2.自由基聚合
- 格式:doc
- 大小:226.51 KB
- 文档页数:10

缩聚和自由基聚合的区别缩聚和自由基聚合是两种常见的聚合反应过程。
它们在化学合成、生物化学以及材料科学等领域中有着广泛的应用。
本文将分别介绍缩聚和自由基聚合的定义、特点、机理以及应用。
一、缩聚缩聚是指通过化学反应将小分子(单体)连接在一起形成高分子化合物的过程。
在缩聚过程中,小分子中的官能团与其他分子中的官能团发生反应,形成化学键,从而形成高分子链。
缩聚反应通常需要外部能量的介入,如热能或光能。
缩聚反应的特点如下:1. 高分子链的长度增加,分子量增大;2. 缩聚反应是一个逆向过程,需要消耗能量;3. 缩聚反应具有选择性,只有具有特定官能团的单体才能发生缩聚反应。
缩聚反应的机理主要有两种:加成聚合和缩合聚合。
1. 加成聚合:加成聚合是指单体中的官能团直接与其他单体中的官能团发生加成反应,形成高分子链。
常见的加成聚合反应有乙烯聚合和苯乙烯聚合等。
2. 缩合聚合:缩合聚合是指单体中的官能团通过缩合反应,失去小分子(如水分子)后连接在一起形成高分子链。
常见的缩合聚合反应有酯交换反应和酰胺形成反应等。
缩聚反应在化学合成、材料科学以及生物化学等领域中有着广泛的应用。
例如,聚合物材料的合成、药物的制备以及生物大分子的合成等都离不开缩聚反应。
二、自由基聚合自由基聚合是指通过自由基反应将单体连接在一起形成高分子化合物的过程。
在自由基聚合过程中,单体分子中的双键被打开产生自由基,自由基与其他单体中的双键发生反应,形成高分子链。
自由基聚合反应通常需要引发剂的作用。
自由基聚合反应的特点如下:1. 高分子链的长度增加,分子量增大;2. 自由基聚合反应是一个自由基链反应,反应速度快;3. 自由基聚合反应具有高度的随机性和非选择性。
自由基聚合反应的机理主要有两种:链引发聚合和自由基聚合。
1. 链引发聚合:链引发聚合是指引发剂通过引发自由基反应,产生大量自由基,引发单体之间的自由基聚合反应。
常见的链引发聚合反应有自由基聚合、阴离子聚合和阳离子聚合等。

自由基聚合的四种方法自由基聚合是一种重要的化学反应,它可以用于合成各种高分子材料。
这种反应的基本原理是将单体分子中的双键开裂,形成自由基,再将自由基与其他单体分子结合,形成高分子链。
这种反应可以通过多种方法实现。
本文将介绍自由基聚合的四种方法,包括自由基引发聚合、自由基链转移聚合、自由基共聚合和自由基接枝聚合。
一、自由基引发聚合自由基引发聚合是最常见的自由基聚合方法。
这种方法需要将引发剂加入到单体中,引发剂可以是过氧化物、硫代硫酸酯等。
在引发剂的作用下,单体分子中的双键开裂,形成自由基。
这些自由基与其他单体分子结合,形成高分子链。
自由基引发聚合是一种高效的方法,可以通过调节引发剂的种类和用量来控制聚合反应的速率和分子量分布。
但是,这种方法容易产生副反应,如引发剂自身的分解和自由基的重组,这些副反应会影响聚合反应的效果。
二、自由基链转移聚合自由基链转移聚合是一种可以控制分子量分布的自由基聚合方法。
这种方法需要将链转移剂加入到单体中,链转移剂可以是醇、硫醇等。
在链转移剂的作用下,自由基聚合链上的氢原子被转移,形成新的自由基,这些自由基与单体结合,形成新的高分子链。
由于链转移剂的作用,聚合反应过程中产生的高分子链会变短,从而控制聚合反应的分子量分布。
自由基链转移聚合是一种可控性较好的聚合方法,可以得到具有狭窄分子量分布的高分子材料。
但是,链转移剂的种类和用量需要进行精确的控制,否则会影响聚合反应的效果。
三、自由基共聚合自由基共聚合是一种将两种或多种单体分子同时参与聚合反应的方法。
这种方法可以得到具有复合性能的高分子材料,如耐热性、耐化学性等。
在共聚反应中,不同单体分子之间的反应速率和选择性不同,需要通过调节反应条件来控制不同单体分子的参与程度,从而得到理想的高分子材料。
自由基共聚合是一种多样性较好的聚合方法,可以得到具有多种性质的高分子材料。
但是,不同单体分子之间的反应速率和选择性需要进行精确的控制,否则会影响聚合反应的效果。

2.自由基聚合2.1引言连锁聚合根据聚合反应机理分类,聚合反应可以分为逐步聚合连锁聚合反应需要活性中心,单体在活性中心上反应形成大分子。
活性中心可以是自由基,也可以是阴、阳离子。
活性中心的性质与化合物共价键断裂的方式有关。
共价键有两种断裂方式:均裂和异裂均裂:共价键上一对电子分属于两个基团,这种带独电子的基团呈电中性,称作自由基或游离基。
异裂:共价键上一对电子全部归属于某一基团,形成阴离子或负离子,则另一缺电子基团称作阳离子或正离子。
自由基、阴离子、阳离子都有可能成为活性中心,可打开烯类单体或羰基单体中的π键,或使环状单体的σ键断裂开环,使之链引发和链增长,分别成为自由基聚合,阴离子聚合,阳离子聚合,和配位聚合,实际上配位聚合也属于离子聚合的范畴。
Eg: 自由基聚合:2.2连锁聚合的单体单体能否聚合,须从热力学和动力学两方面考虑,热力学上能聚合的单体还要求有适当的引发剂、温度等动力学条件,才能保证一定的聚合速度。
从热力学考虑可以进行连锁聚合的单体有:2.2.1适合连锁聚合的单体大致可以分为三类:1.含有碳碳双键的烯类单体:包括单烯类、共轭二烯类,甚至炔烃。
其中:单烯类:乙烯基单体中的碳碳双键中π键可以均裂也可以异裂,因此可以进行自由基聚合或离子聚合。
具体选择哪种聚合方式,由取代基的性质决定。
共轭二烯类:如苯乙烯,丁二烯,异戊二烯等单体处于共轭体系,在外界的影响下,双键的电子云易流动,诱导极化。
因此单体既可以进行自由基聚合,也可以进行离子聚合。
2.羰基化合物如HCHO,CH3CHO,甚至酮类。
Eg: HCHO 羰基的双键有极性,使氧原子带有部分负电荷,而碳原子则带有部分正电荷。
3.杂环化合物羰基化合物和杂环化合物的极性较强,一般不能自由基聚合,只适合于离子聚合。
因此实际上只有碳碳双键的烯类单体可以进行自由基聚合,但也不是所有的都行,其取代基的性质有很大影响。
2.2.2取代基对于乙烯类单体聚合能力的影响。

连锁聚合包括哪些基元反应连锁聚合是指一种重要的聚合物化学反应,通过将一种或多种单体分子以一定的方式连接起来形成长链聚合物的过程。
在连锁聚合中,基元反应是至关重要的,它们负责实现单体之间的结合。
下面将介绍一些常见的基元反应:1. 自由基聚合自由基聚合是一种通过活性自由基进行的聚合反应。
这种反应的特点是单体分子中的一个碳原子上带有未成对电子,通过引发剂的作用,这个未成对电子将被切断并形成自由基,从而引发聚合链的生长。
丙烯酸酯、乙烯、苯乙烯等单体可以通过自由基聚合形成聚合物。
2. 阴离子聚合阴离子聚合是由负离子引发的聚合反应。
负离子引发剂可以将单体分子中的一个离子化的原子或官能团引发离子化,从而形成离子聚合反应。
丙烯酸酯、乙烯基醚等单体可以通过阴离子聚合形成聚合物。
3. 阳离子聚合阳离子聚合是通过正离子引发的聚合反应。
正离子引发剂可以将单体中的一个原子或官能团引发正电荷,从而形成阳离子聚合反应。
环氧乙烷、乙烯基吡咯烷酮等单体可以通过阳离子聚合形成聚合物。
4. 氧化还原聚合氧化还原聚合是通过氧化还原反应进行的聚合反应。
在这种聚合反应中,单体中的一个官能团将被氧化或还原,从而引发聚合过程。
苯醚、聚烯烃等单体可以通过氧化还原聚合形成聚合物。
以上所介绍的基元反应只是连锁聚合中常见的几种类型,实际上还有许多其他类型的基元反应可以用来实现单体的聚合。
通过这些基元反应,单体分子可以在化学键的作用下逐渐连接形成长链结构,最终形成多种类型的聚合物。
连锁聚合在合成高分子材料、药物、涂料等领域具有广泛的应用,而基元反应的种类和条件选择将直接影响到聚合产物的性质和结构。
因此,在设计合成新型聚合物时,深入理解和掌握不同基元反应的特点和规律至关重要。
1。
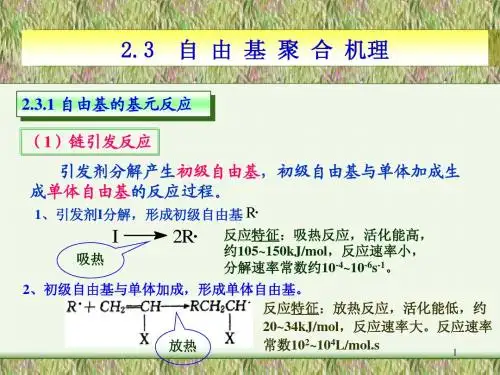

2.自由基聚合能否进行自由基聚合的判断位阻效应判断:1,1—二取代易聚合,除大取代基如—C6H5外1,2—二取代,除取代基为F以外都难聚合双键上电荷密度大,不利于自由基进攻—烯丙基单体取代基吸电性太强也不利于自由基聚合,如CH2=C(CN)2,CH2=CH(NO2) 3.(1)链引发:CH3C NCNC·CH3CH3CH3NCH3CNC CH32CN+N2CH2CHCl·CNCH3CH3C·+CH2CH3CNCCH3(2)链增长:(3)链终止: 偶合:歧化:4.自由基聚合时转化率和分子量随时间变化的特征:转化率随时间逐步提高,中间有自加速现象,分子量随时间变化甚小(短时间后变化很小).与反应机理决定,连锁聚合时RM ·→Mn ·时间极短,没有中间停留阶段。
5.引发剂(1)偶氮二异丁腈(AIBN )、(2)偶氮二异庚腈(ABVN )、(3)过氧化二苯甲酰(BPO )、(4)过氧化二碳酸二乙基己酯(EHP )、(6)过硫酸钾-亚硫酸盐体系、(7)过氧化氢-亚铁盐体系的分解反应式见书本的P26~29,(5)异丙苯过氧化氢的见下面:其中(1)~(5)为偶氮类和有机过氧类,属于油溶性引发剂常用于本体、悬浮和溶液(有机溶剂)聚合,(6)(7)为水溶性氧化-还原体系,适合于水溶液和乳液聚合。
CH 2CHCl CHCl ·CH 2CH 3CN C CH 3CHCl ·CH 3C CH 3+CH 2CH 2CHCl CHCl CH 3CN C CH 3CH 2n-1CH 2CHCl ·CH 2CHCl CH 3CN C CH 3CH 2CHCl CH 3CN C CH 32n 2CHCl CH 3C CH 3CH 2n-1CH 2CHCl ·2CHCl CH 3C CH 3CH 2n-1CH 2CHCl·CHCl CH 2n-1CH 2CH 3CN C CH 3CH 2Cl CHCl CH 2n-1CH 3C CH 3CH CHCl +COOH CO ··OH CH 3C CH 3CH 3C 3+注意:EHP 高活性,配成溶液后低温贮。

自由基聚合反应的特点及分类1、自由基聚合反应:指单体借助于光、热、辐射、引发剂等的作用,使单体分子活化为活性自由基,再与单体分子连锁聚合形成高聚物的化学反应。
2、自由基聚合反应的特点(1)反应可明显的分为链引发、连增长、链终止等基元反应。
(2)反应速度快,单体一经引发,即迅速进行聚合反应,瞬间形成大分子。
(3)体系中始终没有从低分子量到高分子量的中间产物,反应无法停留在中间阶段,也无法分离出稳定的中间产物。
(4)反应是不可逆的反应。
(5)产物分子量大,但分布较窄,即分子量差别不大。
3、分类(1)均聚合反应:同种单体分子间的聚合反应。
(2)共聚合反应:两种以上单体分子间的聚合反应。
4、自由基的产生 (1)自由基化合物的共价键发生均裂反应,形成两个带独电子的中性基团。
R R ∙∙−−→−均裂2R · (2)自由基的相对活性顺序:∙H >3H C ∙>56H C ∙>2H C R ∙>H C R ∙2>∙C Cl 3>∙C R 3>∙C Br 3>HCORC R ∙>HCN C R ∙>HCOOR C R ∙>22H C CH CH ∙=>256H C H C ∙>()H C H C ∙256>l C ∙>()∙C H C 356 考虑:(1)共轭效应:没有共轭效应的自由基活泼,有共轭效应的自由基不活泼。
(2)极性效应:吸电子基团常合自由基稳定,推电子基团使自由基活泼。
(3)空间位阻效应:体积较大基团使自由基活性降低→主导地位 5、自由基的反应特征含有未成键的电子,具有很高的反应活性,可以发生如下反应:(1)自由基的加成反应→→链增长的基础R ·+CH 2=CH 2 R -CH 2-CH 2(2)自由基的夺取原子反应→→链转移和歧化终止的基础a 、夺取其他分子上的氢原子,本身失去活性,同时产生新的自由基。
R ·+R ′SH RH +R ′S ·b 、自由基之间相互夺取原子,同时失去活性,形成一个饱和一个不饱和的化合物。


1.推导自由基聚合动力学方程运用时用了三个基本假设,分别是:稳态、等活性、聚合度足够大且链转移无影响。
假如Rp对[l]的反应级数为0.8,说明兼有单基双基终止,Rp对[M]的反应级数为1.5,说明单体浓度对链引发速率有影响。
2.自由基聚合常用的引发剂可分为偶氮类、有机过氧类、无机过氧类、氧化-还原体系四类,其中氧化-还原体系可以低温引发。
3.单体的相对活性习惯上用竞聚率的倒数判定,自由基的相对活性习惯上用R12判定。
4.苯乙烯与马来酸酐共聚属交替共聚;苯乙烯与丁二烯进行无规共聚得丁苯橡胶SBR。
接枝共聚得高抗冲聚苯乙烯HIPS,嵌段共聚得热塑性弹性体SBS。
5.自由基聚合的方法有本体、溶液、乳液和悬浮。
6.采纳阴离子活性聚合分步加料制备MMA-St嵌段共聚物,其加料次序为先加苯乙烯,后加MMA 。
7.氢卤酸不能(能或不能)用作阳离子聚合的引发剂,缘由是卤离子亲核实力强,易与质子或阳离子共价。
8.阴离子活性聚合可用于制备遥爪聚合物,在活性聚合体系中加入CO2,则聚合物端基为羧基,加入环氧乙烷,则聚合物端基为羟基。
9.开环聚合的推动力为单体的环张力。
10.典型的Ziegler引发剂为TiCI4-AIEt3 ,典型的Natta引发剂为TiCI3-AIEt2 。
11.环氧树脂的固化剂有胺类和酸酐两大类,环氧值的定义为100g树脂中环氧基团的量(mol)。
12.用Carothers方法计算的凝胶点大于(大于、等于、小于)实测值,Flory方法计算的凝胶点小于(大于、等于、小于)实测值。
13.顺丁橡胶采纳硫化交联,二元乙丙橡胶采纳过氧化物自由基交联。
14.聚合度变大的反应通常有接枝、交联、扩链、嵌段。
16.推断引发剂活性的大小可用活化能、残留分率、引发速率、半衰期为标准。
通常引发剂的引发效率达不到100%. 其主要缘由是诱导分解和笼蔽效应。
18.某对单体相聚,r1=0.75,r2=0.20。
其共聚曲线与对角线的交点称为横比点,该点的共聚物组成为F1= 0.76 。

自由基聚合习题参考答案2. 下列烯类单体适于何种机理聚合自由基聚合、阳离子聚合还是阴离子聚合并说明原因。
CH 2=CHCl CH 2=CCl 2 CH 2=CHCN CH 2=C(CN)2 CH 2=CHCH 3 CH 2=C(CH 3)2 CH 2=CHC 6H 5 CF 2=CF 2 CH 2=C(CN)COOR CH 2=C(CH 3)-CH=CH 2答:CH 2=CHCl :适合自由基聚合,Cl 原子是吸电子基团,也有共轭效应,但均较弱。
CH 2=CCl 2:自由基及阴离子聚合,两个吸电子基团。
CH 2=CHCN :自由基及阴离子聚合,CN 为吸电子基团。
CH 2=C(CN)2:阴离子聚合,两个吸电子基团(CN )。
CH 2=CHCH 3:配位聚合,甲基(CH 3)供电性弱。
CH 2=CHC 6H 5:三种机理均可,共轭体系。
CF 2=CF 2:自由基聚合,对称结构,但氟原子半径小。
CH 2=C(CN)COOR :阴离子聚合,取代基为两个吸电子基(CN 及COOR ) CH 2=C(CH 3)-CH=CH 2:三种机理均可,共轭体系。
3. 下列单体能否进行自由基聚合,并说明原因。
CH 2=C(C 6H 5)2 ClCH=CHClCH 2=C(CH 3)C 2H 5 CH 3CH=CHCH 3CH 2=CHOCOCH 3 CH 2=C(CH 3)COOCH 3 CH 3CH=CHCOOCH 3 CF 2=CFCl 答:CH 2=C(C 6H 5)2:不能,两个苯基取代基位阻大小。
ClCH=CHCl :不能,位阻效应,对称结构,极化程度低。
CH 2=C(CH 3)C 2H 5:不能,二个推电子基,只能进行阳离子聚合。
CH 3CH=CHCH 3:不能,位阻效应,结构对称,极化程度低。
CH 2=CHOCOCH 3:醋酸乙烯酯,能,吸电子基团。
CH 2=C(CH 3)COOCH 3:甲基丙烯酸甲酯,能。
自由基聚合的四种方法自由基聚合是高分子化学中最常用的聚合方法之一,它是通过自由基引发剂引发的聚合反应,将单体分子聚合成高分子链的过程。
自由基聚合方法具有操作简单、反应条件温和、适用范围广等优点,在工业生产中得到广泛应用。
本文将介绍自由基聚合的四种方法。
一、自由基聚合反应自由基聚合反应是一种通过引发剂产生自由基,引发单体分子聚合成高分子链的反应。
自由基聚合反应的一般过程如下:1. 引发剂产生自由基2. 自由基引发单体分子聚合3. 高分子链不断增长4. 反应结束,高分子链停止增长自由基聚合反应的引发剂有很多种,常用的有过氧化物、亚硝酸盐、过硫酸盐等。
引发剂的选择要考虑到反应温度、反应速率、反应产物等因素。
二、自由基聚合的溶液聚合法自由基聚合的溶液聚合法是将单体分子溶解在合适的溶剂中,加入引发剂后进行聚合反应。
这种方法适用于聚合物的分子量较低,分子结构较简单的情况。
溶液聚合法的优点是反应条件温和,反应速率较快,但产品纯度较低。
三、自由基聚合的悬浮聚合法自由基聚合的悬浮聚合法是将单体分子悬浮在水中,加入引发剂后进行聚合反应。
这种方法适用于聚合物的分子量较高,分子结构较复杂的情况。
悬浮聚合法的优点是反应条件温和,反应速率较快,产品纯度较高。
四、自由基聚合的乳液聚合法自由基聚合的乳液聚合法是将单体分子和表面活性剂混合,形成乳液后加入引发剂进行聚合反应。
这种方法适用于聚合物的分子量较高,分子结构较复杂的情况。
乳液聚合法的优点是反应条件温和,反应速率较快,产品纯度较高。
此外,乳液聚合法的产品具有较好的分散性和稳定性,可广泛应用于涂料、胶粘剂等领域。
总之,自由基聚合是一种常用的高分子化学方法,具有操作简单、反应条件温和、适用范围广等优点。
不同的自由基聚合方法适用于不同的聚合物分子结构和分子量,选择合适的方法可以提高反应效率和产品质量。
第二章自由基聚合(radical polymerization)【课时安排】2.1 单体的聚合能力2学时2.2 自由基聚合机理4学时2.3 链引发反应3学时2.4 聚合反应动力学2学时2.5 相对分子质量1学时2.6 链转移反应2学时2.7 聚合方法4学时总计18学时【掌握内容】1.单体聚合能力:热力学(△E, △S,T,P);动力学(空间效应-聚合能力,电子效应-聚合类型)2.自由基基元反应每步反应特征,自由基聚合反应特征3.常用引发剂的种类和符号,引发剂分解反应式,表征方法(四个参数),引发剂效率,诱导效应,笼蔽效应,引发剂选择原则4.聚合动力学:聚合初期:三个假设,四个条件,反应级数的变化,影响速率的四因素(M,I,T,P);聚合中后期的反应速率的研究:自动加速现象,凝胶效应,沉淀效应;聚合反应类型5.相对分子质量:动力学链长,聚合度及影响其的四因素(M,I,T,P),6.链转移:类型,聚合度,动力学分析,阻聚与缓聚7.本体,溶液,悬浮,乳液四大聚合方法配方,基本组成,优缺点及主要品种【熟悉内容】1.热、光、辐射聚合。
2.聚合动力学研究方法。
3 自由基聚合的相对分子质量分布。
4 悬浮聚合与乳液聚合所用分散剂种类、聚合过程。
【了解内容】1. 通用单体来源。
2. 自由基聚合进展。
【教学难点】1. 对具体单体聚合热力学与动力学的综合分析2. 终止方式的相对比例及其与体系状态的关系3. 氧化还原类的反应式;笼蔽效应与诱导效应4. 不同条件下反应速率对单体与引发剂浓度的反应级数的推导与分析5. 区别聚合反应速率、动力学链长、平均聚合度的影响因素和变化趋势6. 向不同转移对象的链转移程度的难易分析7. 乳液聚合机理及动力学【教学目标】1. 掌握自由基聚合相关基本概念。
2. 掌握自由基聚合常见单体、引发剂、阻聚剂、聚合方法。
3. 达到如下技能:(1)单体聚合能力的判断与类型的选择(2)引发剂的选择及正确书写引发反应式(3)正确书写任一体系的基元反应式(4)根据动力学方程计算各参数,选择适当方法控制反应进程(5) 根据相对分子质量方程计算各参数,选择适当方法控制产物结构(6)设计聚合工艺,线路与配方2.1 单体的聚合能力【教学内容】2.1.1 聚合热力学一聚合热二聚合熵三聚合温度四小结2.1.2 聚合动力学一连锁聚合种类与活性中心二单体对聚合类型的选择及聚合能力1 取代基对聚合能力的影响(空间效应)2 取代基对聚合类型的选择(电子效应)3 单体共聚能力【授课时间】2学时【教学重点】1影响聚合热的主要因素及其规律2单体对聚合类型的选择及聚合能力【教学难点】1影响聚合热的主要因素及其规律2 对具体单体聚合热力学与动力学的综合分析【教学目标】1 掌握影响聚合热的主要因素及其规律2掌握取代基对单体聚合类型选择及聚合能力的影响规律3 能正确综合分析具体单体的聚合热力学与动力学行为【教学手段】课堂讲授,辅以实例练习【教学过程】聚合能力:化学结构:两个可相互反应官能团常见聚合单体类型两个以上有机官能团单体C=C-X热力学:方向,限度,∆G<0 R-C=O动力学: 聚合方法杂环(O,N,P,S)2.1.1 聚合热力学∆G=∆H-T∆S= ∆E+P∆V-T∆S<0 聚合;=0 达到平衡;>0 解聚一聚合热∆H=∆E+P∆V1 内能变化∆E=∆E f+∆E R+∆E s+∆E’=( E fp - E fm)+( E Rp - E Rm)+( E sp - E sm)+ ∆E’E f------由键能所贡献的内能E R-----由共振效应所贡献的内能E s------由空间张力或位阻效应所贡献的内能∆E’----其它因素引起的内能变化(1) 双键断裂能CH 2=CH 2 -CH 2-CH 2- ∆E f =εm -εp=609.2-2×351.7=-94.2 kJ.mol -1 (实测值∆H=-88.8 kJ.mol -1)(2)共轭效应增强,|—∆H|减小(3)位阻效应增强,|—∆H|减小 (4)氢键与溶剂化作用增强,|—∆H|减小 (5)强电负性取代基的存在使|—∆H|增强(6)需具体综合分析2 压力影响: 压力增大,有利于聚合物进行二 聚合熵 ∆S=-100~-125 kJ.mol -1三 聚合温度1 聚合上限温度∆G=∆H-T ∆S=0→T c =∆H/∆S (不同压力与活度下数值)→T c 有一系列,对应一系列平衡单体浓度→常规定[M] e =1mol/L 时T c 为聚合上限温度→T c =∆H 0/∆S 02 平衡单体浓度eo oM RT S H Tc ]ln[+∆∆= 四 小结增强聚合倾向内因 ∆S 影响不大∆E: 降低共轭效应, 降低位阻效应, 降低氢键与溶剂化作用,增强强电负性取代基 外因 增大压力,降低温度可解释α-甲基苯乙烯(α-MeSt )的聚合现象2.1.2 聚合动力学一 连锁聚合种类与活性中心根据引发活性种与链增长活性中心的不同,链式聚合反应可分为自由基聚合、阳离子聚合、阴离子聚合和配位聚合等二 单体对聚合类型的选择及聚合能力1 取代基对聚合能力的影响(空间效应)(1)单取代能聚合(2)双取代一般可以聚合,但基团太大时难以聚合(3)三、四取代一般不可以聚合,氟取代除外2 取代基对聚合类型的选择(电子效应)(1) 取代基的诱导效应A A 自由基:2A A CH 2CH XA B 阳离子CH =CHX A CH 2H C X离解A +B -δ+B δ-A B 阴离子A CH 2H C X 离解A -B +δ+B δ-带给电子基团的烯类单体易进行阳离子聚合带吸电子基团的烯类单体易进行阴离子聚合与自由基带强给电子基团、强吸电子基团的烯类单体只能分别进行阳离子、阴离子聚合(2) 取代基的共轭效应:流动性大,易诱导极化,可进行多种机理的聚合反应(3) 带不同基团的单体进行几种聚合时的排序 阳离子聚合取代基-X: -NO 2,-CN,-F,-Cl,-COOCH 3,-CONH 2,-OCOR,-CH=CH 2,-C 6H 5,-CH 3,-OR 自由基聚合阴离子聚合3 单体共聚能力:与参与共聚的各种单体均有关2.2 自由基聚合机理【教学内容】2.2.1 自由基2.2.2 自由基聚合的基元反应一 链引发反应(chain initiation )二 链增长反应(chain growth )三 链终止反应(chain termination )四 链转移反应(chain transfer )2.2.3 自由基聚合的反应特征【授课时间】4学时【教学重点】自由基聚合的基元反应;自由基聚合反应特征【教学难点】终止方式的相对比例及其与体系状态的关系【教学目标】1 掌握自由基聚合机理2 掌握自由基聚合反应特征3 能正确写出具体聚合物的基元反应式【教学手段】课堂讲授,配以Flash 动画演示,辅以学生讨论【教学过程】2.2.1 自由基一 分类与产生二 活性1 影响因素:共轭效应大,吸电子诱导效应大,位阻效应强,稳定性强,活性小2 活性顺序三 反应:加成反应,氧化还原反应,偶合反应,脱氢反应,消去反应2.2.2 自由基聚合的基元反应一 链引发反应(chain initiation ) 慢 单体自由基引发剂引发为例二 链增长反应(chain growth ) 快 活性高分子链I 2 Ik I k i I CH 2引发活性种,初级自由基,引发自由基H 2C CHX +CHX M链结构在该步形成:序列结构→头尾为主顺反结构→温度升高有利于顺式结构生成立体结构→无规结构三 链终止反应(chain termination) 速 稳定大分子1 双基终止(均相体系,主要方式) PS,PAN 偶合为主; PMMA 偶合歧化兼有; PVAc 歧化为主问题:k t >>k p , 为何还可得到大分子?2 单基终止四 链转移反应(chain transfer )一定条件下 不同活性的链自由基 2.2.3 自由基聚合的反应特征 1 慢反应,快增长,速终止234 放热反应,低温有利2.3 链引发反应k I CH 2H 2C CH+CH XMCH 2CH CH 2CH 2偶合:CH 2CH CH CH 2歧化:CH 2CH 2CH 2CH 2CH CH X +k k k CH 2+CH S + SCH 2CH 2【教学内容】2.3.1 引发剂类型一 热分解型二 氧化还原类2.3.2 引发剂活性(表征方法)2.3.3 引发剂效率f2.3.4 引发剂的选择【授课时间】2学时【教学重点】典型类型引发剂;引发剂活性表征方法;引发剂效率及影响因素;引发剂的选择原则【教学难点】氧化还原类的反应式;笼蔽效应与诱导效应【教学目标】1 掌握引发剂活性表示方法及其计算方法2掌握引发剂效率、笼蔽效应、诱导效应等基本概念3能正确写出典型引发剂的结构式与引发反应式4 能根据具体要求选择匹配的引发剂【教学手段】课堂讲授,辅以多媒体幻灯图片及实例【教学过程】2.3.1 引发剂类型一 热分解型(Ed=80~140kJ/mol ,中高温使用)1 偶氮类引发剂2 过氧类引发剂(1) 有机过氧类a 烷基过氧化氢(RC-O-O-H):异丙苯过氧化氢(CHP ),叔丁基过氧化氢(t-BHP)b 二烷基过氧化物(R-O-O-R ’):过氧化二异丙苯c 过氧化酯(RCOOCR ’)d 过氧化二酰(RCOOOCOR ’)R 1C R 2N N C R 1R 2R C R N N C R 1R 2对称不对称(X=吸电子取代基)H 3C C CH 3CN N N C CH 3CH 3CN H 3C C CH 3CN 2+ N 2偶氮二异丁腈(AIBN)Ph C O C O Ph 2Ph C O O Ph C O OPh + CO 2过氧化苯甲酰(BPO )e 过氧化二碳酸酯(ROOC-O-O-COOR ’):过氧化二碳酸二异丙酯(IPP)(2) 无机过氧类:S 2O 82─→2 SO 4‧─二 氧化还原类(Ed=40~60kJ/mol ,低温使用)1 水溶性(1) 生成一种R ‧HOOH + Fe 2+→HO ‧+OH ─+Fe 3+S 2O 82─+ Fe 2+ →SO 42─ + SO 4‧─+Fe 3+用量:还原剂<氧化剂,否则Fe 2++‧OH →Fe 3++OH ─白白消耗自由基(2) 生成一种R ‧S 2O 82─+ SO 32─→SO 42─+ SO 4‧─+ SO 3‧─2 油溶性2.3.2 引发剂活性(表征方法)一 分解速率常数kd 越大,引发剂活性越大I k dt I d R d d =-=][二 分解活化能Ed 越小,引发剂活性越大三 半衰期t 1/2越小,引发剂活性越大2/12/][][ln t k I I d o o = 四 残留分率[I]/[I]o 越小,引发剂活性越大2.3.3 引发剂效率f一二 笼蔽效应(Cage Effect)引发剂分解产生的初级自由基,在开始的瞬间被溶剂分子所包围,不能与单体分子接触,无法发生链引发反应。
自由基聚合的四种方法自由基聚合是一种常用的化学合成方法,它可以通过自由基中间体对单体进行链式反应从而合成高分子材料。
在实际应用中,为了获得具有特定性质的高分子材料,需要选择不同的自由基聚合方法来进行合成。
下面,我们将介绍四种典型的自由基聚合方法。
1.自由基串接聚合该方法是以低分子量二乙烯酸乙酯和甲基丙烯酸甲酯为反应物,过氧化苯甲酰作为引发剂,通过双键的形式进行反应,得到大分子化合物。
其反应过程如下:(1)引发剂过氧化苯甲酰分解成自由基;(2)自由基与单体形成新的自由基;(3)新自由基与另一单体形成双自由基;(4)重复步骤(2)(3)直到生成高分子。
2.自由基间隙聚合该方法是选取单体,如苯乙烯、丙烯酸酯类等,选取引发剂,通过当两者符合反应条件,进行自由基间隙聚合生成高分子。
其反应过程如下:(1)引发剂引发双倍半胱氨酸生成自由基;(2)自由基与单体结合生成新自由基;(3)新自由基再次结合,两个自由基形成链式反应;(4)链式反应不断重复,生成高分子。
3.自由基复合聚合该方法是指将两种或两种以上的单体进行反应,形成带有相同或不同的官能团的线性、支化或网状高分子。
反应需要选择复合引发剂,以一定的条件对反应体系深度控制。
其反应过程如下:(1)复合引发剂分解出自由基;(2)自由基与不同单体结合;(3)自由基再次分解自由基,反应物不断增加;(4)反应物分子量逐渐增大,直到得到高分子。
4.交联自由基聚合该方法是将具有交联剂作用的单体添加到反应体系中,形成交联结构的高分子。
它可通过单体中的双键进行反应,或者选择交联剂将其两个丙烯酰基与单体聚合,生成的高分子有较好的拉伸性和弹性。
其反应过程如下:(1)引发剂引发单体形成自由基;(2)单体与交联剂进行自由基反应;(3)自由基结构增加,交联结构不断形成;(4)高分子产生,交联不断增强。
总之,自由基聚合是高分子合成中的基本方法,不同的自由基聚合方法对于不同的高分子材料具有不同的优势,因此在实际应用中需要选择合适的自由基聚合方法。
自由基聚合法是一种常用的高分子合成方法,通过自由基引发剂引发聚合反应,使单体分子在自由基的作用下进行链增长,最终形成高分子聚合物。
下面将对自由基聚合法的原理、特点、影响因素以及应用进行详细介绍。
一、原理自由基聚合法是通过引发剂引发单体分子产生自由基,从而进行链增长的过程。
自由基是由一个未成对电子和空轨道组成的活性分子,具有高度的反应活性。
在聚合过程中,自由基从单体分子上夺取一个氢原子,使单体分子成为自由基,进而进行链增长。
随着反应的进行,不断有新的自由基产生,最终形成高分子聚合物。
二、特点自由基聚合法具有以下特点:1. 聚合反应速度快,可以在较短的时间内获得高分子量聚合物。
2. 可以通过调节引发剂的用量控制聚合反应速率和聚合物分子量。
3. 适用于大多数天然和合成单体的聚合,应用范围广泛。
4. 自由基聚合过程中会产生大量热量,需要进行冷却以避免温度升高对聚合物性能的影响。
三、影响因素自由基聚合法的反应速度和聚合物分子量受到多种因素的影响,主要包括以下几个方面:1. 单体浓度:单体浓度越高,聚合反应速率越快,聚合物分子量越大。
2. 引发剂浓度:引发剂浓度越高,聚合反应速率越快,但引发剂用量过多会导致聚合物分子量降低。
3. 温度:温度升高可以加快聚合反应速率,但过高的温度会导致聚合物分子量降低。
4. 溶剂和介质:溶剂和介质对自由基聚合反应也有影响,不同的溶剂和介质对聚合反应速率和聚合物分子量有不同的影响。
四、应用自由基聚合法在工业上得到了广泛的应用,主要用于合成纤维、橡胶、塑料、涂料、粘合剂等高分子材料。
通过自由基聚合法可以合成不同分子量、不同性能的高分子材料,满足不同领域的需求。
例如,通过自由基聚合法可以合成聚乙烯、聚丙烯、聚氯乙烯等塑料,也可以合成纤维如尼龙、涤纶等。
此外,自由基聚合法还可以用于制备功能高分子材料,如导电聚合物、磁性聚合物、药物载体等。
总之,自由基聚合法是一种常用的高分子合成方法,具有反应速度快、应用范围广泛等特点。
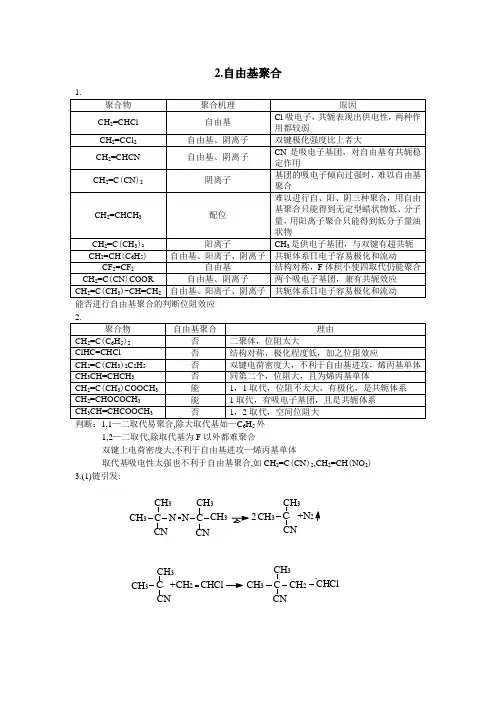
2.自由基聚合能否进行自由基聚合的判断位阻效应判断:1,1—二取代易聚合,除大取代基如—C6H5外1,2—二取代,除取代基为F以外都难聚合双键上电荷密度大,不利于自由基进攻—烯丙基单体取代基吸电性太强也不利于自由基聚合,如CH2=C(CN)2,CH2=CH(NO2) 3.(1)链引发:CH3C NCNC·CH3CH3CH3NCH3CNC CH32CN+N2CH2CHCl CHCl·CNCH3CH3C·+CH2CH3CCH3(2)链增长:(3)链终止: 偶合:歧化:4.自由基聚合时转化率和分子量随时间变化的特征:转化率随时间逐步提高,中间有自加速现象,分子量随时间变化甚小(短时间后变化很小).与反应机理决定,连锁聚合时RM ·→Mn ·时间极短,没有中间停留阶段。
5.引发剂(1)偶氮二异丁腈(AIBN )、(2)偶氮二异庚腈(ABVN )、(3)过氧化二苯甲酰(BPO )、(4)过氧化二碳酸二乙基己酯(EHP )、(6)过硫酸钾-亚硫酸盐体系、(7)过氧化氢-亚铁盐体系的分解反应式见书本的P26~29,(5)异丙苯过氧化氢的见下面:其中(1)~(5)为偶氮类和有机过氧类,属于油溶性引发剂常用于本体、悬浮和溶液(有机溶剂)聚合,(6)(7)为水溶性氧化-还原体系,适合于水溶液和乳液聚合。
CH 2CHCl CHCl ·CH 2CH 3CN C CH 3CHCl ·CH 3C CH 3+CH 2CH 2CHCl CHCl CH 3C CH 3CH 2n-1CH 2CHCl ·CH 2CHCl CH 3C CH 3CH 2CHCl CH 3CN C CH 32n 2CHCl CH 3CN C CH 3CH 2n-1CH 2CHCl ·2CHCl CH 3C CH 3CH 2n-1CH 2CHCl·CHCl CH 2n-1CH 2CH 3C CH 3CH 2Cl CHCl CH 2n-1CH 3CN C CH 3CH CHCl +COOH CO ··OH CH 3C CH 3CH 3C CH 3+注意:EHP 高活性,配成溶液后低温贮。
6.用-ln][][I I =k d t 作图,求k d ,得分解速率k d =0.61hr -1=1.76×10-4s -1 半衰期t 1/2=ln2/k d7.用k d =Aexp (-E d /RT ) lnk d ~1/T 作图,斜率为E d ,得125KJ/mol 。
8.(1)常数A 、B 与频率因子、活化能的关系:t 1/2=dk 2ln k d =A d exp (-E d /RT ) logt 1/2=logln2-logk d =logln2-logA d +(E d /RT )loge比较logt 1/2=A/T-B 与上式得A=(loge /R )E d ,B= log (A d /ln2)(2)当t 1/2=10hr 时,logt 1/2=A/T-B=log10=1,得A/T (t 1/2=10hr )-B=1当t 1/2=1hr 时,logt 1/2=A/T-B=log1=0,得A/ T (t 1/2=1hr )-B=0 则A= T (t 1/2=10hr )T (t 1/2=1hr )/(T (t 1/2=1hr )- T (t 1/2=10hr ))B=T (t1/2=10hr )/(T (t 1/2=1hr )- T (t 1/2=10hr ))由此可见了解半衰期为10hr 和1hr 时的分解温度便于计算A 、B 值,粗略知道反应温度与分解温度的关系,选择引发剂,当t 1/2=1hr 时,反应时间为几小时,温度不限,应注意此时的T 是否在可能的反应温度范围内。
当t 1/2=10hr 时,反应时间为几十小时,此时T 是反应温度的下限。
二者差值(T (t 1/2=1hr )- T (t 1/2=10hr ))大,则反应活化能小。
(3)已知过氧化二碳酸二异丙酯半衰期为10hr 和1hr 时的分解温度分别为45和61℃,得常数A 、B 为A=6.6×103,B=1.99×109.引发剂效率—指引发剂分解生成的自由基中能用于引发聚合的百分数f 。
诱导分解—指链自由基向引发剂的转移反应。
原来的链自由基在形成稳定分子的同时,形成了一个新的自由基。
由于无终止的消耗了一个引发剂分子,故使实际f 降低。
笼蔽效应—由于初级自由基受溶剂分子包围,限制了自由基的扩散,导致初级自由基的偶合(或歧化)终止,使引发剂效率降低。
“溶剂包围之中”由于计算引发效率时不对诱导分解造成的损失校正,故f 包括了诱导分解。
例:笼蔽效应(AIBN )CH 3C N CH 3N CH 3CNC CH 3]+N 2[2CH 3CH 3C ·CH 3CH 3C CH 3N C C CH 3]+N 2CH 3C CNCH 3N CH 3CNN C CH 3[]+N 2笼子偶合加成诱导分解(BPO ):10.光引发聚合:单体在光的激发下形成自由基聚合 直接光引发聚合:即非光敏聚合 光敏聚合(加有光敏剂):光敏直接引发聚合—光敏剂在光照时直接分解的自由基 光敏见解引发聚合—光敏剂吸收光能,传递给单体 11.推导自由基聚合动力学方程时的四个基本假定:(1) 暂不考虑链的转移和支化,链的终止方式为双基终止 聚合度很大,R=R p (2) 等活性 (3) 稳态处理结论:R p ∝[I]1/2,双基终止R i =k d [I]与[M]无关,有关则R ∝[M] 局限:单基终止时则不能用 修正:R t =k t [M •] R=k p [M] R i / k t =2 k p k d / k t [M] [I]热引发时:R i =2k i [M]3=2 k t [M •]2 R p = k p [M](R i / 2k t )1/2R= k p [M](k i [M]3/ k t )1/2= k p (k i / k t )1/2[M]5/2如采用R i =k i [M]3则R= k p (k i / 2k t )1/2[M]5/212.体系几个增长链A 为偶合,1-A 歧化,相对量以动力学链为100%,B 代表每一个大分子含有的引发剂残基。
B=2/(2-A ) 则A=(2B-2)/B 1-A=(2-B )/B A :1-A=(2B-2):(2-B )=(2×1.3-2):(2-1.3)=6:7·CH 2·C OO ·C O O +C O OO O +偶合A=6/13=46.2% 1-A=53.8%其他算法:偶合的分子个数X ,歧化Y , 2X+Y=1.3(X+Y ),从引发剂残基考虑X :Y=3:7 则动力学链为2X :Y=6:7,偶合占6/13,歧化7/13 13.(1)假定[I]不变,则-ln[M]/[M]0=k p (fk d /k t )1/2[I]t k d =ln2/t 1/2=ln2/44×3600=4.38×10-6(S -1) k p =145(L/mol .s ) k t =7.0×107(L/mol .s ) f=0.8[I]=4.0×10-3(mol/L ) [M]/[M]0=50%则t 2122176)100.4()100.71038.48.0(14521ln --⨯⨯⨯⨯=- 则t=94hr 超过了引发剂半衰期(2)推荐使用:)1(][2)()]([)(][][ln 2/2/102/102/102/10--==---⎰t k dt d p t t k t d p d d e k I k fk k dt e I k fk k M M则)1(1038.4)100.4(2)100.71038.48.0(145221ln 2/1038.462/122/1766-⨯-⨯⨯⨯⨯⨯⨯⨯=--⨯----t e 则)e-0.937?(10.693-610-2.19t⨯=t=0.6147×106(s )≈171hr14.苯乙烯 d=0.887g/ml本体聚合:[M]=0.887/104=8.53×10-3(mol/ml )=8.53(mol/l )[I]=(0.109%×0.887)×103/242≈4×10-3(mol/l ) 苯乙烯以偶合终止为主:n x =2ν=2R p /R i(1)R i =2R p /n x =246010255.024-⨯⨯=2.07×10-8(mol/L.s )由R i =2k d f[I] f=0.8(2)k d = R i /2f[I]=381048.021007.2--⨯⨯⨯⨯=3.23×10-6(s -1) (3)自由基寿命τ=[ M ·]/R t =1/2k t [M ·]=k p [M]/ 2 k t R p64p 109.453.882.010255.02][2R --⨯=⨯⨯⨯==M k k t p τ在测定k t 、k p 时,由R p =k p [M][ M ·] R i =2k t [M ·]2得4282422P 21063.833.81007.2)10255.0(2][2R ---⨯=⨯⨯⨯⨯==M R k k I t Pk p =1.76×102(l//mol.s ) k t =3.59×107(l//mol.s )数量级k t > k p » k d(5)[M ·]= 1/2k t τ=1/(2×3.6×10-7×0.82)=1.7×10-8(mol/l )[M] »[M ·](6)R t =[M ·]/τ=1.7×10-8/0.82=2.1×10-8(mol/L.s ) ∴R p »R p =R i15.(1)聚合速率:k=Ae-E/RT][][)(2/12/1M I k fk k R td p p =→一般式 E ˊ=E p +E d /2-E t /2=32.6+125.6/2-10/2=90.4(KJ/mol )(2)2/12/12/12/12][)2(][][)2(][2][][2]][[I k fk M k R M k k M k M k M k M M k R R R R t d p i t p t p t p tp ip ==•=••===ν(n x =2ν)E ˊ=E p -E d /2-E t /2=32.6-125.6/2-10/2=-35.2(KJ/mol )(1) 聚合速率:当T 从50℃→60℃)11(//121212//T T R E RT E RT E ee e k k ----==75.2/)502731602731(31.8104.90123==+-+⨯-ek k当T 从80℃→90℃ 34.2/)802731902731(31.8104.90123==+-+⨯-ek k温度升高,速率增大,在低温区变的更明显 (2) 聚合度674.0)()()502731602731(31.8105.32)11(//12121231212====''==+-+⨯----'-'-eee e k k x x T T R e RT E RT E n n νν718.0)()()802731902731(31.8105.32123==+-+⨯--e x x n n温度升高,聚合度降低,在高温区变的更明显(3) 若为光引发:E d =0 E=E p -E t /2=32.6-10/2=-27.6(KJ/mol )E ˊ=E p -E t /2 (相同)聚合速率:50℃→60℃ 36.1/)502731602731(31.8106.27123==+-+⨯-ek k聚合度:36.1)()(121212===R R x x n n νν k p 、n x 变化一致,温度升高,k p 、n x 将增加80℃→90℃ 29.1/)802731902731(31.8106.27123==+-+⨯-ek k29.1)()(12=n n x x在低温变化将更明显,但总的来说变化不大。