制剂杂质谱对比方式+有关物质方法来源与筛选
- 格式:pdf
- 大小:677.29 KB
- 文档页数:6

罗红霉素原料药及其制剂的有关物质研究摘要目的:对不同厂家罗红霉素原料药及其制剂进行有关物质检查,从而分析多批次罗红霉素的总体水平。
方法:采用HPLC法对原料药及其制剂的杂质的含量进行检测,采用LC-MS法对制剂中的杂质进行归属。
结果:受试的4批原料药与23批制剂,有关物质合格率为100%,制剂中杂质C、E、H、J、I出现概率大。
结论:通过对不同厂家生产的罗红霉素有关物进行考察,可了解各产品的质量情况,提高药品质量,保证用药安全有效。
关键词:罗红霉素;有关物质;HPLC;LC-MSStudy on the Related Substances of roxithromycin API and its preparationsAbstract Objective: The related substances of roxithromycin API and its preparations from different manufacturers were determined, so as to analyze the overall level of multiple batches roxithromycin. Methods: HPLC was used to detect the content of impurities in API and its preparation, and LC-MS was used to classify the impurities in the preparation..Results: The qualified rate of related substances in 4 batches of APIs and 23 batches of preparations was 100%, and the probability of impurity C, E, H, J and I in the preparation is high Conclusion: The related substances of roxithromycin from different manufacturers were investigated, improve the quality of drugs and ensure the safety and effectiveness of drug use.Key words:Roxithromycin ; Related substances; HPLC; LC-MS;罗红霉素为红霉素的醚肟类衍生物,其抗菌作用优于红霉素,不良反应相较于红霉素来说较少,因为它对细胞色素P450的亲和力低[1]。

一致性评价申报资料立卷自查暨审查工作用表填写说明一.品种综合信息1、批准信息(1.1-1.6)应按照申报资料信息填写药品通用名称、商品名、批准文号(包括历年的批准文号\规格、执行标准、药品有效期。
2、申报概况(1.7∙L12)根据《化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求》中概要部分内容填写:药品注册及变更批准证明性文件是否提供、临床信息和不良反应是否提供、再评价品种处方工艺是否变更、自评估报告是否提供、是否按照CTD格式提交资料寄言息。
3、检验报告(1.13)根据(3.2.P.5.4)批检验报告部分内容填写1.13检验报告部分。
二.原研产品及参比制剂信息1、原研产品信息(2.1)根据原研产品上市情况填写相关I言息。
2、参比制剂信息(2.2)按照总局2017年第100号公告以及《普通口服固体制剂参比制剂选择和确定指导原则》等规定的要求,选择和确定参比制剂。
申报资料应明确注明参比制剂信息,并提供所用参比制剂来源证明资料;凡是采用非总局公布目录产品作为参比制剂的,221项应选择“其他说明",同时填写生产企业名称及产品相关信息;如提供了参比品标签/样品照片/说明书中的任意一项,则在相应栏目选"提供";三项均未提供,选"未提供"。
说明书采用网络版本(如PDF格式)打印件,不认可。
每个规格原则上应提供3批(至少1批)参比制剂的考察数据,考察与一致性评价紧密相关的关键质量属性,例如性状、溶出度/释放度、含量、有关物质等(检验报告可列为附件%每个规格的参比制剂原则上应提供3批样品的溶出曲线考察数据f以考察其溶出行为的批内和批间均一性。
对有文献报道或者研究资料表明有光照、高湿、高温、氧化等条件下不稳定的品种,建议考察参比制剂溶出曲线稳定性,为实验室复核结果的重复性提供支持。
三.产品研究信息1、处方组成1.1、原料药及辅料(3.1.1-2):应提供原料药及辅料的批准证明文件、质量标准、检验报告、BSE/TSE风险声明等资料制剂可能含有一个或多个原料药,含有多个原料药的可能存在既有国产来源的原料药也有进口原料药,应根据申报品种处方及提供的证明资料如实填写。
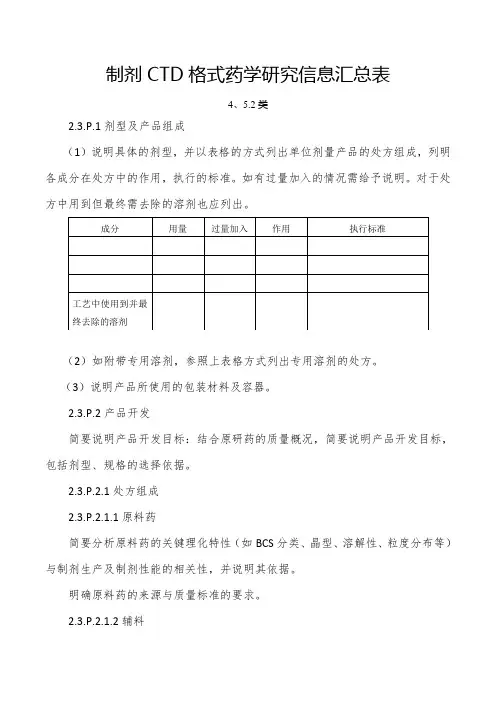
制剂CTD格式药学研究信息汇总表4、5.2类2.3.P.1剂型及产品组成(1)说明具体的剂型,并以表格的方式列出单位剂量产品的处方组成,列明各成分在处方中的作用,执行的标准。
如有过量加入的情况需给予说明。
对于处方中用到但最终需去除的溶剂也应列出。
(2)如附带专用溶剂,参照上表格方式列出专用溶剂的处方。
(3)说明产品所使用的包装材料及容器。
2.3.P.2产品开发简要说明产品开发目标:结合原研药的质量概况,简要说明产品开发目标,包括剂型、规格的选择依据。
2.3.P.2.1处方组成2.3.P.2.1.1原料药简要分析原料药的关键理化特性(如BCS分类、晶型、溶解性、粒度分布等)与制剂生产及制剂性能的相关性,并说明其依据。
明确原料药的来源与质量标准的要求。
2.3.P.2.1.2辅料简述辅料及其用量适合所用的给药途径的依据,结合辅料在处方中的作用简述辅料的与制剂性能相关的关键特性。
简述原料药和辅料的相容性试验的情况,包括试验设计、考察指标、试验结果等。
如未进行原料药和辅料的相容性试验,应提供相应的依据。
2.3.P.2.2 制剂研究2.3.P.2.2.1处方开发过程简述处方研究的主要内容。
包括处方开发的基本思路、试验设计、考察指标和方法、试验结果、与原研药的比较研究情况、处方的放大和调整等。
示例如下:某普通片剂的处方研究小结:参考原研药说明书、原辅料相容性试验情况、相关生产经验等,确定了辅料的基本种类;参考原研药的重量和大小、以及各辅料常规用量,确定了辅料的用量范围,以××××为指标,采用××××方法,对××××的种类和用量进行了比较筛选,对××××处方进行了研究,以原研药为对照药,结果显示××××,根据以上研究确定了初步的处方;在以上研究基础上,进行了影响因素稳定性考察,与原研药进行了××××的质量对比;在批量放大过程中,对××××进行了调整,确定了最终处方。



页眉内容【讨论】浅谈复方制剂的有关物质质量标准的建立应斑竹的邀请,开此贴跟大家讨论一下复方制剂杂质的控制以及分析方法建立的一些相关问题。
本人在分析实验室做了6-7年仿制药申报,负责药物CMC方面的质量研究工作,接触了十几个项目。
基本了解FDA/SFDA审评人员的一些要求和想法。
此贴会结合本人的一些经验,探讨复方制剂有关物质分析的一些注意点,并且会结合产品申报以后FDA给的缺陷信,提示大家如何避免同样的问题。
更希望抛砖引玉,各位版友和专家踊跃参与,共同提高。
废话少说,对于有关物质的研究,最近几年国家法规的要求越来越高,可能大家也越来越重视此项研究。
不过一个不可否认的事实是,我们的杂质研究水平和欧美或者印度等国家差距很大。
我们的绝大部分药品企业基本处于食物链的最底端,以前经常听我们老板说他在美国的药厂工作的时候,要从其他公司买API,很多都是西班牙的,意大利的,或者印度的药厂以极其低廉的价格从国内厂家购买的粗品,然后拿回去做一到两步的提纯,再做一些法规文件,就可以以很多倍的价格卖出去。
我们消耗了主要的人力,物力,牺牲了环境,结果回报只有人家的零头,可悲又可气!所以我们就要具体的看一看到底存在哪些差距。
复方制剂有关物质控制的基本原则和单方一样,你就把他当作是在做两个或者三个产品,只不过是同时分析几个物质。
相关的指导原则大家也应该都知道, ICH Q3A和Q3B; FDA也有相关的工业指南,跟这两个基本差不多。
做杂质研究和其他大部分质量研究工作一样,首先要做的第一步就是去收集资料,包括:1. API厂家的技术资料,一般有DMF的会提供它的open part, 里面会大致列出它的工艺流程,合成路径和所有潜在的杂质。
2.药典的各论,包括USP, EP等等,看看药典有那些要求检测的杂质。
3.查阅文献,看看化合物的潜在杂质和降解产物。
这一点对于制剂的杂质控制有时会提供很有用的信息。
4.国内申报仿制药最好弄到进口注册标准,看看原研厂家是怎样控制杂质的。


高效液相色谱法测定盐酸黄酮哌酯原料和制剂中有关物质探讨摘要】盐酸黄酮哌酯主要呈现出一种白色粉末或者晶体状态,化学方程式为C24H25NO4.HCl;C24H26ClNO4,其分子量为427.92。
本文将针对盐酸黄酮哌酯试药进行详细的分析,其目的是探究出高效液相色谱法测定盐酸黄酮哌酯的策略。
【关键词】高效液相色谱法;盐酸黄酮哌酯【中图分类号】R927.2 【文献标识码】A 【文章编号】2095-1752(2018)21-0361-01就我国现行的盐酸黄酮哌酯质量标准检测手段来说,主要是依照《中国药典(二部)》的相关内容,针对盐酸黄酮哌酯原料和制剂中有关物质进行检定,主要运用TLC法。
但是随着我国药物技术的不断发展,通过高效液相色谱的手段也能够详细的针对盐酸黄酮哌酯的质量以及物质含量进行分析,并且结合我国相关的药物检测标准进行对照,检测出盐酸黄酮哌酯的含量。
客观来说,传统TLC相比HPLC 检测手段来说,更加繁琐,其灵活度也相对较低。
据笔者的亲身统计与观察总结出,传统TLC检测手段,仅控制杂质3-甲基黄酮-8-羧酸,对其他杂质不能进行有效的检测。
本文将针对高效液相色谱法测定盐酸黄酮哌酯原料和制剂中有关物质进行详细的探讨。
1.盐酸黄酮哌酯试药盐酸黄酮哌酯主要用于尿频、尿急、尿痛、排尿困难及尿失禁等泌尿疾病的治疗中。
作为一中国产的口服药物,我国不同制药厂针对盐酸黄酮哌酯的大多检查项目与原研药品的差异并不大,但是药物的溶解与溶出的情况,不同制药厂会存在一定程度上的差异,但是此类差异对盐酸黄酮哌酯的疗效并没有产生直接的影响[1]。
107批样品按照现行质量标准检验,仅有30批样品检出已知杂质,很难针对盐酸黄酮哌酯中出现的其他杂质与未知物质进行检测,说明现行药典有关物质测定方法专属性和灵敏度均较差,检查结果不能反映药品中杂质的真实含量。
2.高效液相色谱法测定盐酸黄酮哌酯的策略2.1 合理的选择色谱条件针对盐酸黄酮哌酯来说,其极性相对较强,很容易被洗脱。


药物杂质研究方法详解药物中的杂质分析是一个非常重要的部分,小析姐就杂质来源分析的重要作用,同时重点探讨了杂质研究过程中分析技术的发展,尤其在结构鉴定中质谱技术的发展,与此同时,根据国外毒性杂质研究的指导原则,明确了杂质毒性研究的方法。
近年来,随着药物研究的不断深入以及杂质研究要求不断提高,杂质的分析技术以及研究方法正发生着重要的改变。
在对杂质建立分析方法时,清晰的杂质研究过程是方法建立的基础,而且选择合适的分析技术也至关重要。
杂质的来源分析药物中的杂质可能来源于药物生产以及销售等各个环节(图1)。
根据ICH指导原则可将药物杂质分为有机杂质、无机杂质、残留溶剂以及其他杂质。
本文主要针对有机杂质进行探讨。
对药物杂质研究时引入“质量源于设计( Quality byDesign,QbD)”的理念,可在药物生产之前根据具体工艺的合成机制、起始物料及各中间体的基本结构,初步勾画出产品的杂质谱。
杂质来源分析是制定药物杂质控制策略的基础,尤其是在对毒性杂质来源分析时,应分析所有合成和生产工艺中的试剂、中间体、副产物,推测可能产生的潜在杂质以及分析实际存在的杂质。
在原料药合成结束后,药物的活性化合物虽然经过毒性分析已不含有“警示结构”(alerting structure),但是在生产过程中使用到含有警示结构的化合物则还需考虑其遗传毒性。
杂质的研究方法在药物研发过程中,药物杂质的分析是关键。
因此,在杂质研究中清晰的杂质结构研思路(如图2)以及合适的杂质分析技术可极大地缩短杂质研究时间,推动着药物研究的快速发展。
1、杂质前处理技术杂质的前处理是伴随着药物活性成分前处理而存在的,然而药物中杂质的含量低且其结构与主成分差异较大,因此常规药物活性成分的前处理和检测方法(如初始流动相溶解后直接进行 HPLC-UV 分析)并不一定适用于药物杂质,应针对不同的样品选择不同的前处理技术。
(1)检测灵敏度低的样品对检测灵敏度低的样品通常使用衍生化的前处理方式,比如引入生色团产生紫外响应,或增加易离子化基团增加离子化效率等。

浅谈国产注射用氨苄西林钠杂质谱及生产工艺发表时间:2018-10-30T15:02:20.363Z 来源:《世界复合医学》2018年第08期作者:王震[导读] 国产注射用氨苄西林钠杂质的含量与生产工艺之间是存在某种联系的,生产工艺甚至影响着杂质的实际数量以及种类,这有助于为以后的临床以及制药研究提供借鉴。
哈药集团制药总厂 150000【摘要】目的:本文为了深入探究国产注射用氨苄西林钠杂质谱和与有关的生产工艺,从而为以后的临床研究和应用提供一些参考。
方法:本文主要运用了色谱检测法、对比法以及文献法等针对190批次的国产注射用氨苄西林钠进行检查,不仅要针对不同的生产工艺所制成的药品进行检验,同时还要进行破坏性的试验,对各种杂质进行鉴别,进而帮助有关人员测定出杂质的成分以及含量。
结果:与其他样品相比,溶媒结晶中的杂质含量最少,而且其数量也相对较少;相反,喷雾干燥样品中的杂质含量则相对最多。
结论:国产注射用氨苄西林钠杂质的含量与生产工艺之间是存在某种联系的,生产工艺甚至影响着杂质的实际数量以及种类,这有助于为以后的临床以及制药研究提供借鉴。
【关键词】注射用氨苄西林钠;杂质谱;生产工艺随着我国经济的快速发展,人们的生活质量得到了显著的改善,相应的需求也有所增加,用药需求就是其中之一,这对于患者身体的健康具有重要的影响。
而随着现代医学的进步与发展,人们也越来越重视用药安全问题。
这就对于制药企业提出了更高的要求。
因此,保障制药的质量就尤为关键。
而其中的生产工艺则起着决定性的作用,而不同药物的生产工艺也有所不同,甚至是同种药物的生产工艺也有可能有所不同。
针对国产注射用氨苄西林钠而言,它的生产工艺就具有多样化,而应用这类药物,能够起到抑制细菌等效果,是比较常见的临床治疗药物。
就目前现状来看,它的生产工艺主要有以下三种,即溶媒结晶法、冷冻干燥法以及喷雾干燥法,针对这几种生产工艺以及其中的杂质就有必要加以深入的探究。
药学主要研究信息汇总要求(制剂)一、剂型及产品组成说明具体的剂型,并以表格的方式列出单位剂量产品的处方组成,列明各成分在处方中的作用,执行的标准。
如有过量加入的情况需给予说明。
对于处方中用到但最终需去除的溶剂也应列出。
如附带专用溶剂,参照上表格方式列出专用溶剂的处方。
说明产品所使用的包装材料及容器。
(一)制剂研究简要对与制剂性能相关的理化性质,如pH,离子强度,溶出度,再分散性,复溶、粒径分布、聚合、多晶型、流变学等进行分析。
提供转入方与转出方药品在处方开发过程中进行的质量特性对比研究结果,例如:(1)口服固体制剂的溶出度:样品批号,对照药品批号和生产厂,溶出条件,取样点,比较结果。
(2)有关物质:样品批号,对照药品批号和生产厂,测定及计算方法,比较结果。
(二)生产工艺的开发汇总研发过程中代表性批次的样品情况,包括:批号、生产时间及地点、批规模、用途(如用于稳定性试验,用于生物等效性试验等)、分析结果(例如有关物质、溶出度以及其他主要质量指标)。
示例如下:批分析汇总包装材料/容器注明详细参见的申报资料项目表“3.药学研究资料”中资料的项目和页码。
注1:关于包材类型,需写明结构材料、规格等。
例如,五层共挤膜输液袋,规格为内层:改性乙烯/丙烯聚合物;第二层:聚乙烯;第三层:聚乙烯;第四层:乙烯甲基丙烯酸酯聚合物;第五层:多酯共聚物。
聚丙烯输液瓶,规格为250ml。
铝塑泡罩包装,组成为: 3.2.PVC/铝、3.2.PVC/3.2.PE/3.2.PVDC/铝、3.2.PVC/3.2.PVDC/铝。
复合膜袋包装,组成为:聚酯/铝/聚乙烯复合膜袋、聚酯/低密度聚乙烯复合膜袋。
注2:表中的配件一栏应包括所有使用的直接接触药品的包材配件。
如:塑料输液容器用组合盖、塑料输液容器用接口等。
(三)相容性简述制剂和附带溶剂或者给药装置的相容性。
注明详细参见的申报资料项目表“3.药学研究资料”中资料的项目和页码。
二、生产(一)生产商生产商的名称(一定要写全称)、地址、电话、传真以及生产场所的地址、电话、传真等。
附件2一致性评价申报资料立卷自查暨审查工作用表填写说明一、品种综合信息1、批准信息(1.1-1.6)应按照申报资料信息填写药品通用名称、商品名、批准文号(包括历年的批准文号)、规格、执行标准、药品有效期。
2、申报概况(1.7-1.12)根据《化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求》中概要部分内容填写:药品注册及变更批准证明性文件是否提供、临床信息和不良反应是否提供、再评价品种处方工艺是否变更、自评估报告是否提供、是否按照CTD格式提交资料等信息。
3、检验报告(1.13)根据(3.2.P.5.4)批检验报告部分内容填写1.13检验报告部分。
二、原研产品及参比制剂信息1、原研产品信息(2.1)根据原研产品上市情况填写相关信息。
2、参比制剂信息(2.2)按照总局2017年第100号公告以及《普通口服固体制剂参比制剂选择和确定指导原则》等规定的要求,选择和确定参比制剂。
申报资料应明确注明参比制剂信息,并提供所用参比制剂来源证明资料;凡是采用非总局公布目录产品作为参比制剂的,2.2.1项应选择“其他说明”,同时填写生产企业名称及产品相关信息;如提供了参比品标签/样品照片/说明书中的任意一项,则在相应栏目选“提供”;三项均未提供,选“未提供”。
说明书采用网络版本(如PDF格式)打印件,不认可。
每个规格原则上应提供3批(至少1批)参比制剂的考察数据,考察与一致性评价紧密相关的关键质量属性,例如性状、溶出度/释放度、含量、有关物质等(检验报告可列为附件)。
每个规格的参比制剂原则上应提供3批样品的溶出曲线考察数据,以考察其溶出行为的批内和批间均一性。
对有文献报道或者研究资料表明有光照、高湿、高温、氧化等条件下不稳定的品种,建议考察参比制剂溶出曲线稳定性,为实验室复核结果的重复性提供支持。
三、产品研究信息1、处方组成1.1、原料药及辅料(3.1.1-2):应提供原料药及辅料的批准证明文件、质量标准、检验报告、BSE/TSE风险声明等资料制剂可能含有一个或多个原料药,含有多个原料药的可能存在既有国产来源的原料药也有进口原料药,应根据申报品种处方及提供的证明资料如实填写。
(19)中华人民共和国国家知识产权局(12)发明专利(10)授权公告号 (45)授权公告日 (21)申请号 201910592961.9(22)申请日 2019.07.03(65)同一申请的已公布的文献号 申请公布号 CN 110398548 A (43)申请公布日 2019.11.01(73)专利权人 山西仟源医药集团股份有限公司地址 037010 山西省大同市经济技术开发区湖滨大街53号(72)发明人 赵树军 茅仁刚 罗小妹 朱银飞 岳唯唯 葛轲 (74)专利代理机构 上海九泽律师事务所 31337代理人 周云(51)Int.Cl.G01N 30/02(2006.01)(56)对比文件EP 1762573 A1,2007.03.14刘畅等.液相色谱-电喷雾离子阱质谱法分析磷霉素氨丁三醇及其有关物质.《药物分析杂志》.2009,(第12期),第2081-2084页.赵晓冬等.浊度法测定磷霉素氨丁三醇的效价.《药物分析杂志》.2009,第29卷(第07期),第1233-1235页.刘佳等.高效液相色谱法测定磷霉素氨丁三醇散中磷霉素氨丁三醇的含量.《中国药物评价》.2015,(第3期),第139-141页.Liu , H等.Determination of fosfomycintrometamol and its related substances inthe bulk drug by ion-pair HPLC withevaporative light scattering detection.《JOURNAL OF LIQUID CHROMATOGRAPHY &RELATED TECHNOLOGIES》.2006,第29卷(第1期),第15-24页.审查员 徐心田 (54)发明名称一种磷霉素氨丁三醇原料药及其制剂中有关杂质的高效液相色谱分离测定方法及应用(57)摘要本发明公开了磷霉素氨丁三醇原料药及制剂中杂质高效液相色谱分离测定方法及应用,用于分离杂质;用氨丙基硅烷键合硅胶为填充剂的色谱柱,以示差检测器检测;以磷酸盐缓冲液和甲醇‑乙腈混为流动相;1、系统适用性溶液制备:取原料药或制剂用水润湿,加热,加流动相溶解并稀释作溶液A;取原料药或制剂用A溶解并稀释为系统适用性溶液;2、供试品溶液制备:取原料药或制剂加流动相溶解并稀释成磷霉素氨丁三醇设定含量作供试品溶液;3、对照溶液制备:取供试品溶液用流动相稀释至含磷霉素氨丁三醇相当于供试品溶液质量浓度0.3~0.5%的溶液为对照溶液;4、测定法:取上述三种溶液分别注入液相色谱仪,用主成分自身对照法计算原料药中各杂质含量。
药品研发申报基本要求-—--质量部分摘要:药品研发申报资料基本要求是结合CDE审评技术要求制定的基本规范(保证不大补、不退审为根本目的),格式满足CTD要求及综合美学原则,内容旨在体现科学有效质控模式和方法,保证产品和原研一致性,保证分析方法经济、环保、专属、精密、准确。
主要内容包括:格式形式(格式排版、内容表达形式、表格、附图、附件)质量标准(一般口服原料、注射用原料、一般口服制剂、一般注射剂、脂肪乳、NDDS、组合包装、起始物料、中间体、关键辅料)分析方法(方法基本要求)分析方法验证(具体技术要求)起草说明(具体技术要求)杂质分析(具体技术要求)批检验报告(具体技术要求)对照品(具体技术要求)稳定性(具体技术要求)1. 目的:为了规范药品分析研发设计、文件审核与资料撰写,保证申报资料规范性要求,特制订本要求。
2. 适用范围:本要求适用于分析部项目研发全过程(立项准备、方案设计、方法开发与验证、分析方法转移、质量标准起草、稳定性设计与研究、过程节点审核、申报资料撰写与审核)。
3。
职责:3。
1 部门负责人:根据本要求对项目研发进行全程有效管控(效率和质量的度)。
3。
2 项目负责人(总负责人、质量负责人、专项负责人):根据本要求进行立项启动准备、试验方案设计、方法开发与验证、方法转移交接、质量标准起草(起始物料、中间体、关键辅料、API、制剂)、稳定性设计与试验、含量对照品及杂质对照品鉴定与标定、杂质分析、资料撰写与修改。
3。
3 试验复核/审核人员:根据本要求对试验方案、试验结果、申报资料、质量标准进行复核、审核.4. 申报资料基本要求4。
1 格式形式4。
1。
1 资料格式排版字体字号:宋体小四;标题加粗;行距段距:1。
5倍行距,表格后第一段文字增加0.5倍行距,其他段间不需额外段距,首行缩进2字;页码规定:所有资料均编页码,首页“0”不打印,打印位置:页面低端、居中;页边距离:适中:上、下:2.54cm,左、右:1.91cm。