture from materials of any heading,except that of the productex1701Cane or beet sugar and chemically puresucrose,in solid form,containing addedflavouring or colouring matter Manufacture in which the value of all the materials of Chapter17used does not exceed30%of the ex-works price of the product1702Other sugars,including chemically purelactose,maltose,glucose and fructose,insolid form;sugar syrups not containingadded flavouring or colouring matter;artificial honey,whether or not mixedwith natural honey;caramel:–Chemically-pure maltose and fructose Manufacture from materials of anyheading,including other materials ofheading1702–Other sugars in solid form,containing added flavouring or colouring matter Manufacture in which the value of all the materials of Chapter17used does not exceed30%of the ex-works price of the product–Other Manufacture in which all the materialsused are originatingex1703Molasses resulting from the extraction orrefining of sugar,containing addedflavouring or colouring matter Manufacture in which the value of all the materials of Chapter17used does not exceed30%of the ex works price1704Sugar confectionery(including whitechocolate),not containing cocoaManufacture:–from materials of any heading,exceptthat of the product,and–in which the value of all the materialsof Chapter17used does not exceed30%of the ex-works price of theproductChapter18Cocoa and cocoa preparations Manufacture:–from materials of any heading,exceptthat of the product,and–in which the value of all the materialsof Chapter17used does not exceed30%of the ex-works price of theproduct1901Malt extract;food preparations of flour,groats,meal,starch or malt extract,notcontaining cocoa or containing less than40%by weight of cocoa calculated on atotally defatted basis,not elsewherespecified or included;food preparationsof goods of headings0401to0404,notcontaining cocoa or containing less than5%by weight of cocoa calculated on atotally defatted basis,not elsewherespecified or included:–Malt extract Manufacture from cereals of Chapter10–Other Manufacture:–from materials of any heading,exceptthat of the product,and–in which the value of the materials ofeach of Chapters4and17used doesnot exceed30%of the ex-works priceof the product1902Pasta,whether or not cooked or stuffed(with meat or other substances)orotherwise prepared,such as spaghetti,macaroni,noodles,lasagne,gnocchi,ravioli,cannelloni;couscous,whether ornot prepared:–Containing20%or less by weight of meat,meat offal,fish,crustaceans or Manufacture in which all the cereals and derivatives(except durum wheat and its1902 (cont'd)–Containing more than20%by weightof meat,meat offal,fish,crustaceans ormolluscsManufacture in which:–all the cereals and their derivatives(except durum wheat and itsderivatives)used are wholly obtained,andall the materials of Chapters2and3used are wholly obtained–all the materials of Chapters2and3used are wholly obtained1903Tapioca and substitutes therefor preparedfrom starch,in the form of flakes,grains,pearls,siftings or similar forms Manufacture from materials of any heading,except potato starch of heading11081904Prepared foods obtained by the swellingor roasting of cereals or cereal products(for example,corn flakes);cereals(otherthan maize(corn))in grain form or inthe form of flakes or other workedgrains(except flour,groats and meal),pre-cooked or otherwise prepared,notelsewhere specified or included Manufacture:–from materials of any heading,except those of heading1806,–in which all the cereals and flour (except durum wheat and Zea indurata maize,and their derivatives)used are wholly obtained,and–in which the value of all the materials of Chapter17used does not exceed 30%of the ex-works price of the product1905Bread,pastry,cakes,biscuits and otherbakers'wares,whether or not containingcocoa;communion wafers,emptycachets of a kind suitable forpharmaceutical use,sealing wafers,ricepaper and similar products Manufacture from materials of any heading,except those of Chapter11ex Chapter20Preparations of vegetables,fruit,nuts orother parts of plants;except for:Manufacture in which all the fruit,nuts or vegetables used are wholly obtainedex2001Yams,sweet potatoes and similar edibleparts of plants containing5%or moreby weight of starch,prepared orpreserved by vinegar or acetic acid Manufacture from materials of any heading,except that of the productex2004and ex2005Potatoes in the form of flour,meal orflakes,prepared or preserved otherwisethan by vinegar or acetic acidManufacture from materials of anyheading,except that of the product2006Vegetables,fruit,nuts,fruit-peel andother parts of plants,preserved by sugar(drained,glacéor crystallised)Manufacture in which the value of all the materials of Chapter17used does not exceed30%of the ex-works price2007Jams,fruit jellies,marmalades,fruit ornut purée and fruit or nut pastes,obtained by cooking,whether or notcontaining added sugar or othersweetening matter Manufacture:–from materials of any heading,except that of the product,and–in which the value of all the materials of Chapter17used does not exceed 30%of the ex-works price of the productex2008–Nuts,not containing added sugar orspirits Manufacture in which the value of all the originating nuts and oil seeds of headings0801,0802and1202to1207 used exceeds60%of the ex-works price of the product–Peanut butter;mixtures based on cereals;palm hearts;maize(corn)Manufacture from materials of any heading,except that of the product–Other except for fruit and nuts cooked otherwise than by steaming or boiling in water,not containing added sugar, frozen Manufacture:–from materials of any heading,except that of the product,and–in which the value of all the materials of Chapter17used does not exceed 30%of the ex-works price of the product2009Fruit juices(including grape must)andvegetable juices,unfermented and notcontaining added spirit,whether or notcontaining added sugar or othersweetening matter Manufacture:–from materials of any heading,except that of the product,and–in which the value of all the materials of Chapter17used does not exceed 30%of the ex-works price of the productex Chapter21Miscellaneous edible preparations,exceptfor:Manufacture from materials of any heading,except that of the product2101Extracts,essences and concentrates,ofcoffee,tea or matéand preparationswith a basis of these products or with abasis of coffee,tea or maté;roastedchicory and other roasted coffeesubstitutes,and extracts,essences and Manufacture:–from materials of any heading,except that of the product,and–in which all the chicory used is。