杂环化合物的反应
- 格式:ppt
- 大小:809.00 KB
- 文档页数:51





有机化学基础知识点杂环化合物的合成与反应有机化学是研究含碳的化合物以及其反应机理的学科。
杂环化合物是其中一类重要的有机化合物,由多个不同的原子构成的环状结构赋予其特殊的性质和活性。
本文将重点介绍杂环化合物的合成与反应。
一、杂环化合物的合成1. 环状结构的直接合成直接合成是指通过无需过多中间步骤,直接将杂环结构形成的方法。
最常见的有两种:环内缩合与环外缩合。
环内缩合是通过分子内的反应实现环状结构的形成。
例如,可以通过两个官能团的内部反应,如酰胺和酰胺之间的内酰胺化反应,形成含有杂环结构的化合物。
环外缩合是通过分子间的反应实现环状结构的形成。
例如,可以通过偶氮化物和亲电试剂的反应,形成含有杂环结构的化合物。
2. 环状结构的间接合成间接合成是指通过多步反应,将不同的官能团转化为杂环结构。
这种方法更加灵活,可以根据具体需求选择不同的反应路径。
常见的方法有:(1) 拉曼反应:通过烷基金属物与芳香酮之间的反应,将芳香酮上的羰基还原成羟基,形成杂环结构。
(2) 脱水环化反应:通过脱水反应形成环状结构。
最常见的是使用酸催化剂将醇或酸上的羟基与相邻的官能团上的氢原子进行消除反应,形成杂环结构。
(3) 杂环化合物的可溶性和稳定性增大,可使用催化剂或光催化反应进行合成。
二、杂环化合物的反应1. 变性反应杂环化合物可以通过一系列的变性反应进行官能团的转换。
例如,通过酸催化或碱催化的酯水解反应,将酯转化为醇或酸;通过羟胺或胺与酸酐或酰氯的反应,形成酰胺或酰脲。
2. 变位反应变位反应是杂环化合物中常见的反应之一,通过杂环结构上的元素进行位置的变化。
例如,通过环内亲电试剂的攻击,实现环内碳-氧的位置变化,形成环内醇或环内醚。
3. 开环反应通过开环反应,可以将杂环化合物打开,形成更加简单的化合物。
其中最常见的是酸性水解和碱性水解反应,将杂环结构上的官能团裂解成独立的官能团。
综上所述,杂环化合物的合成与反应是有机化学中重要的研究领域。
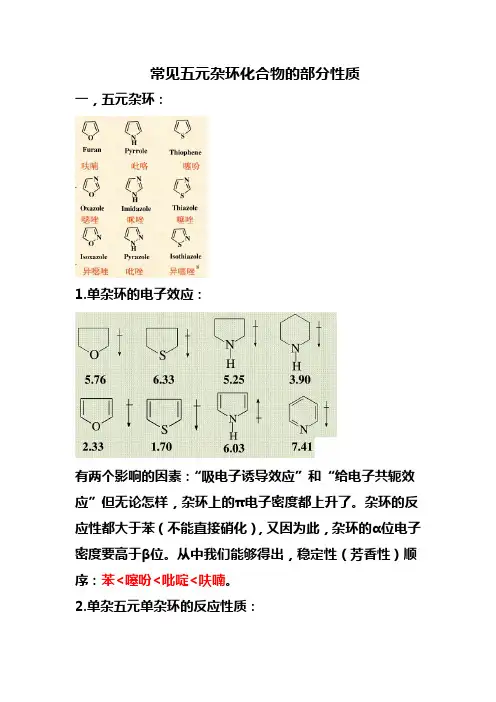
常见五元杂环化合物的部分性质一,五元杂环:1.单杂环的电子效应:有两个影响的因素:“吸电子诱导效应”和“给电子共轭效应”但无论怎样,杂环上的π电子密度都上升了。
杂环的反应性都大于苯(不能直接硝化),又因为此,杂环的α位电子密度要高于β位。
从中我们能够得出,稳定性(芳香性)顺序:苯<噻吩<吡啶<呋喃。
2.单杂五元单杂环的反应性质:加成反应:苯<噻吩<吡啶<呋喃亲电取代:苯<噻吩<呋喃<吡啶(取代考虑的具体因素应该是α位的电子云密度问题,而不是整个环的稳定性。
)·呋喃太容易实行加成,在溴水/甲醇中得到只有用二氧六环溴合物才能得到正常的溴代产物,当然,钝化基团的加入能够使反应变得较为温和。
·与苯炔反应时,呋喃生成D-A产物,而吡咯生成苯炔的加成产物(1-苯基吡咯),噻吩则不能反应。
3.双杂五元单杂环的反应性质:咪唑能够互变,通常4,5位混杂,不过在有基团时并不相等,例如“4(5)-硝基咪唑”绝绝绝大部分都为4位。
咪唑分子间有氢键(20个分子左右),沸点异常地高。
相比之下吡唑一般二聚。
唑环的电子云密度比相对应的单杂环要低,其亲电取代的顺序为:苯>氮杂>硫杂>氧杂其反应时,取代位通常为三级氮的间位。
机理上先是氮的质子化(噻唑能够在较弱条件下硝化,而噻吩不能够)。
弱的亲电试剂不能够和唑环反应,例如F-C。
虽然唑环硝化和磺化时反应活性比苯环低,但是卤化时却比苯环高。
因为存有三级氮,唑环还能够发生亲核取代(在其邻对位)。
4.单杂五元苯并杂环:5.吲哚吲哚合成法:1>Fischer:苯腙+酮其机理如下:2>Reisset:邻硝基甲苯+草酸二乙酯3>Nenitzescu:对苯醌+β-氨基巴豆酸乙酯And more。
吲哚和苯并噻吩亲电取代在3位,而苯并呋喃在二位。
在酸性下吲哚发生质子化,杂环的密度将小于苯环,取代在苯环上实行(硝化则在5位实行)。

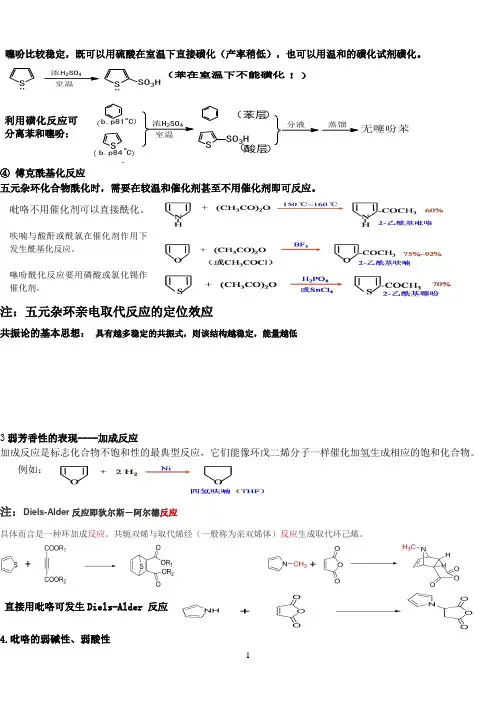
注:五元杂环亲电取代反应的定位效应
Diels-Alder反应即狄尔斯-阿尔德反应直接用吡咯可发生Diels-Alder 反应
氮上孤电子对参与环内共轭,减弱了对H+吸引能力。
氮上电子云少,H 易离解,显一定的酸性。
吡咯表面上是个仲胺,但实际上吡咯是一个很弱的碱,碱性比苯胺弱得多,它只能慢慢地溶解在冷的稀酸溶液中。
三五元杂环化合物
1呋喃、噻吩、吡咯的结构与芳香性
呋喃、噻吩、吡咯都是平面结构,环上所有原子都是SP2 杂化,各原子均以SP2 杂化轨道重叠形成σ键。
碳未杂化的P 轨道中有一个电子,杂原子的P 轨道中有一对电子,P 轨道互相平行重叠,形成闭合的共轭体系。
二几个常
见的杂环
1.吡咯`呋喃`
噻吩均具有
芳香性
②硝化反应
五元杂环化合物与硝酸(强氧化剂)反应,得不到预期产物,而是芳环被破坏。
呋喃, 噻吩和吡咯易氧化, 一
般不用硝酸直接硝化; 通常用比较温和的非质子硝化试剂,如:乙酰基硝酸酯,反应在低温下进行。
③磺化反应
吡咯、呋喃不能直接用硫酸磺化,因为它们在浓硫酸中不稳定,会发生聚合,通常用一种温和的磺化剂——吡。

2005年第25卷有机化学V ol. 25, 2005第6期, 634~640 Chinese Journal of Organic Chemistry No. 6, 634~640ygzhou@*E-mail:Received August 2, 2004; revised October 25, 2004; accepted November 23, 2004.No. 6卢胜梅等:芳香杂环化合物不对称催化氢化反应的研究进展635坏稠环的芳香性比完全破坏单环的芳香性所需能量低. 另外, 芳香杂环化合物的氢化比非芳香杂环化合物容易, 这一方面因为杂原子对所在的环有活化作用; 另一方面, 杂原子上的孤对电子可参与和催化剂的金属原子配位, 使催化活性中心靠近底物从而发生氢化反应. 所以在芳香稠杂环化合物氢化时, 一般都是含杂原子的环被氢化[5].在均相催化体系中, 第一例报道的芳香杂环化合物的氢化是在1987年, Murata 等[8]使用原位产生的(+)-(DIOP)RhH 作催化剂, 乙醇作溶剂, 室温下对2-位取代的喹喔啉1进行不对称氢化(Eq. 1), 反应需36~72 h, 产物2-甲基-1,2,3,4-四氢喹喔啉只有3%的对映选择性(Table 1, Entry 1). 虽然ee 值很低, 但毕竟实现了对芳香杂环化合物均相不对称氢化, 为后来致力于研究芳香杂环化合物不对称氢化的工作者开辟了道路.1998年, Bianchini 研究小组[9]利用邻位金属化铱的二氢复合物fac -exo -(R )-[IrH 2{C 6H 4C*H(Me)N(CH 2CH 2- PPh 2)2}] (L1) 作催化剂, 实现了对2-甲基喹喔啉(1)的高对映选择性氢化, 取得了高达90%的ee 值(Table 1, Entry 2), 但转化率只有54%, 当转化率为97%时, ee 值为73% (Table 1, Entry 3), 反应要在100 ℃进行, 甲醇和异丙醇是最好的溶剂选择. 这是目前对2-甲基喹喔啉氢化取得的最好结果. 同一研究组在2001年又报道了用[(R ,R )-BDPBzPIr(COD)]OTf 和[(R ,R )-BDPBzPRh(NBD)]- OTf 作催化剂, 对2-甲基喹喔啉(1)进行氢化[10], 但ee 值不理想, 分别为23%和11% (Table 1, Entries 4 and 5). 在反应中, 他们发现铑的活性比铱的高, 但对映选择性低.2003年, Henschke 和Casy 等使用Noyori 的RuCl 2-氢化为模型反应, 50 ℃, 3.0 MPa 的氢气压力下, 对一系列的手性双磷配体和手性二氨的组合进行了筛选,结果发现(S )-xyl-hexaPHEMP (L3)和(S ,S )-DACH 的组合取得了较好的结果(73% ee ) (Table 1, Entry 6), 所有反应20 h 内转化率都在94%以上, 且S /C 为1000/1[11]. 该催化体系的活性很好, 但对映选择性只是中等.表1 2-甲基喹喔啉的不对称氢化Table 1 Asymmetric hydrogenation of 2-methylquinoxaline Entry Catalyst Yield/%ee /%1 (+)-(DIOP)RhH 72.0 32 L1 53.7 90a 3L196.5 73b4 [L2Ir(COD)]OTf 40.7 23a5 [L2Rh(NBD)]OTf 93.2 11a6 RuCl 2/L3/(S ,S )-DACH 99.0 73caCH 3OH 作溶剂; b i -PrOH 作溶剂; c t -BuOH 作溶剂.2000年, Ito 等[12]首次报道了对N -Ac 和Boc 保护的2-位取代吲哚进行不对称催化氢化(Eq. 2), 反应在60 ℃下完成, 取得了最高为95%的ee 值. 他们使用的是一个反式鳌合配位的二茂铁双磷配体L4, 金属前体是[Rh(NBD)2]SbF 6. 这一催化体系对2-位取代的N -Ac 保护的吲哚, 无论是收率或对映选择性都取得了令人满意的结果, 碱碳酸铯的加入是取得高对映选择性所必须的. 对N -Boc 保护的吲哚氢化对映选择性不如N -Ac. 但对于3-位取代的N -Ac 保护的吲哚2在上面标准条件下, 反应不能转化完全, 除了所要的氢化产物3外, 还得到了N 上Ac 被脱除的产物4 (Eq. 3).636有 机 化 学 V ol. 25, 2005为了提高3-位取代吲哚类化合物氢化的选择性, 同一研究组又用同一催化体系对3-位取代吲哚的氢化进行了深入研究, 他们考察了用N -Boc, N -Ts, N -Ms, N -Tf 代替N -Ac 对反应的转化率和对映选择性的影响, 结果发现N -Ts 保护的3-位取代吲哚5给出最好的结果(Eq. 4), 其转化率能达到100%, 并且最高能获得98%的ee 值[13].在2003年, 周永贵等[14]首次实现了对2-位取代喹啉的对映选择性氢化(Eq. 5), 他们使用的是[Ir(COD)Cl]2/ L5/I 2/Toluene 的催化体系, 在室温下即可以进行反应, 并取得了最高为96%的ee 值. 这一催化体系对羟基和酯基等官能团无影响, 对3-位或4-位取代的喹啉的氢化活性低, 且产物基本是消旋的. 碘的存在是取得高活性和高对映选择性所必须的, 如果没有碘, 反应不能进行. 利用这一催化氢化的方法学, 可以方便地合成一系列2-位取代的1,2,3,4-四氢喹啉类的天然产物6, 7 [15]和一些药物8的关键中间体.对于稠环其它类型的芳香杂环化合物如异喹啉、苯并呋喃、苯并噻吩等均相的不对称氢化还未见报道. 1.2 芳香单杂环化合物的不对称催化氢化氢化芳香单杂环化合物比氢化稠环的要困难, 因为完全破坏一个单环的芳香性比部分破坏一个稠环的芳香性所需能量更多, 因此, 对单环杂环芳香化合物的不对称氢化更具有挑战性. 文献报道均相体系中氢化芳香单杂环化合物第一例是1997年, Fuchs [16]利用[Rh(NBD)- Cl]2/L*/MeOH 的催化体系对2-位取代的吡嗪羧酸衍生物进行了氢化(Eq. 6), 使用的手性配体是二茂铁衍生的双磷化合物L6, 对N -叔丁基吡嗪酰胺(9a )最高取得了78%的ee 值, 对于简单的2-吡嗪羧酸甲酯(9b )的氢化, 只获得3.6%的ee 值.2000年, Studer [17]尝试对单取代的吡啶17 (Eq. 7)和呋喃18 (Eq. 8)进行不对称氢化.经过一系列的条件优化, 他们发现Rh(NBD)2BF 4为最佳的金属前体, 催化剂用量为5%, 反应在10.0 MPa, 60 ℃下进行, 当用DIOP 作配体时, 2-吡啶甲酸乙酯取得了最高为27%的ee 值(Table 2, Entry 1), 但转化率只有41%; 当BINAP 作配体时, 转化率为100%, 但ee 值只有25% (Table 2, Entry 2). 对3-位取代吡啶的氢化显得更加困难, 收率低, 对映选择性也低. 如3-吡啶甲酸乙酯, 除了所要的完全氢化产物外, 还有较多的部分氢化的产物, ee 值最高仅为17% (Table 2, Entry 6). 对2-呋喃甲醇的氢化虽然收率高, 但产物几乎是消旋的, 最高ee 值为仅7% (Table 3, Entry 1). Rh(NBD)2BF 4/PPF-P(t -Bu)2的组合对2-呋喃甲酸取得了最高为24%的ee 值, 但收率只有3% (Table 3, Entry 4).当Cy 2PF-PCy 2作配体时, 收率为100%, 但ee 值仅为1% (Table 3, Entry 5). 甲醇或乙醇是该类反应的最好溶剂, 反应需在60 ℃, 10 MPa 的氢气压力下进行, 条件比较剧烈, 催化剂用量高, 且对映选择性较低.均相体系中, 对芳香单杂环化合物的不对称氢化的例子不多, 而且结果不好, 这是一个有待于进一步深入探索的研究领域.No. 6卢胜梅等:芳香杂环化合物不对称催化氢化反应的研究进展637表2 取代吡啶羧酸及酯的不对称催化氢化Table 2 Asymmetric hydrogenation of substituted pyridine carboxylic acids and esters EntryR L * Yield/%ee /%1 2-CO 2Et DIOP 4127 2 2-CO 2Et BINAP 96 25 3 2-CO 2Et BDPP 97 9 4 2-CO 2H Cy 2PF-PPh 2 100 25 5 3-CO 2Et DIOP 52 126 3-CO 2Et BDPP 45 177 3-CO 2H Cy 2PF-PCy 2 8 17 8 3-CO 2HPPF-P(t -Bu)2 5 6表3 取代呋喃的不对称催化氢化Table 3 Asymmetric hydrogenation of substituted furan EntryR L*Yield/% ee /% 1 CH 2OH BINAP 91 7 2 CH 2OH PROPHOS 98 5 3 CH 2OH DIOP 98 44 COOH PPF-P(t -Bu)2 3 245 COOHCy 2PF-PCy 2 100 12 非均相的不对称催化氢化非均相的催化氢化体系与均相相比具有催化剂回收方便、操作简单等优点, 从而引起了人们广泛的研究兴趣. 近年来已经有一些科学家把非均相的催化氢化体系应用到芳香杂环化合物的不对称氢化中. 1992年Brunner [18]等发现在硅胶上Rh(I)的金属前体和手性双磷配体反应生成的手性催化体系, 在NaH 2PO 4/Na 2HPO 4 (pH 7)组成的缓冲溶液中, 能选择性地氢化叶酸的杂环部分, 所得到的5,6,7,8-四氢叶酸在DNA 碱前体的生物合成中起着重要的作用. 接着, 他们又经过对金属前体、手性配体和担载物的优化后, 发现能与[Rh(COD)- Cl]2形成七元环的配体(-)-BPPM 和(-)-DIOP 的效果最好, 硅胶Merckosorb SI60 是最佳的担载物, 取得了最高为92.3%的de 值[19](Eq. 9). 这一非均相催化体系对其它类似底物的氢化未见报道.Hegedus [20]用手性脯氨酸甲酯作手性诱导试剂,10% Pd/C 作催化剂, 加热下对3-吡啶甲酸进行非对映选择性氢化. 溶剂对此反应速度有较大影响, 但对选择性影响不大. 甲醇和乙酸乙酯都是较好的溶剂. 在室温,即使10 MPa 的氢气压力下也不反应. 高温有利于转化率提高, 但选择性降低.经过条件优化, 他们在50 ℃,5.0 MPa 的氢气压力下, 反应10.5 h, 对2-位取代的吡啶羧酸10的氢化可获得79%的de 值(Eq. 10), 对3-位取代的吡啶羧酸11的氢化可获得94%的de 值(Eq. 11), 对2-位取代吡啶盐12的氢化可获得98%的de 值(Eq. 12), 但后经证实, 此结果并没有重复性, 可重复的最高de 值为30%[21a].638有 机 化 学 V ol. 25, 2005Pinel 和Besson 等[21]利用手性脯氨酸酯及其衍生物作辅助试剂, 先与2-甲基-3-吡啶甲酸反应, 然后用Rh/C 和Rh/Al 2O 3作催化剂, 对所生成的2-甲基-3-吡啶甲酸衍生物进行氢化(Eq. 13). 他们考察了催化剂、温度、溶剂、手性诱导试剂等对氢化反应的影响, 经过一系列实验, 他们发现Rh/Al 2O 3 (3.8%)和Rh/C (4.2%)的活性最好, 但前者的选择性好. 用他们作催化剂时, 随着温度升高, 反应速度增加, 但前者选择性降低, 而后者则不受影响. 甲醇是较好的溶剂, 酸的加入并没有提高选择性. 手性的泛酸内酯是最好的诱导试剂, 诱导的de 值最高为35%. 后来他们又尝试用其它方法来提高选择性, 结果并不理想[21b].Studer 等[22]使用10,11-二氢辛可尼定改性的钯催化剂对3-位取代的吡啶羧酸酯进行氢化(Eq. 14), 获得了手性的哌啶, 但首先要经过一步Pd/C 氢化获得四氢化物. 他们也尝试了Rh/C, Rh/PtO 2等金属催化剂, 但结果都不好. 经过一系列的筛选, 他们发现5% Pd/TiO 2在DMF/H 2O/AcOH (1∶1∶0.001, 体积比)中取得了最好的对映选择性(24% ee 值), 但收率仅有10% (Table 4, Entry 3), 且此结果重复性不好. 此反应需在50 ℃, 13.0 MPa 的氢气压力下进行, 催化剂的用量大(S /C =10/3), 催化体系不稳定. 尽管如此, 但这是第一例非均相体系中对映选择性氢化取代吡啶的报道.表4 取代吡啶的非均相不对称氢化Table 4 Asymmetric hydrogenation of substituted pyridine carboxylic estersEntry Catalyst Product/% ee /% 1 10% Pd/C 12 19a 2 10% Pd/C 81 2.5b 3 5% Pd/TiO 2 10 24c4 5% Rh/C 46 1.5b5 Rh/Pt oxide97 3baDMF 作溶剂; b n -Hexane 作溶剂; c DMF/H 2O/AcOH (1∶1∶0.001, V ∶V ∶V )作溶剂.在此基础上Thomas 和Johnson 等[23]利用中孔MCM-41固载二茂铁/Pd 的非均相催化剂对3-位取代的吡啶羧酸酯进行一步氢化(Eq. 15), 取得了最高为17%的ee 值, 转化率超过50%, 反应条件(40 ℃, 2.0 MPa H 2)比前者(50 ℃, 13.0 MPa H 2)温和. 中孔MCM-41孔外壁先用Ph 2SiCl 2去活化, 这样, 手性的金属配体只与孔内的活性部位反应, 整个手性环境被限制在孔内, 有利于底物的对映选择性氢化, 而它的均相体系给出消旋的产物. 虽然产物的ee 值较低, 但这为设计非均相催化剂提供了一种新的方法.Baiker 等[24]使用辛可尼啶改性的Pd/Al 2O 3的催化体系对取代的呋喃羧酸和苯并呋喃羧酸进行了非均相的不对称氢化, 对苯并呋喃羧酸13氢化后获得50%的ee 值, 但收率只有29% (Eq. 16). 对2-呋喃羧酸(14)在收率为95%情况下获得最高为32%的ee 值(Table 5, Entry 1);对二取代的呋喃羧酸来说, 在室温, 3.0 MPa 的氢气压力下, 全部得到的是顺式异构体, 但ee 值低(Table 5, En-tries 2 and 4 ) (Eq. 17). 底物中羧基是取得高对映选择性所必须的, 在同样条件下当把羧基换成酯基时, 则没有选择性. 推测原因可能是羧基和辛可尼啶的羟基和桥头氮之间可以形成氢键, 这种氢键相互作用一方面使底物靠近手性中心, 另一方面稳定过渡态复合物的结构, 而酯基不能形成氢键, 因此没有选择性. 在此催化条件下, 辛可尼啶能被部分氢化(Eq. 18), 因此在反应中, 需要不断地加入适量的辛可尼啶才能保证反应顺利进行.No. 6卢胜梅等:芳香杂环化合物不对称催化氢化反应的研究进展639表5 取代呋喃羧酸的非均相不对称氢化Table 5 Asymmetric hydrogenation of substituted furan car-boxylic acids Entry X Y Yield/% ee /% de /% 1 COOH H 95 32 — 2 COOH CH 3615 100 3 H COOH 100 23—4CH 3 COOH 222 1002004年, Glorius 等[25]在取代吡啶的2-位引入手性的唑烷基酮, 在无质子存在下, 由于偶极矩最小化合物最稳定, 因此构象16为主要存在形式. 但在酸性条件下, 因为存在氢键作用, 以构象19为主. 利用这种氢键作用可以控制底物的构象, 又利用唑烷基酮上的手性基团挡住一个面, 这样, 氢化只能从另一个面进行, 因此, 可以很好地控制产物的非对映选择性. 在10.0 MPa 的氢气压力下, 以乙酸作溶剂, Pd(OH)2/C, Rh/C 或Rh/Pd/C 等都能对其进行氢化, 在脱掉手性辅助试剂后, 可获得最高为98%的ee 值, 如果吡啶环上有多个取代基, 氢化后可同时产生多个手性中心. 这是在非均相体系中对吡啶类底物不对称氢化取得的最好结果(Scheme 1).3 展望综上所述, 在均相体系中对一些稠环的芳香杂环化合物的不对称催化氢化虽已取得了一些成绩, 但对其它类型的稠环芳香杂环化合物, 如异喹啉、苯并呋喃、苯并噻吩及其衍生物等的氢化还未有报道; 对单环的芳香杂环化合物的氢化结果目前都不令人满意. 在非均相体系中单环的芳香杂环化合物的不对称催化氢化研究较多, 但好的结果较少, 只有Glorius 利用底物诱导对取代吡啶取得了好的结果; 对稠环的芳香杂环化合物除了苯并呋喃羧酸外, 其它的都还未见报道. 研究对芳香杂环化合物的不对称催化氢化, 无论是均相的还是非均相的, 都是一个非常有意义和发展前景的课题. 新的均相和非均相催化剂的开发是将来芳香杂环化合物不对称催化氢化领域研究的重点.Scheme 1References1 (a) Ojima, I. Catalytic Asymmetric Synthesis , VCH Publish-ers, New York, 1999.(b) Noyori, R. Asymmetric Catalytysis in Organic Synthesis , Wiley, New York, 1994.(c) Jacobensen, E. N.; Pfaltz, A.; Yamamoto, H. Compre-hensive Asymmetric Catalysis , Springer, Berlin, 1999, Vol. 1.2 Elliott, M. A.; McNeil, D. Chemistry of Coal Utilization ,second Suppl. Vol. Wiely, New York, 1981, p. 1003.3 Barton, D.; Nakanishi, K.; Meth-Cohn, O. ComprehensiveNatural Products Chemistry , Elsevier, Oxford, 1999, Vol. 1~9.640有机化学V ol. 25, 2005thesis, Academic Press, New York 1979, p. 175.5 Bird, C. W. Tetrahedron Lett. 1992, 48, 335.6 (a) Murahashi, S.-I.; Imada, Y. Bull. Chem. Soc. Jpn. 1989,62, 2968.(b) Murahashi, S.-I.; Imada, Y.; Hirai, Y. Tetrahedron Lett.1987, 28, 77.7 (a) Fish, R. H.; Thormodsen, A. D.; Gremer, G. A. J. Am.Chem. Soc. 1982, 104, 5234.(b) Fish, R. H.; Tan, J. L.; Thormodsen, A. D. J. Org.Chem. 1984, 49, 4500.(c) Fish, R. H.; Tan, J. L.; Thormodsen, A. D. Or-ganometallics1985, 4, 1743.(d) Baralt, E.; Smith, S. J.; Hurwitz, J.; Horvath, I. T.; Fish,R. H. J. Am. Chem. Soc. 1992, 114, 5187.8 Murata, S.; Sugomoto, T.; Matsuura, S. Heterocycles1987,26, 763.9 Bianchini, C.; Barbaro, P.; Scapacci, G.; Farnetti, E.;Graziani, M. Organometallics1998, 17, 3308.10 Bianchini, C.; Barabro, P.; Scapacci, G. J. Organomet.Chem. 2001, 621, 26.11 (a) Cobley, C. J.; Henschke, J. P. Adv. Synth. Catal. 2003,345, 195.(b) Henschke, J. P.; Burk, M. J.; Malan, C. G.; Herzberg,D.; Peterson, J. A.; Wildsmith, A. J.; Cobley, C. J.; Casy, G.Adv. Synth. Catal. 2003, 345, 300.12 Kuwano, R.; Sato, K.; Kurokawa, T.; Karube, D.; Ito, Y. J.Am. Chem. Soc. 2000, 122, 7614. 13 Kuwano, R.; Kaneda, K.; Ito, T.; Sato, K.; Kurokawa, T.;Ito, Y. Org. Lett. 2004, 13, 2213.14 Wang, W. B.; Lu, S. M.; Yang, P. Y.; Han, X. W.; Zhou, Y.G. J. Am. Chem. Soc. 2003, 125, 10536.15 Yang, P. Y.; Zhou, Y. G. Tetrahedron: Asymmetry2004, 15,1145.16 Fuchs, R. EP 803502,1997[Chem. Abstr. 1998, 128,13286].17 Studer, M.; Wedemeyer-Exl, C.; Spindler, F.; Blaser, H. U.Monatsh. Chem. 2000, 131, 1335.18 Brunner, H.; Huber, C. Chem. Ber. 1992, 125, 2085.19 Brunner, H.; Bublak, P.; Helget, M. Chem. Ber. 1997, 130,55.20 Hegedus, L.; Hada, V.; Tungler, A.; Mathe, T.; Szepesy, L.Appl. Catal., A2000, 201, 107.21 (a) Douja, N.; Besson, M.; Gallezot, P.; Pinel, C. J. Mol.Catal. A: Chem.2002, 186, 145.(b) Douja, N.; Malacea, R.; Banciu, M.; Besson, M.; Pinel,C. Tetrahedron Lett. 2003, 44, 6991.22 Blaser, H.-U.; Honig, H.; Studer, M.; Wedemeyer-Exl, C. J.Mol. Catal. A: Chem.1999, 139, 253.23 Raynor, S. A.; Thomas, J. M.; Raja, R.; Johnson, B. F. G.;Bell, R. G.; Mantle, M. D. Chem. Commun. 2000, 1925.24 Maris, M.; Huck, W.-R.; Mallat, T.; Baiker, A. J. Catal.2003, 219, 52.25 Glorius, F.; Spielkamp, N.; Holle, S.; Goddard, R.; Lehman,C. W. Angew. Chem., Int. Ed. 2004, 43, 2850.(Y0408023 QIN, X. Q.; LING, J.)。

杂环化合物脱磺酸基的条件
杂环化合物脱磺酸基是有机合成中常见的一种反应,通常使用还原剂来实现。
常见的条件包括使用金属催化剂或还原剂。
其中,亚硫酸钠和硫酸氢钠是常用的脱磺酸基试剂。
在有机合成中,通常会选择适当的还原剂和溶剂来实现脱磺酸基的反应。
另外,还有一些其他的条件也可以实现杂环化合物脱磺酸基的反应,例如氢气和催化剂的存在下进行催化加氢反应,或者使用亚砜等还原剂来实现脱磺酸基的反应。
此外,还可以利用还原性金属如锌、铁等来实现脱磺酸基的反应。
需要注意的是,不同的杂环化合物结构可能需要不同的条件来实现脱磺酸基的反应,因此在具体的有机合成中,需要根据具体化合物的结构和反应条件进行选择合适的脱磺酸基条件。
同时,在进行有机合成实验时,也需要考虑反应的选择性、收率和副反应的控制等因素,以确保脱磺酸基反应的高效进行。
总的来说,杂环化合物脱磺酸基的条件可以根据具体的化合物结构和实验要求选择适当的还原剂和催化剂,以实现高效的脱磺酸基反应。

封线教研室主 任教务处验收人班级 学 号 姓 名1.写出下列反应的主要有机产物或所需之原料、试剂(如有立体化学问题请注明)。
(1.0分) 2.写出下列反应的主要有机产物或所需之原料、试剂(如有立体化学问题请注明)。
(1.0分) 3.写出下列反应的主要有机产物或所需之原料、试剂。
(如有立体化学问题请注明)(1.0分) 4.写出下列反应的主要有机产物或所需之原料、试剂(如有立体化学问题请注明)。
(1.0分) 5.写出下列反应的主要有机产物,如有立体化学问题,也应注明。
(1.0分)封线教研主 教务验收班级 学 号 姓 名6.写出下列反应的主要有机产物或所需之原料、试剂(如有立体化学问题请注明)。
(1.0分)7.写出下列反应的主要有机产物或所需之原料、试剂(如有立体化学问题请注明)。
(1.0分)8.写出下列反应的主要有机产物或所需之原料、试剂。
(如有立体化学问题请注明)(1.0分)9.写出下列反应的主要有机产物或所需之原料、试剂。
(如有立体化学问题请注明)(1.0分)10.写出下列反应的主要有机产物,如有立体化学问题,也应注明。
(1.0分)11.写出下列反应的主要有机产物或所需之原料、试剂(如有立体化学问题请注明)。
(1.0分)封试 间主考班级 学 号 姓 名12.写出下列反应的主要有机产物或所需之原料、试剂(如有立体化学问题请注明)。
(1.0分)13.写出下列反应的主要有机产物或所需之原料、试剂。
(如有立体化学问题请注明)(1.0分)14.写出下列反应的主要有机产物或所需之原料、试剂。
(如有立体化学问题请注明)(1.0分)15.写出下列反应的主要有机产物或所需之原料、试剂。
(如有立体化学问题请注明)(1.0分)16.写出下列反应的主要有机产物或所需之原料、试剂(如有立体化学问题请注明)。
(1.0分)17.写出下列反应的主要有机产物或所需之原料、试剂(如有立体化学问题请注明)。
(1.0分)密考 时 总主班级 学 号 姓 名18.写出下列反应的主要产物。
最简单的杂环反应引言杂环反应是有机化学中一类重要的反应,指在分子中形成一个或多个环状结构的化学反应。
其中,最简单的杂环反应是一种将两种不同原子(通常是碳和氮)连接成一个五元环的反应。
本文将详细介绍最简单的杂环反应。
一、定义最简单的杂环反应是指将一个含有一个碳原子和一个氮原子的分子转化为一个含有五个原子(三个碳原子和两个氮原子)的环状分子。
二、机理1.亲核进攻:由于氮原子上带负电荷,它可以作为亲核攻击碳原子上带正电荷的电离质中间体。
2.脱离羧基:在亲核进攻后,羧基会脱离分子,并释放出一个质子。
3.质子转移:在脱离羧基后,氮原子上剩余的负电荷会向相邻碳原子转移。
4.芳香性恢复:由于芳香性对稠合五元环非常重要,因此在形成新键后必须恢复芳香性。
这可以通过氢离去来实现。
5.断裂C-N键:在芳香性恢复后,必须断裂C-N键,以便形成五元环。
三、实例以下是一些最简单的杂环反应的实例:1.吡咯的合成吡咯是一种常用的杂环化合物,可以通过将1,4-丁二酸和乙醇胺反应而成。
该反应需要加热,并使用盐酸作为催化剂。
2.嘧啶的合成嘧啶是DNA和RNA中重要的碱基之一,可以通过将丙二酸二乙酯和氨水反应而成。
该反应需要使用碘离子作为催化剂。
3.吡啶的合成吡啶是一种常用的有机溶剂,可以通过将1,5-戊二酸和氨水反应而成。
该反应需要使用过量的氨水,并加热。
结论最简单的杂环反应指将一个含有一个碳原子和一个氮原子的分子转化为一个含有五个原子(三个碳原子和两个氮原子)的环状分子。
该反应机理包括亲核进攻、脱离羧基、质子转移、芳香性恢复和断裂C-N键等步骤。
吡咯、嘧啶和吡啶是常见的杂环化合物,可以通过最简单的杂环反应合成。
杂环化合物的反应
杂环化合物是一类有机化合物,其环状结构中至少含有一个杂原子(如氮、氧、硫等)。
这类化合物具有丰富的生物活性和药理活性,因此在药物化学和天然产物化学等领域具有广泛的应用价值。
杂环化合物的反应主要包括:
环的稳定性:杂环化合物通常具有稳定的环状结构,这使得它们在化学反应中不易破裂。
杂原子的反应性:杂环化合物中的杂原子(如氮、氧、硫等)可以参与多种化学反应,如亲核取代反应、亲电子反应、还原反应等。
环的反应性:杂环化合物中的环也可以发生多种化学反应,如开环反应、闭环反应、环加成反应等。
例如,含氮杂环化合物可以发生亲核取代反应,亲核试剂进攻氮原子,氮原子上的电子云密度降低,离去基团离去,生成新的化合物。
此外,含氮杂环化合物还可以发生还原反应,例如霍夫曼降解反应和柯尔伯-施密特反应等。
N-杂环卡宾催化的Stetter反应N-杂环卡宾催化的Stetter反应1.引⾔1.1 卡宾简介卡宾⼜名碳烯,是亚甲基及其衍⽣物的总称,是有机反应过程中形成的不同于正、负碳离⼦的另⼀类缺电⼦的活性中间体。
虽然光谱研究已经证明了游离卡宾的存在,但是由于其在⼤多数条件下反应活性⾼、寿命短,因⽽难以分离和表征。
此外,游离卡宾的⾼活性和低反应选择性也常常限制了其在有机化学中的应⽤。
[1] ⽽N-杂环卡宾(N-H eterocyclic carbene, NHC)是⼀类纯有机化合物,碳原⼦以sp2形式杂化,卡宾碳原⼦周围只有六个电⼦,是⼀个缺电⼦体系。
卡宾碳原⼦上的⼀对电⼦处在σ轨道上。
从共轭效应考虑,两个氮原⼦p轨道上的孤对电⼦和卡宾碳原⼦上的空p轨道可以发⽣给电⼦的共轭效应,这样可以有效的降低卡宾碳原⼦的缺电⼦性。
从诱导效应考虑,两个电负性较⼤的氮原⼦与卡宾碳原⼦相连,由于氮原⼦的吸电⼦作⽤能够使卡宾碳原⼦上的孤对电⼦趋于稳定,同时,C=C双键参与的共轭作⽤,也为N-杂环卡宾结构体系的稳定做出了贡献。
[2]在卡宾家族中, NHC由于其结构的特殊性,⼀直是卡宾化学⼯作者的关注热点[3]。
NHC 具有⽐普通碳卡宾更稳定的化学结构,并且具有毒性⼩、给电⼦能⼒强、空间和电⼦效应很容易通过改变氮原⼦上取代基进⾏调控等特点, 其性质类似于富电⼦的膦配体, 且具有良好的热稳定性、耐⽔性和耐氧化性, 在许多情况下可以代替不稳定的膦配体应⽤于催化反应中。
[4]1.2 N-杂环卡宾的发展20世纪60年代,Wanzlick对N-杂环卡宾已经有所研究,但当时并未成功分离得到N-杂环卡宾。
1968 年,Wanzlick等合成了N-杂环卡宾的⾦属络合物, 但他们并未分离出游离的NHC。
[4] [5] 1991 年, Arduengo[6]等⾸次成功分离得到了第⼀个稳定的N-杂环卡宾-咪唑-2-碳烯,推动了NHC化学的迅速发展。
1995年Enders⼩组⾸次合成了三唑衍⽣的N-杂环卡宾。