2013-第六章 配合物的反应动力学解析
- 格式:ppt
- 大小:621.00 KB
- 文档页数:60


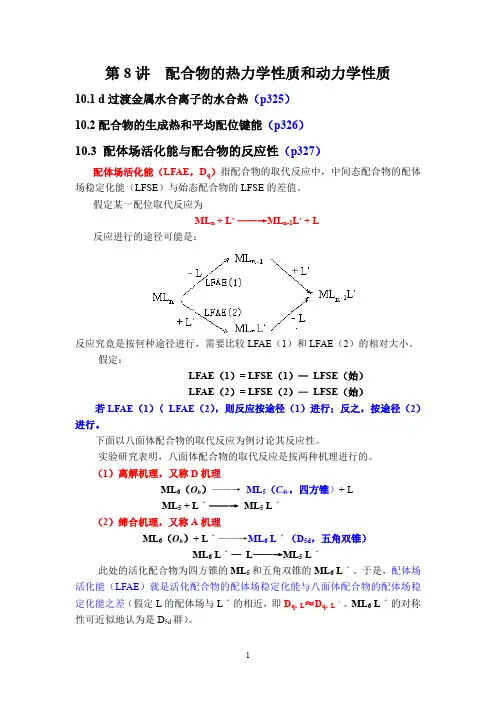
第8讲配合物的热力学性质和动力学性质10.1 d过渡金属水合离子的水合热(p325)10.2配合物的生成热和平均配位键能(p326)10.3 配体场活化能与配合物的反应性(p327)配体场活化能(LFAE,D q)指配合物的取代反应中,中间态配合物的配体场稳定化能(LFSE)与始态配合物的LFSE的差值。
假定某一配位取代反应为ML n + L,——→ML n-1L,+ L反应进行的途径可能是:反应究竟是按何种途径进行,需要比较LFAE(1)和LFAE(2)的相对大小。
假定:LFAE(1)= LFSE(1)—LFSE(始)LFAE(2)= LFSE(2)—LFSE(始)若LFAE(1)〈LFAE(2),则反应按途径(1)进行;反之,按途径(2)进行。
下面以八面体配合物的取代反应为例讨论其反应性。
实验研究表明,八面体配合物的取代反应是按两种机理进行的。
(1)离解机理,又称D机理ML6(O h)——→ML5(C4v,四方锥)+ LML5 + Lˊ——→ML5 Lˊ(2)缔合机理,又称A机理ML6(O h)+ Lˊ——→ML6 Lˊ(D5d,五角双锥)ML6 Lˊ—L——→ML5 Lˊ此处的活化配合物为四方锥的ML5和五角双锥的ML6 Lˊ。
于是,配体场活化能(LFAE)就是活化配合物的配体场稳定化能与八面体配合物的配体场稳定化能之差(假定L的配体场与Lˊ的相近,即D q,L ≈D q,Lˊ。
ML6 Lˊ的对称性可近似地认为是D5d群)。
表1 离解机理的配体场活化能(D q)离子构型强场弱场四方锥八面体LFAE 四方锥八面体LFAEd00 0 0 0 0 0 d1-4.57 -4.00 -0.57 -4.57 -4.00 -0.57 d2-9.14 -8.00 -1.14 -9.14 -8.00 -1.14 d3-10.00 -12.00 2.00 -10.00 -12.00 2.00 d4-14.57 -16.00 1.43 -9.14 -6.00 -3.14 d5-19.14 -20.00 0.86 0 0 0 d6-20.00 -24.00 4.00 -4.57 -4.00 -0.57 d7-19.14 -18.00 -1.14 -9.14 -8.00 -1.14 d8-10.00 -12.00 2.00 -10.00 -12.00 2.00 d9-9.14 -6.00 -3.14 -9.14 -6.00 -3.14 d100 0 0 0 0 0表2 缔合机理的配体场稳定化能(D q)离子构型强场弱场五角双锥八面体LFAE 五角双锥八面体LFAEd00 0 0 0 0 0 d1-5.28 -4.00 -1.28 -5.28 -4.00 -1.28 d2-10.56 -8.00 -2.56 -10.56 -8.00 -2.56 d3-15.84 -12.00 -3.84 -7.74 -12.00 4.16 d4-21.12 -16.00 -5.12 -4.93 -6.00 1.07 d5-18.30 -20.00 1.70 0 0 0 d6-15.48 -24.00 8.52 -5.28 -4.00 -1.28 d7-10.56 -18.00 7.45 -10.56 -8.00 -2.56 d8-7.74 -12.00 4.26 -7.74 -12.00 4.16 d9-4.92 -6.00 1.08 -4.92 -6.00 1.08 d100 0 0 0 0 0事实上,D 机理和A 机理仅是两种极端情况。








配合物催化反应机理研究配合物催化反应机理研究是化学领域中一项重要的研究课题。
催化反应是通过引入催化剂来加速反应速率的过程。
而配合物催化剂是由中心金属离子与配体形成的配合物。
在催化反应中,配合物催化剂通过与底物发生相互作用,改变反应的活化能,从而提高反应速率。
本文将探讨配合物催化反应机理的研究方法和应用前景。
一、催化反应机理的研究方法1. 实验方法研究配合物催化反应机理的实验方法主要包括动力学研究、核磁共振(NMR)研究和X射线晶体学研究等。
动力学研究通过测量反应速率随时间的变化,得到反应级数和速率常数等信息。
这种方法可以揭示反应的速率控制步骤和催化剂的作用机理。
NMR研究可以通过观察反应物和产物在催化剂作用下的化学位移变化,揭示催化剂与反应物之间的相互作用。
同时,NMR还可以用于研究配合物催化剂的结构和构象变化。
X射线晶体学研究可以通过解析催化剂的晶体结构,揭示催化剂与反应物之间的空间排布和相互作用。
这种方法对于理解催化剂的活性中心和反应机理有着重要的意义。
2. 计算方法除了实验方法外,理论计算方法也是研究配合物催化反应机理的重要手段。
量子化学计算方法可以通过计算配合物催化剂的电子结构和能量变化,预测反应的活化能和反应路径等信息。
常用的计算方法包括密度泛函理论(DFT)、分子力场(MM)和分子动力学(MD)等。
这些计算方法可以帮助研究者预测催化剂与反应物之间的相互作用和反应机理,为实验研究提供理论指导。
二、配合物催化反应机理的应用前景配合物催化反应机理的研究在有机合成、能源转化和环境保护等领域具有广泛的应用前景。
在有机合成领域,配合物催化反应机理的研究可以帮助合成有机化合物的高效方法。
例如,金属有机配合物催化剂可以用于不对称催化合成手性化合物,从而在药物合成和生物活性研究中具有重要意义。
在能源转化领域,配合物催化反应机理的研究可以用于开发新型能源材料和催化剂。
例如,金属配合物催化剂可以用于氢气产生和氧气还原反应,从而实现高效能源转化。