First-Principles Study of La Doping Effects on the Electronic Structures and Photocatalyti
- 格式:pdf
- 大小:2.46 MB
- 文档页数:7

第50卷第2期2021年2月人 工 晶 体 学 报JOURNALOFSYNTHETICCRYSTALSVol.50 No.2February,2021S La共掺杂ZnO性能的第一性原理研究赵旭东,崔瑞瑞,邓朝勇(贵州大学大数据与信息工程学院,贵阳 550025)摘要:采用基于广义梯度近似的第一性原理方法,研究了纯ZnO、S单掺、La单掺和S La共掺ZnO的能带结构、态密度和光学性质。
S单掺ZnO后,价带和导带同时向低能量转移,导致带隙减小。
La单掺ZnO后在导带底产生杂质能级使得带隙减小。
SLa共掺ZnO导致La的局部化减弱,表明La形成的施主能级由于S的3p态的影响变得更浅,从而减小了带隙。
和纯ZnO相比,掺杂后的ZnO吸收系数和反射系数都减小,导致透射能力增强,为ZnO作为透明导电氧化物应用于太阳能电池提供了潜在的理论依据。
关键词:第一性原理;ZnO;光学性质;电子结构;S La共掺ZnO中图分类号:TN36;O471.5 文献标志码:A 文章编号:1000 985X(2021)02 0266 07First PrinciplesStudyonPropertiesofS LaCo DopedZnOZHAOXudong,CUIRuirui,DENGChaoyong(CollegeofBigDataandandInformationEngineering,GuizhouUniversity,Guiyang550025,China)Abstract:Thebandstructure,densityofstatesandopticalpropertiesofpureZnO,Ssingle doped,Lasingle dopedandS Laco dopedZnOwerestudiedbyfirst principlesmethodbasedonthegeneralizedgradientapproximation.AfterZnOdopedwithS,thevalencebandandconductionbandtransfertolowenergyatthesametime,whichleadstothereductionofbandgap.ThebandgapofZnOdopedwithLareducesbecauseoftheimpurityenergylevelatthebaseoftheconductionband.ThelocalizationofS Laco dopedZnOresultsintheweakeningofLalocalization,whichindicatesthatthedonorenergylevelofLabecomesshallowerduetotheinfluenceofS3pstate,thusreducingthebandgap.Afterdoping,thebandgapofZnOdecreasesobviously,whichincreasestheabsorptionabilityofZnOinvisiblelightregion,andfurtherproducesmorephoto electricchargecarrierstoimprovethephotocatalyticactivityofZnO.ComparedwithpureZnO,thedopedZnOhaslowerabsorptioncoefficientandreflectioncoefficient,andstrongertransmissionability,whichprovidesapotentialtheoreticalbasisfortheapplicationofZnOastransparentconductiveoxideinsolarcells.Keywords:first principle;ZnO;opticalproperty;electronicstructure;S Laco dopedZnO 收稿日期:2020 11 29 基金项目:国家自然科学基金(51762010);贵州省高层次创新型人才百层次人才(2015 4006) 作者简介:赵旭东(1997—),男,山西省人,硕士研究生。

第53卷第2期2024年2月人㊀工㊀晶㊀体㊀学㊀报JOURNAL OF SYNTHETIC CRYSTALSVol.53㊀No.2February,2024Ni,Cu,Zn掺杂四方相PbTiO3力学性能㊁电子结构与光学性质的第一性原理研究王云杰1,2,张志远1,2,文杜林1,2,吴侦成1,2,苏㊀欣1,2(1.伊犁师范大学物理科学与技术学院,伊宁㊀835000;2.伊犁师范大学新疆凝聚态相变与微结构实验室,伊宁㊀835000)摘要:采用第一性原理研究了四方相钙钛矿PbTiO3以及Ni㊁Cu㊁Zn掺杂PbTiO3的力学性能㊁电子结构和光学性质㊂力学性能计算结果表明,Ni掺杂PbTiO3的体积模量㊁剪切模量及弹性模量在三种掺杂体系中最大㊂Ni掺杂体系德拜温度最高㊂G/B为材料的脆㊁韧性判据,Zn掺杂PbTiO3的G/B值最大,说明化学键定向性最高㊂Ni㊁Zn掺杂体系的G/B 范围为0.56<G/B<1.75,均为脆性材料,而本征PbTiO3和Cu掺杂体系G/B值小于0.56,均为韧性材料㊂通过电子结构分析,发现掺杂体系相比于本征体系带隙变窄,跃迁能量减小㊂Ni掺入使得PbTiO3费米能级处出现杂质能级,而Cu㊁Zn掺杂PbTiO3价带顶上移,费米能级进入价带,使得Cu㊁Zn掺杂PbTiO3呈现p型导电特性㊂从复介电函数㊁光学反射谱和吸收谱分析中发现,掺杂体系的静介电常数相较于本征体系有所提升㊂Ni㊁Cu㊁Zn的掺杂使得PbTiO3吸收范围扩展到红外波段,且增强了可见光波段的吸收强度,Cu掺杂PbTiO3材料的光催化特性在本征PbTiO3和三种单掺PbTiO3材料中是最好的㊂关键词:第一性原理;PbTiO3;掺杂;力学性能;电子结构;光学特性中图分类号:O561㊀㊀文献标志码:A㊀㊀文章编号:1000-985X(2024)02-0258-09 First Principles Study on Mechanical Properties,Electronic Structure and Optical Properties of Ni,Cu,Zn Doped Tetragonal PbTiO3WANG Yunjie1,2,ZHANG Zhiyuan1,2,WEN Dulin1,2,WU Zhencheng1,2,SU Xin1,2(1.School of Physical Science and Technology,Yili Normal University,Yining835000,China;2.Xinjiang Laboratory of Phase Transitions and Microstructures of Condensed Matter Physics,Yili Normal University,Yining835000,China) Abstract:The mechanical property,electronic structure,and optical properties of tetragonal perovskite PbTiO3and Ni,Cu, Zn-doped PbTiO3were studied by first principles.The mechanical property calculations show that Ni-doped PbTiO3exhibits the highest values for volume modulus,shear modulus,and elastic modulus among the three doping systems.Notably,the Ni-doped system also has the highest Debye temperature.The G/B ratio represents the material s brittleness and toughness, which is highest for Zn-doped PbTiO3,indicating the highest degree of chemical bond orientation.The G/B range for Ni and Zn-doped systems is0.56<G/B<1.75,indicating brittle materials,while the intrinsic PbTiO3and Cu-doped systems have G/B values less than0.56,indicating ductile materials.The electronic structure reveals that the doped systems have narrower band gaps and reduced transition energies compared to the intrinsic system.The introduction of Ni introduces impurity levels at the Fermi energy level in PbTiO3,while Cu and Zn doping shifts the valence band maximum upwards,causing the Fermi level to enter the valence band and resulting in p-type conductivity for Cu and Zn-doped PbTiO3.The doping of Ni,Cu and Zn expands the absorption range of PbTiO3to the infrared region and enhances the absorption intensity in the visible light range.Among the intrinsic PbTiO3and three single-doped PbTiO3materials,Cu-doped PbTiO3exhibits the best photocatalytic properties.Key words:first principle;PbTiO3;doping;mechanical property;electronic structure;optical property㊀㊀收稿日期:2023-08-02㊀㊀基金项目:伊犁师范大学科研专项提升重点项目(22XKZZ21);伊犁师范大学科研项目(2022YSZD004);伊犁师范大学大学生创新训练项目(S202110764006,YS2022G018);新疆伊犁科技计划(YZ2022Y002);新疆维吾尔自治区天山英才计划第三期(2021-2023)㊀㊀作者简介:王云杰(1999 ),男,新疆维吾尔自治区人,硕士研究生㊂E-mail:1575469121@㊀㊀通信作者:苏㊀欣,博士,副教授㊂E-mail:suxin_phy@㊀第2期王云杰等:Ni,Cu,Zn掺杂四方相PbTiO3力学性能㊁电子结构与光学性质的第一性原理研究259㊀0㊀引㊀㊀言PbTiO3(PTO)作为一种典型的钙钛矿型铁电氧化物,在居里温度(763K)以下为四方相,当处于居里温度(763K)以上时,PTO的相由四方相转变为立方相[1-2]㊂四方相PTO铁电性能较为优异,广泛应用于存储器㊁电换能器㊁微电子㊁无线通信用电介质等设备㊂此外,四方相PTO还具有较大的电光系数和较高的光折变灵敏度[3-5],因此可以用于光学传感器㊁光转换器和光调制器等[6-9]㊂除TiO2催化剂外,Ti基钙钛矿(例如CaTiO3㊁SrTiO3)还参与了自然污染物的光催化脱色和光催化水分解制氢㊂与TiO2一样,这些钙钛矿型催化剂也受到宽禁带的限制,这使得其可见光反应非常困难,光催化能力被减弱[10]㊂钙钛矿晶体结构提供了一个极好的框架,可根据特定光催化反应的要求修改带隙值,以允许可见光吸收和带边能量㊂此外,钙钛矿晶体化合物中的晶格畸变强烈影响光生载流子的分配㊂PTO由于高光催化活性,受到了广泛关注[11]㊂PTO是典型的钙钛矿型铁电氧化物,通常用于电子器件,很少用作光催化剂[12-13]㊂近年来,研究人员发现通过合理的合成方法和材料改性对PTO光催化性能进行改善㊂Hussin等和Niu 等[14-15]基于第一性原理,分别研究了La和N掺杂体系PTO的电子结构,发现La掺杂体系的带隙比本征带隙窄,N掺杂体系的PTO的费米能级进入价带顶部,使得N掺杂体系材料呈现出p型导电特性,能带结构的禁带宽度减小,对于光催化能力有一定的改善,但是关于光学性质方面并没有进行报道㊂李宏光等[16]基于第一性原理,研究了N掺杂体系的光学性质,发现光学吸收能力在可见光区域并没有较大的改善,并且Ti的氧化物进行非金属掺杂时,需要高温处理[17-18],从能量消耗的角度来说是不利的㊂综上所述,确定掺杂位置以及掺杂量成为改善PTO光催化性能的关键㊂而二价金属Ni㊁Cu㊁Zn离子更容易取代Ti4+,使O的电负性变弱,更容易改善PTO性能[19]㊂在文献调研中发现关于PTO力学性能的系统报道大多是基于本征体系[20-22],对掺杂体系的力学性能报道是罕见的,因此有必要对掺杂体系PTO光催化性能研究的同时,也对掺杂体系力学性能的改善进行系统地讨论㊂本文的主要内容是采用密度泛函理论对本征以及单掺Ni㊁Cu㊁Zn四方相PTO(PTOʒNi㊁PTOʒCu㊁PTOʒZn)的力学性能和光电性能展开系统地讨论,以期PTO能够在力学性能以及光催化方面得到更大的改善㊂1㊀理论模型与计算方法四方相PTO晶体是典型的钙钛矿结构,属于P4mm空间群[23],建立共包含40个原子的2ˑ2ˑ2超胞结构,掺杂浓度为12.5%的掺杂体系结构如图1所示,考虑到边界条件的影响,用一个Ni㊁Cu㊁Zn分别去取代超胞中的Ti原子,在超胞中有8个Ti原子的位点,根据晶体的对称性所示这8个位点为等效位点,所以不同的掺杂位置对体系没有影响㊂基于密度泛函理论的第一性原理平面波赝势方法[24-25]应用MaterialsStudio8.0[26]计算了原子各轨域的电子态密度,选择基组为广义梯度近似(general gradient approximate,GGA)下的PBE(Perdew-Burke-Ernzerhof)[27-28]交换-关联泛函,使用超软赝势(ultra-soft pseudopotential,USP)计算本征以及掺杂体系PTO 的力学性能㊁电子结构和光学性质㊂将能量㊁自洽场以及能带的收敛精度均定为5ˑ10-6eV/atom;作用于原子上的最大力为0.01eV/Å,内应力收敛精度为0.02GPa,最大位移收敛精度为5ˑ10-5Å㊂截止能为400eV,在布里渊区积分采用4ˑ4ˑ4的Monkhost-Pack型K点网格进行迭代设置[29]㊂图1㊀超晶胞掺杂模型Fig.1㊀Supercell doping model260㊀研究论文人工晶体学报㊀㊀㊀㊀㊀㊀第53卷2㊀结果与讨论2.1㊀几何结构分析表1为几何结构优化后的本征以及掺杂体系PTO超胞的晶格常数和体积的变化㊂由表1可知,本征PTO的晶格常数计算值为a=b=7.688Å,c=9.567Å,理论值为a=b=7.759Å,c=8.572Å[30],两项数据对比,晶格常数c相差约1Å,但是理论值和计算值的c/a近似,说明选用参数的可靠性㊂与本征PTO相比, Ni㊁Cu掺杂PTO的晶格常数a㊁b㊁c减小,晶胞体积减小㊂Zn掺杂PTO的晶格常数a㊁b减小,c增大,晶胞体积增大㊂表1㊀Ni㊁Cu㊁Zn掺杂的PTO超胞晶格常数㊁密度和体积Table1㊀Lattice constants,density and volume of PTO supercell doped with Ni,Cu and Zn Sample a=b/Åc/ÅVolume/Å3Density/(g㊃cm-3)c/a PTO(Experimental)7.7598.572516.0537.802 1.1 PTO(Calculated)7.6889.567565.3527.122 1.2Ni doping7.6759.396553.4507.307 1.2Cu doping7.6559.515557.6037.268 1.2Zn doping7.6639.688568.9617.127 1.22.2㊀缺陷形成能分析缺陷形成能是表征掺杂体系稳定性和原子掺入体系难易程度的物理变量㊂基于几何结构优化后的体系总能量和不同原子的化学势计算相应结构的形成能㊂各掺杂体系的形成能E f满足以下公式[31-32]:E f=E doped-E perfect-lμX+nμTi(1)式中:E doped表示各掺杂体系的总能量,E perfect表示纯PbTiO3超晶胞体系总能量,系数l㊁n分别表示掺入的原子和替代的原子数,μX表示掺入原子(X=Ni㊁Cu㊁Zn)的化学势,μTi表示被替换的Ti原子化学势㊂由于材料的缺陷形成能与其生长制备的条件有密切关系,本文计算了富氧且富铅状态下各掺杂体系的形成能㊂从表2可以看出,Ni㊁Cu㊁Zn单掺PbTiO3体系在富O(O-rich)和富Pb(Pb-rich)条件下的形成能均为负㊂这意味着在O-rich和Pb-rich条件下,Ni㊁Cu㊁Zn原子可以融入PTO中,可在实验中制造Ni㊁Cu㊁Zn单掺PbTiO3材料㊂表2㊀Ni㊁Cu㊁Zn掺杂的PTO的缺陷形成能Table2㊀Defect formation energy of PTO doped with Ni,Cu and ZnSubstitute form O-rich and Pb-rich defect formation energy/eVNi doping-14.905Cu doping-13.336Zn doping-18.6542.3㊀力学性能基于密度泛函理论,结合当前应用最普遍的有限应变方法[33],通过计算应力应变的线性得到弹性系数6个独立分量,得到6ˑ6的弹性张量矩阵㊂根据晶格点阵的空间对称性,部分分量相等,部分分量为零㊂计算所得本征以及掺杂体系PTO晶格常数变化结构的特征弹性系数矩阵元,在优化晶体结构的基础上计算出本征以及掺杂体系PTO的弹性常数C ij,如表3所示㊂同时,基于Voigt-Reuss-Hill近似[34-36]得到体积模量㊁剪切模量㊁弹性模量㊁泊松比㊁Pugh比㊁维氏硬度㊁德拜温度θD,如表4所示㊂本文B和G取Hill值,通过弹性常数分别计算下限值B V㊁G V和上限值B R㊁G R,然后求平均值得出㊂这里弹性模量可由下面公式给出[37]B=(B V+B R)/2(2)G=(G V+G R)/2(3)其中,G V=(1/15)[C11+C22+C33+3(C44+C55+C66)-2(C12+C13+C23)],B R=Δ[C11(C22+C33+C23)+C22(C33-2C13)-C33C12+C12(2C23-C12)+C13(2C12-C13)+C23(2C13-C23)]-1,㊀第2期王云杰等:Ni,Cu,Zn掺杂四方相PbTiO3力学性能㊁电子结构与光学性质的第一性原理研究261㊀G R=15{4[C11(C22+C33+C23)+C22(C33+C13)+C33C12-C12(C12+C23)-C13(C12+C13)-C23(C13+ C23)]/Δ+3[(1/C44)+(1/C55)+(1/C66)]-1,Δ=C13(C12C23-C13C22)+C23(C12C13-C11C23)+C33(C11C22-C12C12)㊂弹性模量E和泊松比分别依照下列公式(4)和(5)计算得出E=9BG/(3B+G)(4)μ=(3B-E)/(6B)(5)采用Chen-Niu模型[38],得到维氏硬度H V公式为H V=2(k2G)0.585-3(6)其中Pugh比[39]k=G/B㊂对于本征以及掺杂体系PTO的弹性常数满足Born弹性稳定性判据[30]:C11(C22+C33)ȡ2C212,C22ȡC23, C44ȡ0,C55ȡ0,说明这四种结构是力学稳定的㊂体积模量是衡量材料是否容易被压缩的标志,Ni掺杂PTO 体积模量(80.034GPa)最大,所以相较于其他三种结构更不容易被压缩㊂剪切模量可以衡量材料硬度,Ni 掺杂PTO具有最大的剪切模量,对应最大的维氏硬度10.411GPa㊂弹性模量是标志材料刚度的重要物理量,Ni掺杂PTO的弹性模量最大,所以相较于其他三种结构刚性最高㊂G/B=1.75是区分脆性材料和延展性材料分界点,G/B=0.56是区分材料韧性/脆性分界点㊂由表4可以看出,G/B的值都小于1.75,Ni㊁Zn掺杂PTO大于0.56,都是脆性材料,本征以及Cu掺杂PTO小于0.56,属于是韧性材料㊂而泊松比反映了材料在形变下体积所发生的变化,说明四种结构形变时体积变化不大,泊松比的变化规律与Pugh比的正好相反㊂众所周知,德拜温度与材料的很多物理性质,如熔点㊁弹性㊁硬度㊁比热等基本物理量密切相关㊂采用以下公式[33]求得德拜温度θD=h kB34πV a[]1/3v m(7)式中:h为普朗克常量,k B为玻尔兹曼常量,V a为原子体积,v m为平均声速,由下式求出v m=132v3t +1v31()[]-1/3(8)式中:v1与v t分别为纵波㊁横波速度,可由下面的公式求得v1=3B+4G3ρ()1/2(9)v t=Gρ()1/2(10)式(9)和(10)中,ρ为密度,已由表1给出㊂本征以及掺杂体系PTO德拜温度的计算结果见表4㊂从表4给出的结果可以看出,Ni掺杂体系的德拜温度(201.506K)最高,与它有最大的C11(196.541GPa)㊁C23(63.626GPa)㊁C66(82.707GPa),最大的体积模量(80.034GPa),最大的剪切模量(45.499GPa)和最大的弹性模量(114.752GPa)密切相关㊂由表4可知,掺杂体系的剪切模量㊁弹性模量㊁Pugh比㊁维氏硬度和德拜温度均大于本征体系㊂其中Ni 掺杂体系的体积模量要大于本征体系,Cu㊁Zn掺杂体系的小于掺杂体系,说明除Cu㊁Zn掺杂体系在抗压性低于本征体系外,在硬度和刚性等力学性能均强于本征体系㊂可见二价金属Ni㊁Cu㊁Zn的掺杂,有助于改善四方相PTO的力学性能㊂表3㊀本征以及掺杂体系PTO的弹性常数C ijTable3㊀Elastic constants C ij of PTO in intrinsic and doped systemsCompound C11/GPa C12/GPa C13/GPa C22/GPa C23/GPa C33/GPa C44/GPa C55/GPa C66/GPa PTO172.44690.23880.526217.93161.95560.58151.59247.50381.781 Ni doping196.54190.00955.858210.65263.62661.79045.25745.19982.707 Cu doping183.37769.41847.886189.35455.26166.79630.10341.91071.456 Zn doping163.76165.71541.457163.76141.45766.02635.17035.17064.722262㊀研究论文人工晶体学报㊀㊀㊀㊀㊀㊀第53卷表4㊀本征以及掺杂体系PTO的体积模量(B)㊁剪切模量(G)㊁弹性模量(E)㊁泊松比(μ)㊁Pugh比(G/B)㊁维氏硬度(H V)和德拜温度θDTable4㊀Bulk modulus(B),shear modulus(G),elastic modulus(E),Poisson ratio(μ),Pugh ratio(G/B), Vickers hardness(H V),Debye temperature(θD)of PTO in intrinsic and doped systems Compound B/GPa G/GPa E/GPaμG/B H V/GPaθD/K PTO78.43539.170100.7400.2860.4998.389188.293 Ni doping80.03445.499114.7520.2610.56810.411201.506 Cu doping75.25140.052101.7410.2750.5328.977189.392 Zn doping68.30740.606101.6710.2520.5949.880190.852 2.4㊀能带结构分析图2是本征PbTiO3以及掺杂体系的能带结构图㊂为便于分析,范围选取-5~5eV,包含费米能级,在四种体系中除Ni掺杂PbTiO3为间接带隙外,其他均为直接带隙㊂图2(a)是本征PbTiO3的能带结构图,禁带宽度为2.007eV,与实验值3.6eV相较偏低[40],所以采用剪刀算符[41]修正其带隙值(剪刀算符为1.6eV),修正后的带隙为3.607eV㊂图2(b)~(d)分别是Ni㊁Cu㊁Zn掺杂PTO的能带结构图,掺杂体系的跃迁形式所需的能量,相较于本征结构降低,并且区间处于0~1eV能带条数增多,Cu㊁Zn掺杂PbTiO3带隙值分别为1.930㊁1.936eV,价带顶有所上移,费米能级进入价带顶,使得Cu㊁Zn掺杂PbTiO3呈现出p型导电特性㊂Ni 掺杂PbTiO3价带顶到导带底的间距是1.678eV,在2eV附近出现受主能级,价带顶处出现多余的空穴载流子,这有利于电子吸收极少的能量由价带顶跃迁至受主能级,再由受主能级跃迁至导带底,或者实现受主能级之间的跃迁,从而能够大幅改善PbTiO3材料的光催化特性和导电性㊂李宏光等[16]关于N掺杂PbTiO3的研究中,能带结构出现受主能级,且价带顶下移,出现p型半导体特性,但是电子跃迁性能并不比Ni㊁Cu㊁Zn 掺杂PbTiO3更强㊂图2㊀本征PTO及三种掺杂体系的能带结构分布Fig.2㊀Band structures of intrinsic PTO and three doping systems2.5㊀态密度分析图3是本征PTO以及三种掺杂体系的总态密度图和分波态密度图㊂图3(a)是本征PTO的态密度图,㊀第2期王云杰等:Ni,Cu,Zn掺杂四方相PbTiO3力学性能㊁电子结构与光学性质的第一性原理研究263㊀Ti-3d轨道是构成导带部分的总态密度主要部分㊂价带能量处于-19~-14eV的总态密度主要由Pb-5d和O-2s轨道提供,在-8eV至费米能级的总态密度主要由O-2p以及Pb-6s轨道贡献,这与相关研究结果一致[16]㊂图3(b)~(d)分别是Ni㊁Cu㊁Zn掺杂PTO的态密度图㊂掺杂体系Pb㊁Ti和O对总态密度的贡献基本与本征态一致㊂区别在于在费米面附近,主要由O-2p及Ni㊁Cu㊁Zn的3d态之间进行杂化贡献,表现出强大的局域性㊂当Ni㊁Cu㊁Zn掺杂到PTO之后,由于掺入的Ni㊁Cu㊁Zn对总态密度贡献相对较小而不易被观察,但可以从O-2p轨道的变化进行说明,使得O-2p轨道在费米能级附近出现自由电子㊂2价金属Ni㊁Cu㊁Zn 的掺杂使得Pb㊁Ti和O之间的杂化发生变化,进而影响态密度的整体分布情况㊂掺杂体系的电子从价带顶跃迁到导带底的过程变得容易,与能带结构情况吻合㊂图3㊀本征PTO及三种掺杂体系的态密度曲线Fig.3㊀Density of states curves of intrinsic PTO and three doping systems2.6㊀光学性质分析本征以及三种掺杂体系的PTO复介电函数实部曲线和虚部曲线如图4所示,图4(a)中PTO㊁PTOʒNi㊁PTOʒCu和PTOʒZn的静态介电常数分别为2.307㊁3.305㊁3.411和4.513㊂PTOʒCu在低能区介电函数实部随着光子能量的增大而增大,并到达峰值5.714(光子能量为1.38eV),从态密度图看出这是由Cu-3d轨道向O-2p轨道的电子跃迁引起的㊂图4(b)显示PTOʒNi㊁PTOʒCu和PTOʒZn的介电函数虚部主要集中在0~10eV 的低能区,而本征PTO在虚部低能区(ɤ3eV)虚部值很小,接近零,而Ni㊁Cu㊁Zn掺杂PTO体系在虚部1.5eV左右形成新的次级主峰,PTOʒCu在低于2eV的低能区具有压倒性数值㊂可见,Ni㊁Cu㊁Zn掺杂PTO 体系光谱吸收范围扩展到红外区域,且PTOʒCu更具有优势,在可见光波段的能量吸收效果较强,说明PTOʒCu在低能区的吸收效果在三种掺杂体系中是最强的㊂图4(c)是本征以及三种掺杂体系的PTO体系的反射光谱㊂可知,本征PTO在5.77㊁7.41㊁9.74eV出现三个峰值㊂Ni㊁Cu掺杂PTO体系在可见光区域能量值大于本征PTO㊂在红外光区,Ni㊁Cu㊁Zn掺杂PTO的反射值大于本征PTO体系,PTOʒCu对可见光区域和红外光区的利用率较高,这与复介电函数图所得的结果一致㊂图4(d)是含Ni㊁Cu㊁Zn掺杂的PTO的吸收光谱㊂本征PTO只吸收紫外波段,对红外部分不吸收,本征264㊀研究论文人工晶体学报㊀㊀㊀㊀㊀㊀第53卷PTO的禁带宽度决定了Ni㊁Cu㊁Zn掺杂的PTO体系吸收主要集中在紫外波段㊂同时,掺杂使得电子跃迁变得容易,Ni㊁Cu㊁Zn掺杂的PTO体系吸收范围扩展到红外波段㊂在可见光波段,PTOʒCu吸收效果最好,并且吸收边640nm所对应的频率为1.94eV,这表明电子是从价带内跃迁到导带的,说明PTOʒCu具有潜在的光催化能力㊂在红外以及远红外波段,PTOʒZn吸收效果和PTOʒCu相近,并且比李宏光等[16]报道的N掺杂的PTO在红外远红外区域吸收效果更好㊂吸收光谱与介电㊁反射光谱的变化趋势是一致的㊂图4㊀本征PTO及三种掺杂体系的光学图谱㊂(a)复介电函数实部;(b)复介电函数虚部;(c)反射光谱;(d)吸收光谱Fig.4㊀Optical spectra of intrinsic PTO and three doping systems.(a)Real part of complex dielectric function;(b)imaginary part of complex dielectric function;(c)reflection spectra;(d)absorption spectra3㊀结㊀㊀论1)Ni掺杂PTO的体积㊁剪切和弹性模量最大,这是Ni掺杂PTO德拜温度最高的重要原因㊂体积模量的大小是衡量材料是否容易被压缩的标志,体积模量越高,材料越不容易被压缩;高剪切模量是高硬度的基本条件,最大的剪切模量使得Ni掺杂PTO有最大的维氏硬度;弹性模量是标志材料刚度的重要物理量,表明四种材料中Ni掺杂PTO的刚性最高㊂2)Zn掺杂PTO的G/B值是四种材料中最大的,说明此结构中原子间的化学键的定向性最高㊂3)Ni㊁Zn掺杂PTO的G/B大于0.56,都是脆性材料,本征以及Cu掺杂PTO的G/B小于0.56,是韧性材料㊂泊松比反映了材料在形变下体积的变化,本征以及掺杂体系的泊松比都在0.25~0.5,表明本征及掺杂体系PTO形变时体积将不会发生较大的变化㊂4)掺杂体系较于本征体系跃迁能量减小,Ni掺入PTO材料的费米能级处出现杂质能级㊂Cu㊁Zn掺杂的PTO费米能级进入价带顶,使得Cu㊁Zn掺杂PTO材料呈现出p型导电特性㊂5)Ni㊁Cu㊁Zn的掺杂使得PTO吸收范围扩展到红外波段,且增强了可见光波段的吸收强度,四种结构中PTOʒCu材料的光催化性能最好㊂参考文献[1]㊀ZHANG S J,LI F,JIANG X N,et al.Advantages and challenges of relaxor-PbTiO3ferroelectric crystals for electroacoustic transducers:a review[J].㊀第2期王云杰等:Ni,Cu,Zn掺杂四方相PbTiO3力学性能㊁电子结构与光学性质的第一性原理研究265㊀Progress in Materials Science,2015,68:1-66.[2]㊀LIU Y,NI L H,REN Z H,et al.First-principles study of structural stability and elastic property of pre-perovskite PbTiO3[J].Chinese PhysicsB,2012,21(1):016201.[3]㊀SUNTIVICH J,GASTEIGER H A,YABUUCHI N,et al.Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuelcells and metal-air batteries[J].Nature Chemistry,2011,3(7):546-550.[4]㊀黄㊀建,张学伍,赵㊀程,等.钛酸铅系功能陶瓷改性的研究现状及改性陶瓷的应用现状[J].机械工程材料,2021,45(6):94-98.HUANG J,ZHANG X W,ZHAO C,et al.Research status of modification of lead titanate series functional ceramics and application of modified ceramics[J].Materials for Mechanical Engineering,2021,45(6):94-98(in Chinese).[5]㊀邓鹏星,文志勤,马㊀博,等.体积应变对立方钛酸铅电子结构和光学性质的影响[J].人工晶体学报,2022,51(1):85-91.DENG P X,WEN Z Q,MA B,et al.Effect of volume strain on electronic structure and optical properties of cubic lead titanate[J].Journal of Synthetic Crystals,2022,51(1):85-91(in Chinese).[6]㊀SCOTT J F,PAZ DE ARAUJO C A.Ferroelectric memories[J].Science,1989,246(4936):1400-1405.[7]㊀HOSSEINI S M,MOVLAROOY T,KOMPANY A.First-principle calculations of the cohesive energy and the electronic properties of PbTiO3[J].Physica B:Condensed Matter,2007,391(2):316-321.[8]㊀ZHU Z Y,WANG B,WANG H,et al.First-principle study of ferroelectricity in PbTiO3/SrTiO3superlattices[J].Solid-State Electronics,2006,50(11/12):1756-1760.[9]㊀GE F F,WU W D,WANG X M,et al.The first-principle calculation of structures and defect energies in tetragonal PbTiO3[J].Physica B:Condensed Matter,2009,404(20):3814-3818.[10]㊀CHEN X,TAN P F,ZHOU B H,et al.A green and facile strategy for preparation of novel and stable Cr-doped SrTiO3/g-C3N4hybridnanocomposites with enhanced visible light photocatalytic activity[J].Journal of Alloys and Compounds,2015,647:456-462. [11]㊀GRABOWSKA E.Selected perovskite oxides:characterization,preparation and photocatalytic properties:a review[J].Applied Catalysis B:Environmental,2016,186:97-126.[12]㊀OHNO T,TSUBOTA T,NAKAMURA Y,et al.Preparation of S,C cation-codoped SrTiO3and its photocatalytic activity under visible light[J].Applied Catalysis A:General,2005,288(1/2):74-79.[13]㊀MORET M P,DEVILLERS M A C,WÖRHOFF K,et al.Optical properties of PbTiO3,PbZr x Ti1-x O3,and PbZrO3films deposited bymetalorganic chemical vapor on SrTiO3[J].Journal of Applied Physics,2002,92(1):468-474.[14]㊀HUSSIN N H,TAIB M F M,HASSAN O H,et al.Study of geometrical and electronic structure of lanthanum doped PbTiO3and PbZrTiO3:firstprinciples calculation[C]//AIP Conference Proceedings.Ho Chi Minh,Vietnam.Author(s),2018.[15]㊀NIU P J,YAN J L,MENG D L.The effects of N-doping and oxygen vacancy on the electronic structure and conductivity of PbTiO3[J].Journalof Semiconductors,2015,36(4):043004.[16]㊀李宏光,闫金良.N掺杂位置对四方相PbTiO3电子结构和光学性能的影响[J].材料科学与工程学报,2017,35(1):14-18.LI H G,YAN J L.Electronic structures and optical properties of N-doped tetragonal PbTiO3with different doping sites[J].Journal of Materials Science and Engineering,2017,35(1):14-18(in Chinese).[17]㊀ASAHI R,MORIKAWA T,OHWAKI T,et al.Visible-light photocatalysis in nitrogen-doped titanium oxides[J].Science,2001,293(5528):269-271.[18]㊀OKUNAKA S,TOKUDOME H,ABE R.Facile water-based preparation of Rh-doped SrTiO3nanoparticles for efficient photocatalytic H2evolutionunder visible light irradiation[J].Journal of Materials Chemistry A,2015,3(28):14794-14800.[19]㊀XIN H,PANG Q,GAO D L,et al.Mn ions'site and valence in PbTiO3based on the native vacancy defects[J].Condensed Matter Physics,2021,24(2):23705.[20]㊀KUMA S,WOLDEMARIAM M M.Structural,electronic,lattice dynamic,and elastic properties of SnTiO3and PbTiO3using density functionaltheory[J].Advances in Condensed Matter Physics,2019,2019:1-12.[21]㊀HACHEMI A,HACHEMI H,FERHAT-HAMIDA A,et al.Elasticity of SrTiO3perovskite under high pressure in cubic,tetragonal andorthorhombic phases[J].Physica Scripta,2010,82(2):025602.[22]㊀LI Z,GRIMSDITCH M,FOSTER C M,et al.Dielectric and elastic properties of ferroelectric materials at elevated temperature[J].Journal ofPhysics and Chemistry of Solids,1996,57(10):1433-1438.[23]㊀SÁGHI-SZABÓG,COHEN R E,KRAKAUER H.First-principles study of piezoelectricity in tetragonal PbTiO3and PbZr1/2Ti1/2O3[J].Physical Review B,1999,59(20):12771-12776.[24]㊀PERDEW J P,WANG Y E.Accurate and simple analytic representation of the electron-gas correlation energy[J].Physical Review B,1992,45(23):13244-13249.[25]㊀SEGALL M D,LINDAN P J D,PROBERT M J,et al.First-principles simulation:ideas,illustrations and the CASTEP code[J].Journal ofPhysics:Condensed Matter,2002,14(11):2717-2744.[26]㊀CLARK S J,SEGALL M D,PICKARD C J,et al.First principles methods using CASTEP[J].Zeitschrift Für Kristallographie-Crystalline266㊀研究论文人工晶体学报㊀㊀㊀㊀㊀㊀第53卷Materials,2005,220(5/6):567-570.[27]㊀ERNZERHOF M,BURKE K,PERDEW J P.Density functional theory,the exchange hole,and the molecular bond[M]//Theoretical andComputational Chemistry.Amsterdam:Elsevier,1996:207-238.[28]㊀PERDEW J P,ERNZERHOF M,ZUPAN A,et al.Nonlocality of the density functional for exchange and correlation:physical origins andchemical consequences[J].The Journal of Chemical Physics,1998,108(4):1522-1531.[29]㊀MONKHORST H J,PACK J D.Special points for brillouin-zone integrations[J].Physical Review B,1976,13(12):5188-5192.[30]㊀TAIB M F M,YAAKOB M K,BADRUDIN F W,et al.First-principles comparative study of the electronic and optical properties of tetragonal(P4mm)ATiO3(A=Pb,Sn,Ge)[J].Integrated Ferroelectrics,2014,155(1):23-32.[31]㊀WANG Q J,WANG J B,ZHONG X L,et al.Magnetism mechanism in ZnO and ZnO doped with nonmagnetic elements X(X=Li,Mg,andAl):a first-principles study[J].Applied Physics Letters,2012,100(13):673-677.[32]㊀CHEN H,LI X C,WAN R D,et al.A DFT study on modification mechanism of(N,S)interstitial co-doped rutile TiO2[J].Chemical PhysicsLetters,2018,695:8-18.[33]㊀BOUHEMADOU A.First-principles study of structural,electronic and elastic properties of Nb4AlC3[J].Brazilian Journal of Physics,2010,40(1):52-57.[34]㊀CHEN X Q,NIU H Y,LI D Z,et al.Modeling hardness of polycrystalline materials and bulk metallic glasses[J].Intermetallics,2011,19(9):1275-1281.[35]㊀VOIGT W.Lehrbuch der kristallphysik(mit ausschluss der kristalloptik),edited by bg teubner and jw edwards,leipzig berlin[J].Ann Arbor,Mich,1928.[36]㊀REUSS A.Berechnung der fließgrenze von mischkristallen auf grund der plastizitätsbedingung für einkristalle[J].ZAMM-Journal of AppliedMathematics and Mechanics,1929,9(1):49-58.[37]㊀HILL R.The elastic behaviour of a crystalline aggregate[J].Proceedings of the Physical Society Section A,1952,65(5):349-354.[38]㊀WATT J P.Hashin-Shtrikman bounds on the effective elastic moduli of polycrystals with monoclinic symmetry[J].Journal of Applied Physics,1980,51(3):1520-1524.[39]㊀PUGH S F.XCII.Relations between the elastic moduli and the plastic properties of polycrystalline pure metals[J].The London,Edinburgh,and Dublin Philosophical Magazine and Journal of Science,1954,45(367):823-843.[40]㊀YADAV H O.Optical and electrical properties of sol-gel derived thin films of PbTiO3[J].Ceramics International,2004,30(7):1493-1498.[41]㊀高㊀妍,董海涛,张小可,等.(Al x Ga1-x)2O3结构㊁电子和光学性质的第一性原理研究[J].人工晶体学报,2023,52(9):1674-1680+1719.GAO Y,DONG H T,ZHANG X K,et al.First-principle study on structure,electronic and optical properties of(Al x Ga1-x)2O3[J].Journal of Synthetic Crystals,2023,52(9):1674-1680+1719(in Chinese).。

分类号:密级:U D C:编号:学位论文有序α-(Ga1-x Fe x)2O3结构与性质的第一性原理研究赵辉指导教师姓名:宋庆功教授河北工业大学申请学位级别:硕士学科、专业名称:理论物理论文提交日期: 2012 年 11 月 论文答辩日期: 2012 年 12 月 学位授予单位:河北工业大学答辩委员会主席:评阅人:2012 年 12 月Dissertation Submitted toHebei University of TechnologyforThe Master Degree ofTheoretical PhysicsFIRST-PRINCIPLES STUDIES ON STRUCTURES ANDPROPERTIES OF ORDERED α-(Ga1-x Fe x)2O3 SYSTEMbyZhao HuiSupervisor: Prof. Song Qing-GongDecember 2012This work was supported by the National Natural Science Foundation of China (No.60979008)河北工业大学硕士学位论文有序α-(Ga1-x Fe x)2O3结构与性质的第一性原理研究摘 要本文采用了基于密度泛函理论的第一性原理计算方法,对 (Ga1-x Fe x)2O3系列晶体的晶体结构、电子性质和光学性质进行研究。
以(Ga1-x Fe x)2O3晶格参量的实验值作为参考,建立了晶胞结构,采用GGA+U方案,对α-Ga2O3、α-Fe2O3、GaFeO3-A、GaFeO3-B、α-(Ga1/4Fe3/4)2O3、α-(Ga3/4Fe1/4)2O3和GaFeO3-C晶体进行几何结构优化。
以优化后的晶体结构为基础,对晶体的总能量、电子性质和光学性质进行计算研究和对比分析,获得的主要结论如下:七种晶体结构优化后的晶格参量与实验值相符合。
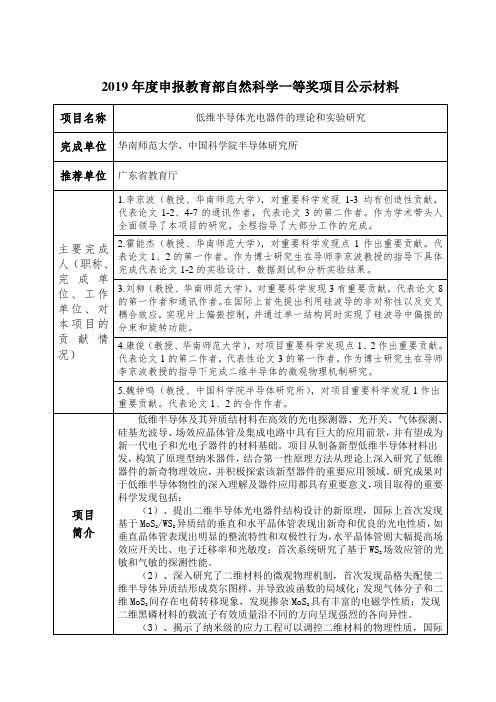
2019年度申报教育部自然科学一等奖项目公示材料项目名称低维半导体光电器件的理论和实验研究完成单位华南师范大学、中国科学院半导体研究所推荐单位广东省教育厅主要完成人(职称、完成单位、工作单位、对本项目的贡献情况)1.李京波(教授、华南师范大学),对重要科学发现1-3均有创造性贡献,代表论文1-2、4-7的通讯作者,代表论文3的第二作者。
作为学术带头人全面领导了本项目的研究,全程指导了大部分工作的完成。
2.霍能杰(教授、华南师范大学),对重要科学发现点1作出重要贡献。
代表论文1、2的第一作者。
作为博士研究生在导师李京波教授的指导下具体完成代表论文1-2的实验设计、数据测试和分析实验结果。
3.刘柳(教授、华南师范大学),对重要科学发现3有重要贡献,代表论文8的第一作者和通讯作者。
在国际上首先提出利用硅波导的非对称性以及交叉耦合效应,实现片上偏振控制,并通过单一结构同时实现了硅波导中偏振的分束和旋转功能。
4.康俊(教授、华南师范大学),对项目重要科学发现点1、2作出重要贡献。
代表论文1的第二作者,代表性论文3的第一作者。
作为博士研究生在导师李京波教授的指导下完成二维半导体的微观物理机制研究。
5.魏钟鸣(教授、中国科学院半导体研究所),对项目重要科学发现1作出重要贡献。
代表论文1、2的合作作者。
项目简介低维半导体及其异质结材料在高效的光电探测器、光开关、气体探测、硅基光波导、场效应晶体管及集成电路中具有巨大的应用前景,并有望成为新一代电子和光电子器件的材料基础。
项目从制备新型低维半导体材料出发,构筑了原理型纳米器件,结合第一性原理方法从理论上深入研究了低维器件的新奇物理效应,并积极探索该新型器件的重要应用领域。
研究成果对于低维半导体物性的深入理解及器件应用都具有重要意义,项目取得的重要科学发现包括:(1)、提出二维半导体光电器件结构设计的新原理,国际上首次发现基于MoS2/WS2异质结的垂直和水平晶体管表现出新奇和优良的光电性质,如垂直晶体管表现出明显的整流特性和双极性行为,水平晶体管则大幅提高场效应开关比、电子迁移率和光敏度;首次系统研究了基于WS2场效应管的光敏和气敏的探测性能。

First-principles calculations of electronic structure and optical properties of Boron-doped ZnO with intrinsicdefectsYen-Chun Peng,Chieh-Cheng Chen,Hsuan-Chung Wu ⇑,Jong-Hong LuDepartment of Materials Engineering,Ming Chi University of Technology,New Taipei 24301,Taiwana r t i c l e i n f o Article history:Received 11August 2014Received in revised form 27October 2014Accepted 27October 2014Available online 15November 2014Keywords:First principles B-doped ZnO Intrinsic defectElectronic structure Optical propertya b s t r a c tThis study adopted first-principles calculations to evaluate the effects of intrinsic defects on the elec-tronic structure and optical properties of Boron-doped ZnO (BZO).Four types of defect were considered:non-defective (B Zn ),Zn vacancies (V Zn ),O vacancies (V O ),and interstitial Zn (Zn i ).Calculations of forma-tion energy illustrate that O-rich conditions tend to induce V Zn ,while O-poor conditions tend to induce V O and Zn i .With respect to electric properties,V Zn defects in BZO decrease carrier concentration as well as mobility,which consequently decreases the conductivity of BZO.The existence of V O or Zn i defects in BZO leads to n-type conductive characteristics and increases the optical band gap.The existence of Zn i defects in BZO also increases the effective mass,which decreases the mobility and conductivity of BZO.As for the optical properties,the introduction of V Zn to BZO leads to an increase in transmittance in the visible light region,but a decrease in the UV region.The introduction of intrinsic V O and Zn i defects to BZO leads to a significant decrease in transmittance in the visible as well as UV regions.The calculated results were also compared with experimental data from the literature.Ó2014Elsevier B.V.All rights reserved.1.IntroductionZnO is an abundant,non-toxic material with a wide band gap (3.37eV)and transparent properties under visible light.ZnO has recently attracted considerable attention as an alternative for Tin-doped In 2O 3(ITO),which is currently the most common choice of transparent conductive oxide for a variety of applications [1,2].The resistivity of pure ZnO is on the order of 10À2X -cm,which is far higher than that of ITO (10À4X -cm order).A great deal of research has gone into enhancing the conductivity of ZnO through the addition of various dopants,which can mainly be divided into metals [3–5]and non-metals [6,7].B-doped ZnO (BZO)thin film shows considerable promise for its superior photoelectric proper-ties and stability [8,9].Many groups have investigated the effects of process parameters on the electric and optical properties of BZO thin film,with the aim of optimizing performance [7–13].Miyata et al.[7]indicated that the transmittance of BZO thin film could be improved through the introduction of O 2gas from 0sccm to 10sccm.David et al.[10]reported that annealing temperature and atmosphere strongly affect the conductivity of BZO.Yang et al.[11]concluded that the low oxygen partial pressure during deposition increases the carrier density of oxygen vacancies,which leads to a strong decline in resistivity.However,resistivity in sam-ples produced under the high oxygen partial pressure is far higher than in samples deposited under low oxygen partial pressure,which suggests the existence of p-type carriers of Zinc vacancies in films grown under high oxygen partial pressure.Patil et al.[12]synthesized B-doped ZnO powders using a mechanochemical method.The photoluminescence (PL)spectra at room temperature is an indication that a greater number of oxygen vacancies exist in nonmetal-doped ZnO,compared to pure ZnO.In the fabrication of BZO microrods,Yılmaz et al.[13]investigated the influence of B diffusion doping on optical emission and defect formation.PL spec-tra results revealed that the intensity of the deep level visible band emission increases with an increase in annealing time,which implies a significant increase in the concentration of intrinsic defects.As outlined above,various process conditions influence the type and number of intrinsic defects with a subsequent influence on the electric and optical properties of BZO.Gaining a comprehensive understanding of the electric and optical characteristics of BZO would require in-depth study into the effects of intrinsic defects on the properties of BZO.First-principles calculations can provide information concerning materials at the microscopic scale to eluci-date the connection between structure and properties.It is well known that the use of conventional density functional theory/10.1016/j.optmat.2014.10.0580925-3467/Ó2014Elsevier B.V.All rights reserved.⇑Corresponding author at:Department of Materials Engineering,Ming Chi University of Technology,84Gungjuan Road,Taishan,New Taipei 24301,Taiwan.Tel.:+8862290898994675;fax:+886229084091.E-mail address:hcwu@.tw (H.-C.Wu).(DFT)leads to a considerable underestimation of the calculated band gap in ZnO [14–16].In our previous study [17],we used the DFT plus Hubbard U (DFT +U)method to avoid underestimat-ing the band gap.This approach reduced the differences in calcu-lated band gap and lattice constant to within 1%of the experimental values.The current study extended the utilization of the DFT +U method to calculate and analyze the effects of intrin-sic defects (V Zn ,V O ,and Zn i )on the formation energy,crystal struc-ture,electronic structure,and optical properties of BZO.These results clarify the connections among the fabrication process,structure,and properties of BZO,for use in determining the criteria for future material designs.2.Calculation methodsThis study considered a 2Â2Â2supercell of a Wurtzite ZnO,including 16Zn atoms and 16O atoms,as shown in Fig.1.A B-monodoping model was constructed by substituting one Zn atom (number 1site)with one B atom (B Zn model),which correspond to the B concentrations of 6.25at.%.We also considered three intrinsic defects in the B Zn structure,in which Zn vacancies(B Zn V Zn ),O vacancies (B Zn V O ),and interstitial Zn (B Zn Zn i )are repre-sented as 2,3,and 4,respectively.The V Zn ,V O ,and Zn i concentra-tions corresponds to doping levels of 6.25, 6.25,and 5.88at.%,respectively.The defect concentration could be reduced using a larger supercell for the real systems;however,this study was lim-ited with regard to computer resources.Therefore,the properties of the defects calculated from a 2Â2Â2ZnO supercell such as this could be used as qualitative analysis.1432ZnO BTable 1Formation energy and optimized structure of BZO with varying intrinsic defects.Formation energy (eV)Optimized structure O-richO-poor Zn–O (Å)B–O (Å)4V (%)ZnO –– 1.981––B Zn3.750.39 1.996 1.526À3.1B Zn V Zn 5.68 5.81 1.993 1.530À3.3B Zn V O 7.550.70 1.995 1.521À5.3B Zn Zn i10.513.662.0031.5174.74.5 eV2.15 eV3.25 eV4.68 eV4.41 eV(a)(b)(c)(d)Band structures of B-doped ZnO for (a)B Zn ,(b)B Zn V Zn ,(c)B Zn V O models.Y.-C.Peng et al./Optical Materials 39(2015)34–3935All models presented in this study were developed using CASTEP software [18].Structural optimization was performed on each model before calculating properties.The Monkhorst–Pack scheme [19]K-points grid sampling in the supercells was set at 4Â4Â2.Electron–ion interactions were modeled using the ultrasoft pseudo-potential method [20].The valence configurations of the atoms were 4s 23d 10for Zn,2s 22p 4for O,and 2s 22p 1for B.The elec-tron wave functions were expanded in plane wave with an energy cutoff of 380eV.In the structural optimization process,the change in energy,maximum force,maximum stress,and maximum displacement tolerances were set at 10À5eV/atom,0.03eV/Å,0.05GPa,and 0.001Å,respectively.The energy convergence crite-rion for the self-consistent field was set at 10À6eV.To describe the electronic structures more accurately,we adopted the DFT +U d +U p method [21],in which the U d value for Zn-3d and the U p value for O-2p orbitals were set at 10and 7eV,respectively.The band structures,band gaps,and Zn-3d orbital locations of pure ZnO,which were used for the selection of U d and U p values,can be referenced in our previous research [17,22].3.Results and discussion 3.1.Optimized structureThe average bond lengths and volume difference ratio,as obtained from geometric optimization,are summarized in Table 1.In pure ZnO,each Zn atom is bonded to its three horizontal and one vertical oxygen neighbors.The average bond length of Zn-O is 1.981Åand optimized lattice constants are a =b =3.249Åandc =5.232Å,which are in agreement with the experimental values of a =b =3.249,c =5.206Å[23].Following the substitution of one B atom for one Zn atom (B Zn model),the Zn–O bond length is longer than that of B–O (1.526Å).This is because the B 3+radius (0.27Å)is smaller than that of Zn 2+(0.74Å)[24].Therefore,the cell volume of B Zn model shrinks,which is consistent with the experi-mental results [25].Clearly,the presence of Zn or O vacancies in BZO also leads to a shrinkage in volume.Conversely,the presence of interstitial Zn leads to a longer Zn–O length and expansion in volume.3.2.Formation energyTo examine the relative stability of BZO with intrinsic defects in neutral charge state,the defect formation energy can be expressed as follows:[26,27]E f ðD Þ¼E tot ðD ÞÀE tot ðZnO ÞþXn i l ið1Þwhere E tot (ZnO)and E tot (D )are the total energy in pure ZnO and in the defective systems,respectively.n i is the number of i atoms removed from or added to the supercell.If an atom is removed from the supercell,n i is positive,otherwise is negative.l i is the chemical potential of atom i .Formation energy depends on the growth envi-ronment during the preparation process,which can be O-rich or O-poor (Zn-rich).In thermo-dynamic equilibrium,D l Zn +D l O =D H f (ZnO),where D H f (ZnO)represents the formation enthalpy of ZnO.For the chemical potential of B,this study adopted the relation of 2D l B +3D l O 6D H f (B 2O 3)under O-rich conditions and l B =l B(bulk)under O-poor conditions,where D H f (B 2O 3)represents the(a)(b)(c)(d)V ZnV OZn iBZnOO density difference for (a)B Zn ,(b)B Zn V Zn ,(c)B Zn V O ,and (d)B Zn Zn i models.The red,orange,yellow,green,and interpretation of the references to color in this figure legend,the reader is referred to the web version 36Y.-C.Peng et al./Optical Materials 39(2015)34–39formation enthalpy of B2O3.D l i represents the chemical potential of atom i referred to as the elemental solid/gas of l i(bulk/molecule).It is well known that a defective structure with lower formation energy forms more readily and denotes an increased occurrence of defects.Table1presents a summary of the calculated formation energy of BZO with various intrinsic defects,based on the neutral charge state.With the existence of B Zn,E f(B Zn V Zn)<E f(B Zn V O)<E f (B Zn Zn i)under O-rich conditions,implying that O-rich conditions are more likely to induce the formation of V Zn,followed by V O and Zn i.Under O-poor conditions,E f(B Zn V O)<E f(B Zn Zn i)<E f (B Zn V Zn),which implies that O-poor conditions are more likely to induce the formation of V O.As a result,process conditions,such as O2gasflow rate and substrate temperature,largely determine the type of intrinsic defects that form in BZO during preparation. The occurrence of V O is far more likely under a low-O atmosphere, and V Zn is more likely to occur under a high-O atmosphere.For the sake of comparison,we also calculated the formation energy of a single intrinsic defect(V Zn and V O)in pure ZnO.The calculated values of E f(V Zn)and E f(V O)are3.09and4.33eV under O-rich conditions and6.58and0.84eV under O-poor conditions. Thus,we can see that the formation energy of a Zn vacancy from pure ZnO(E f(V Zn))is greater than that obtained from BZO(E f(B Zn V Zn)ÀE f(B Zn)=1.93eV under O-rich conditions and5.42eV under O-poor conditions).This demonstrates that Zn vacancies form more easily in BZO than in ZnO.These results are similar to those calculated for O vacancies,which implies that B-doping facilitates the formation of V Zn and V O.Previous studies [12]obtained similar results,indicating that a greater number of oxygen vacancies or defects exist in BZO than in pure ZnO.3.3.Electronic structureTo clarify the influence of intrinsic defects on the electronic structure of BZO,we calculated the band structure,difference in charge density,and density of states(DOS),as shown in Figs.2–4,respectively.In our previous study[17,22],the calculated band structure revealed a band gap of3.37eV in pure ZnO,which is in excellent agreement with values obtained in experiments.In the present study,we focus on the properties of BZO with intrinsic defects.Fig.2presents the band structures for BZO with various intrin-sic defect models.The Fermi level indicated by the dotted line was set to zero.Fig.2(a)shows the situation in which a Zn atom in pure ZnO is replaced by a B atom,in which the Fermi level shifts from the valence band(VB)maximum to the bottom of the conduction band(CB),resulting in a shallow donor level at the bottom of the CB.The shallow donor level at B doping causes an increase in the optical band gap to4.5eV at B concentration of6.25at.%,which is well known as the Burstein-Moss effect[28].The definitionof Fig.4.Density of states for(a)B Zn,(b)B Zn V Zn,(c)B Zn V O,and(d)B Zn Zn i models.optical band gap is from the top offor n-type semiconducting materials and the bottom of conduction band for materials.Similar tendencies were experiment-based studies[29,30].Asin the vicinity of B impurities appears atoms.The calculated Mulliken bond and B–O bonds are0.371and0.658, that a B–O bond is more covalent than Mulliken bond population represents characteristics).As shown in Fig.4(a),to the bottom of CB are the Zn-4s anda few O-2s and O-2p orbitals.The main extra electron tofill up the CBM.According to the results calculated regarded as an intrinsic defect under the B Zn V Zn model(Fig.2(b)),when donor levels coexist,the empty states produced trons from the B Zn donor level,resulting level as well as the formation of p-type band gap of B Zn V Zn can be narrowed to eration of conduction electrons requires energy from the Fermi level to the CB, be required in the B Zn model.Thus,in may lead to a decrease in the carrier known that mobility is related to the time.The relaxation time could not be software and was assumed as a defects in BZO.The followingeffects of the effective mass on thenear the Fermi level appear nearlyof carriers with a smaller curvature The larger effective mass is related toTherefore,V Zn defects in BZO reduce both carrier concentration as well as mobility,which consequently increases the resistivity of BZO.Fig.3(b)shows that the O atoms surrounding a Zn vacancy gain fewer electrons(green color),implying the occurrence of a number of empty states of O atoms.These empty states are O-2p orbitals near the Fermi level,as shown in Fig.4(b).V O and Zn i can be regarded as intrinsic defects in an O-poor environment.Fig.2(c)and(d)show the band structures in B Zn V O and B Zn Zn i models,in which n-type conductive characteristics appear and the optical band gap increases to4.68eV and4.41eV, respectively.One shallow donor state and one deep donor state occur in these two models.In the B Zn V O model,the deep donor level is probably the charge remaining in the oxygen vacancy (Fig.3(c));in the B Zn Zn i model,it is probably the covalence charge in the vicinity of the interstitial Zn atom(Zn i)(Fig.3(d)).Fig.4(c) and(d)show that the shallow donor level in both models origi-nated from B doping,whereas the deep donor level in the B Zn V O and B Zn Zn i models originated from the addition of V O and Zn i, respectively.The deep donor level in the B Zn V O model comprises mainly Zn and O atoms;however,in the B Zn Zn i model,it also includes B atom(B-2s and B-2p states).The shallow donor states provide conduction electrons;however,the deep donor states may contribute less to the increase in carrier concentration.Qual-itatively,the curvature of the energy band near the Fermi level in the B Zn Zn i model is smaller than that in the B Zn V O model.There-fore,Zn i defects present in BZO increase the effective mass,which may consequently decrease the mobility and conductivity of BZO.3.4.Optical propertiesThe optical properties can be described via the dielectric func-tion e(x)=e1(x)+i e2(x)[31].The imaginary part of the dielectric function e2(x)is calculated as follows:e2¼2e2pX e0Xk;v;cu ckuÁrj j u v k2d E ckÀE vkÀxÀÁð2Þwhere e is the electronic charge;X is the unit cell volume;u is the vector defining the polarization of the incident electricfield;x is the frequency of light;and u c k and u v k are the wave functions of the conduction and valence bands,respectively.Fig.5(a)shows the e2(x)of BZO with various intrinsic defects. In the B Zn model,a blue-shift in the intrinsic absorption edge occurred due to an enlarged optical band gap as compared with ZnO.The shallow donor levels mentioned in Section3.3resulted in an absorption peak at1.2eV.The absorption peaks in the intrin-sic defect models were as follows:B Zn V Zn(0.3eV),B Zn V O(1.5eV), and B Zn Zn i(0.9eV).These peaks resulted in enhanced absorption in the visible range.The peak of B Zn V Zn is the lowest,which can probably be attributed to the transition between occupied states and unoccupied states near the Fermi level.In the B Zn V O,and B Zn Zn i models,the absorption in the visible range may be the result of a shift from the shallow and deep donor occupied states to the unoccupied states of the conduction band.Fig.5(b)presents the transmittance of BZO under various defec-tive models.Table2presents the calculated values for average transmittance associated with each model under UV and visible light.It is should be noted that the calculated results were based on the doping levels of B(6.25at.%),V Zn(6.25at.%)V O(6.25at.%), and Zn i(5.88at.%).The average transmittance of pure ZnO is89.2 %in the visible region and65.6%in the UV region.Fig.5(b)shows that the incorporation of B into ZnO decreased transmittance in the range of800–1200nm(infrared region)and400–800nm(visible light region),but increased transmittance in the range of200–400nm(UV region),compared with pure ZnO.When V Zn was introduced to BZO,transmittance in the visible light region was Optical properties of BZO with varying intrinsic defects.(a)Imaginary dielectric function,(b)Transmittance.38increased,but transmittance in the UV region decreased,compared with BZO.When V O or Zn i was introduced to BZO,the transmit-tance of visible as well as UV light declined significantly,which implies that transmittance could be reduced by employing a low-O environment for fabrication.This may explain why trans-mittance is enhanced by an increase in oxygen pressure during processing[7].4.ConclusionsThis study used the DFT+U method to investigate the influence of intrinsic defects on the formation energy,crystal structure,elec-tronic structure,and optical properties of BZO.Our results revealed that the formation energy of V Zn is lowest under O-rich conditions and the formation energy of V O is lowest under O-poor conditions. V Zn defects in BZO may decrease carrier concentration as well as mobility,which increases transmittance in the visible light region but decreases transmittance in the UV region.V O or Zn i defects in BZO lead to the appearance of n-type conductive characteristics, increasing the optical band gap,and decreasing transmittance in the visible light and UV regions.In addition,Zn i defects increase the effective mass,which may consequently decrease the mobility and conductivity of BZO.Conflict of interestsThe authors declare that there is no conflict of interests regard-ing the publication of this article.AcknowledgementsThis work was supported by the National Science Council in Taiwan(NSC102-2221-E-131-008),for which the authors are grateful.We also acknowledge the National Center for High-performance Computing for computer time and the use of its facilities.Reference[1]T.Minami,Semicond.Sci.Technol.20(2005)S35–S44.[2]L.Zhao,G.Shao,S.Song,X.Qin,S.Han,Rare Metals30(2011)175–182.[3]V.Bhavanasi,C.B.Singh,D.Datta,V.Singh,K.Shahi,S.Kumar,Opt.Mater.35(2013)1352–1359.[4]C.Huang,M.Wang,Z.Deng,Y.Cao,Q.Liu,Z.Huang,Y.Liu,W.Guo,Q.Huang,J.Mater.Sci.–Mater.Electron21(2010)1221–1227.[5]J.L.Zhao,X.W.Sun,H.Ryu,Y.B.Moon,Opt.Mater.33(2011)768–772.[6]L.Cao,L.P.Zhu,W.F.Chen,Z.Z.Ye,Opt.Mater.35(2013)1293–1296.[7]T.Miyata,Y.Honma,T.Minami,J.Vac.Sci.Technol.A25(2007)1193–1197.[8]X.L.Chen,B.H.Xu,J.M.Xue,Y.Zhao,C.C.Wei,J.Sun,Y.Wang,X.D.Zhang,X.H.Geng,Thin Solid Films515(2007)3753–3759.[9]D.W.Kang,J.Y.Kwon,D.J.Lee,M.K.Han,J.Electrochem.Soc.159(2012)H61–H65.[10]C.David,T.Girardeau, F.Paumier, D.Eyidi, croix,N.Papathanasiou,B.P.Tinkham,P.Gu´erin,M.Marteau,J.Phys.:Condens.Matter23(2011)334209.[11]H.Yang,X.Xu,X.Zhou,Y.Ma,J.Dong,T.Wang,J.Miao,Y.Jiang,J.Mater.Sci.47(2012)6513–6516.[12]A.B.Patil,K.R.Patil,S.K.Pardeshi,J.Solid State Chem.184(2011)3273–3279.[13]S.Yılmaz,J.Nisar,Y.Atasoy,E.McGlynn,R.Ahuja,M.Parlak,E.Bacaksız,Ceram.Int.39(2013)4609–4617.[14]L.Li,W.Wang,H.Liu,X.Liu,Q.Song,S.Ren,J.Phys.Chem.C113(2009)8460–8464.[15]X.Qu,W.Wang,S.Lv,D.Jia,Solid State Commun.151(2011)332–336.[16]G.Ji,Z.Gu,M.Lu,J.Zhou,S.Zhang,Y.Chen,Physica B405(2010)4948–4950.[17]H.C.Wu,Y.C.Peng,C.C.Chen,Opt.Mater.35(2013)509–515.[18]M.D.Segall,P.J.D.Lindan,M.J.Probert,C.J.Pickard,P.J.Hasnip,S.J.Clark,M.C.Payne,J.Phys.:Condens.Matter.14(2002)2717–2744.[19]H.J.Monkhorst,J.D.Pack,Phys.Rev.B13(1976)5188–5192.[20]D.Vanderbilt,Phys.Rev.B41(1990)7892–7895.[21]X.Ma,B.Lu,D.Li,R.Shi,C.Pan,Y.Zhu,J.Phys.Chem.C115(2011)4680–4687.[22]H.C.Wu,Y.C.Peng,T.P.Shen,Materials5(2012)2088–2100.[23]R.D.Vispute,V.Talyansky,S.Choopun,R.P.Sharma,T.Venkatesan,Appl.Phys.Lett.73(1998)348–350.[24]D.R.Lide,CRC Handbook of Chemistry and Physics,87th ed.,Taylor andFrancis,Philadelphia,2006.[25]X.G.Xu,H.L.Yang,Y.Wu,D.L.Zhang,S.Z.Wu,Appl.Phys.Lett.97(2010)232502.[26]A.Janotti, C.G.Van de Walle,Phys.Rev.B:Condens.Matter.76(2007)165202.[27]T.Guo,G.Dong,Q.Chen,X.Diao,F.Gao,J.Phys.Chem.Solids75(2014)42–47.[28]P.V.Kamat,N.M.Dimitrijevic, A.J.Nozik,J.Phys.Chem.93(1989)2873–2875.[29]L.Gao,Y.Zhang,J.M.Zhang,K.W.Xu,Appl.Surf.Sci.257(2011)2498–2502.[30]B.N.Pawar,S.R.Jadkar,M.G.Takwale,J.Phys.Chem.Solids66(2005)1779–1782.[31]R.Chowdhury,S.Adhikari,P.Rees,Physica B405(2010)4763–4767.Table2Average transmittance of BZO with varying intrinsic defects.UV region(%)Visible region(%)ZnO65.689.2B Zn91.175.6B Zn V Zn68.186.8B Zn V O52.053.2B Zn Zn i59.256.9Y.-C.Peng et al./Optical Materials39(2015)34–3939。

表面技术第52卷第8期Y掺杂MgZn2稳定性、电子结构和力学性能的第一性原理计算陈敬昶1,刘建国2,陆绍敏1,钟丽辉1,陈丽萍1,王远1(1.西南林业大学,昆明 650224;2.云南建投第七建设有限公司,昆明 650100)摘要:目的稀土元素Y掺杂是改善7xxx系铝合金断裂韧性的重要途径,然而因其掺杂量极低,通过实验很难测定微量Y对7xxx系铝合金析出相及强韧机制产生的作用,限制了7xxx系铝合金的进一步发展。
采用第一性原理计算方法探究Y掺杂对7xxx系铝合金中重要析出相MgZn2的影响机理,为7xxx系铝合金的微合金化强韧机理研究提供理论依据。
方法构建适于第一性原理计算、Mg/Zn的原子数分数比为1∶2的晶体模型,Y原子通过替换Mg或Zn原子的方式进行掺杂,通过能量计算、电子计算和弹性常数计算等分析Y掺杂对MgZn2能量稳定性、电子结构和力学性能的影响机理。
结果经Y掺杂后,形成3种固溶体Mg3Zn8Y、Mg4Zn7Y-1和Mg4Zn7Y-2,它们的形成热均小于0,即它们均可自发形成且稳定存在。
通过结合能计算发现,3种固溶体的结合能都小于MgZn2的结合能,说明Y掺杂促进了MgZn2的稳定性。
通过电子结构分析发现,Y掺杂后与Mg、Zn原子形成强的共价键,增强了体系的稳定性,Mg-Zn原子间形成了强离子键,MgZn2中Zn-Zn原子间的共价键变为强离子键。
力学性能计算结果表明,经Y掺杂后MgZn2的硬度降低、韧性上升,即Y掺杂增强了7xxx系铝合金中重要弥散析出相MgZn2的韧性,从而提升了7xxx 系铝合金的断裂韧性和抗疲劳能力。
结论基于计算结果分析得出,Y掺杂提升了MgZn2的稳定性、键合强度和断裂韧性,相关计算分析为微量Y掺杂增强7xxx系铝合金断裂韧性的实验分析提供了指导。
关键词:MgZn2;Y掺杂;电子结构;力学性能;第一性原理计算中图分类号:TG135 文献标识码:A 文章编号:1001-3660(2023)08-0444-07DOI:10.16490/ki.issn.1001-3660.2023.08.040First-principles Calculations of Stability, Electronic Structure andMechanical Properties of Y-doped MgZn2CHEN Jing-chang1, LIU Jian-guo2, LU Shao-min1, ZHONG Li-hui1, CHEN Li-ping1, WANG Yuan1(1. Southwest Forestry University, Kunming 650224, China; 2. YYCIN No.7 Construction Co., Ltd., Kunming 650100, China)ABSTRACT: Micro-alloying of rare earth element Y is an important way to strengthen the fracture toughness of 7xxx series收稿日期:2022-07-20;修订日期:2023-02-16Received:2022-07-20;Revised:2023-02-16基金项目:国家自然科学基金(51301144);云南省教育厅科学研究基金(2022Y574)Fund:National Natural Science Foundation of China (51301144); Scientific Research Foundation of Education Department of Yunnan Province (2022Y574)作者简介:陈敬昶(1996—),男,硕士,主要研究方向为材料表面改性。
first principles of instruction -回复题目:初探“指导教学的第一原理”引言:在教育领域,有一种教学设计方法被广泛应用,它被称作“指导教学的第一原理”(First Principles of Instruction),这种方法强调学习者参与、指导者引导和基于真实任务的学习。
本文将从定义、原理和实施步骤三个方面来初步探索“指导教学的第一原理”。
一、定义指导教学的第一原理(First Principles of Instruction)是由美国教育家David Merrill在1996年提出的一种教学设计模式。
该模式根据科学原理、认知心理学和学习理论等学科的研究成果,结合实践经验,以提高学习者的学习效果为目标。
此模式强调学习者参与、指导者引导和基于真实任务的学习。
二、原理1. 学习者参与(Engagement):学习者要积极参与学习过程,通过实际操作、解决问题和交流合作等方式,来激发兴趣和提高学习效果。
2. 指导者引导(Guidance):指导者在学习过程中扮演重要角色,他们提供指导、支持和反馈,帮助学习者构建知识结构和解决问题。
3. 基于真实任务的学习(Authentic Tasks):学习活动应该与现实生活中的任务和情境相关,让学习者能够将所学知识和技能应用于实践,从而提高学习效果。
三、实施步骤1. 确定学习目标:明确学习者需要掌握的知识、技能和态度,并将其转化为具体的学习目标。
2. 设计真实任务:根据学习目标,设计与现实生活相关的任务和情境,让学习者能够通过实际操作和解决问题来学习。
3. 提供指导和支持:指导者应该提供适当的指导和支持,帮助学习者理解任务要求、组织学习过程,并提供必要的反馈和激励。
4. 促进合作与交流:鼓励学习者进行交流合作,分享经验和观点,促进彼此之间的学习和发展。
5. 管理学习过程:指导者应该管理学习过程,及时调整教学策略和方法,确保学习者能够达到学习目标。
a rXiv:mtrl -th/9522v14Fe b1995First-principle study of excitonic self-trapping in diamond Francesco Mauri ∗and Roberto Car Institut Romand de Recherche Num´e rique en Physique des Mat´e riaux (IRRMA)IN-Ecublens 1015Lausanne,Switzerland Abstract We present a first-principles study of excitonic self-trapping in diamond.Our calculation provides evidence for self-trapping of the 1s core exciton and gives a coherent interpretation of recent experimental X-ray absorption and emission data.Self-trapping does not occur in the case of a single valence exciton.We predict,however,that self-trapping should occur in the case of a valence biexciton.This process is accompanied by a large local relaxation of the lattice which could be observed experimentally.PACS numbers:61.80.−x,71.38.+i,71.35+z,71.55.−iTypeset using REVT E XDiamond presents an unusually favorable combination of characteristics that,in connection with the recent development of techniques for the deposition of thin diamondfilms,make this material a good candidate for many technological applications.Particularly appealing is the use of diamond in electronic or in opto-electronic devices,as e.g.UV-light emitting devices.Moreover,diamond is an ideal material for the construction of windows that operate under high power laser radiation or/and in adverse environments.It is therefore interesting to study radiation induced defects with deep electronic levels in the gap,since these can have important implications in many of these applications.Excitonic self-trapping is a possible mechanism for the formation of deep levels in the gap.The study of such processes in a purely covalent material,like diamond,is interesting also from a fundamental point of view.Indeed,excitonic self-trapping has been studied so far mostly in the context of ionic compounds,where it is always associated with,and often driven by,charge transfer effects.In a covalent material the driving mechanism for self-trapping is instead related to the difference in the bonding character between the valence and the conduction band states.Both experimental data and theoretical arguments suggest the occurrence of self-trapping processes in diamond.In particular,a nitrogen(N)substitutional impurity induces a strong local deformation of the lattice[1–3]that can be interpreted as a self-trapping of the donor electron.The structure of a1s core exciton is more controversial[4–9].Indeed the similarity between an excited core of carbon and a ground-state core of nitrogen suggests that the core exciton should behave like a N impurity.However,the position of the core exciton peak in the diamond K-edge absorption spectra is only0.2eV lower than the conduction band minimum[4,7,8],while a N impurity originates a deep level1.7eV below the conduction band edge[10].On the other hand,emission spectra[8]suggest that a1s core exciton should self-trap like a N impurity.Finally,we consider valence excitations.In this case experimental evidence indicates that a single valence exciton is of the Wannier type,i.e.there is no self-trapping.To our knowledge,neither experimental nor theoretical investigations on the behavior of a valence biexciton in diamond have been performed,although simple scalingarguments suggest that the tendency to self-trap should be stronger for biexcitons than for single excitons.In this letter,we present a detailed theoretical study of excitonic self-trapping effects in diamond.In particular,we have investigated the Born-Oppenheimer(BO)potential energy surfaces corresponding to a core exciton,a valence exciton and a valence biexciton in the context of density functional theory(DFT),within the local density approximation(LDA) for exchange and correlation.Our calculation indicates that the1s core exciton is on a different BO surface in absorption and in emission experiments.Indeed X-ray absorption creates excitons in a p-like state as required by dipole selection rules.Subsequently the system makes a transition to an s-like state associated to a self-trapping distortion of the atomic lattice,similar to that found in the N impurity case.These results provide a coherent interpretation of the experimental data.In addition,our calculation suggests that self-trapping should also occur for a valence biexciton.This is a prediction that could be verified experimentally.Let us start by discussing a simple model[11,12].In diamond,the occupied valence and the lower conduction band states derive from superpositions of atomic sp3hybrids having bonding and antibonding character,respectively.Thus,when an electron,or a hole,or an electron-hole pair is added to the system,this can gain in deformation energy by relaxing the atomic lattice.Scaling arguments suggest that the deformation energy gain E def∝−1/N b, where N b is the number of bonds over which the perturbation is localized.This localization,due to quantum confinement.The in turn,has a kinetic energy cost E kin∝+1/N2/3bbehavior of the system is then governed by the value of N b that minimizes the total energy E sum=E def+E kin.Since the only stationary point of E sum is a maximum,E sum attains its minimum value at either one of the two extrema N b=1or N b=∞.If the minimum occurs for N b=1,the perturbation is self-trapped on a single bond which is therefore stretched.If the minimum occurs for N b=∞,there is no self-trapping and the perturbation is delocalized.When N p particles(quasi-particles)are added to the system,one can showthat,for a given N b,E def scales as N2p,while E kin scales as N p.As a consequence,the probability of self-trapping is enhanced when N p is larger.This suggests that biexcitons should have a stronger tendency to self-trap than single excitons[12,13].In order to get a more quantitative understanding of self-trapping phenomena in dia-mond,we performed self-consistent electronic structure calculations,using norm-conserving pseudopotentials[14]to describe core-valence interactions.The wave-functions and the electronic density were expanded in plane-waves with a cutoffof35and of140Ry,respec-tively.We used a periodically repeated simple cubic supercell containing64atoms at the experimental equilibrium lattice constant.Only the wave-functions at theΓpoint were con-sidered.Since the self-trapped states are almost completely localized on one bond,they are only weakly affected by the boundary conditions in a64atom supercell.The effect of the k-point sampling was analysed in Ref.[3]where similar calculations for a N impurity were performed using the same supercell.It was found that a more accurate k-point sampling does not change the qualitative physics of the distortion but only increases the self-trapping energy by20%compared to calculations based on theΓ-point only[3].In order to describe a core exciton we adopted the method of Ref.[15],i.e.we generated a norm conserving pseudopotential for an excited carbon atom with one electron in the1s core level andfive electrons in the valence2s-2p levels.In our calculations for a valence exciton or biexciton we promoted one or two electrons,respectively,from the highest valence band state to the lowest conduction band state.Clearly,our single-particle approach cannot account for the(small)binding energy of delocalized Wannier excitons.However our approach should account for the most important contribution to the binding energy in the case of localized excitations.Structural relaxation studies were based on the Car-Parrinello(CP) approach[16].We used a standard CP scheme for both the core and the valence exciton, while a modified CP dynamics,in which the electrons are forced to stay in an arbitrary excited eigenstate[12,17],was necessary to study the BO surfaces corresponding to a valence biexciton.All the calculations were made more efficient by the acceleration methods of Ref.[18].Wefirst computed the electronic structure of the core exciton with the atoms in the ideal lattice positions.In this case the excited-core atom induces two defect states in the gap:a non-degenerate level belonging to the A1representation of the T d point group,0.4eV below the conduction band edge,and a3-fold degenerate level with T2character,0.2eV below the conduction band edge.By letting the atomic coordinates free to relax,we found that the absolute minimum of the A1potential energy surface correponds to an asymmetric self-trapping distortion of the lattice similar to that found for the N impurity[3].In particular, the excited-core atom and its nearest-neighbor,labeled a and b,respectively,in Fig.1, move away from each other on the(111)direction.The corresponding displacements from the ideal sites are equal to10.4%and to11.5%of the bond length,respectively,so that the (a,b)-bond is stretched by21.9%.The other atoms move very little:for instance the nearest-neighbor atoms labeled c move by2.4%of the bond length only.This strong localization of the distortion is consistent with the simple scaling arguments discussed above.As a consequence of the atomic relaxation,the non-degenerate level ends up in the gap at1.5eV below the conduction band edge,while the corresponding wavefunction localizes on the stretched bond.The3-fold degenerate level remains close to the conduction band edge,but since the distortion lowers the symmetry from T d to C3v,the3-fold degenerate level splits into a2-fold degenerate E level and a non-degenerate A1level.In Fig.2we report the behavior of the potential energy surfaces corresponding to the ground-state,the A1and the T2core exciton states as a function of the self-trapping dis-tortion.Notice that the distortion gives a total energy gain of0.43eV on the A1potential energy surface.The same distortion causes an increase of the ground-state energy of1.29 eV.Our calculation indicates that the core-exciton behaves like the N impurity[3],support-ing,at least qualitatively,the validity of the equivalent core approximation.The similar behavior of the A1level in the core exciton and in the N impurity case was also pointed out recently in the context of semi-empirical CNDO calculations[9].The differences between the core exciton and the impurity[3]are only quantitative:in particular,the relaxationenergy and especially the distance of the A1level from the conduction band edge are smaller for the core exciton than for the N impurity.Our results suggest the following interpretation of the experimental data of Refs.[4,8]: (i)During X-ray absorption the atoms are in the ideal lattice positions.Dipole transitions from a1s core level to a A1valence level are forbidden,but transitions to the T2level are allowed.In our calculation the T2level is0.2eV lower than the conduction band edge,in good agreement with the core exciton peak observed in X-ray absorption spectra[4,8].(ii) On the T2BO potential energy surface the lattice undergoes a Jahn-Teller distortion which lowers its energy(see Fig.2).(iii)Since the LO phonon energy in diamond(0.16eV)is comparable to the energy spacing between the A1and the T2surfaces,which is less than 0.2eV after the Jahn-Teller distortion,the probability of a non-adiabatic transition from the T2to the A1surface is large.(iv)On the A1level the system undergoes a strong lattice relaxation resulting in a localization of the exciton on a single bond.(v)The self-trapping distortion induces a Stokes shift in the emitted photon energy.If the atomic relaxation were complete the Stokes shift would be equal to1.9eV,which correponds(see Fig.2) to the energy dissipated in the T2-A1transition(0.2eV),plus the energy gained by self trapping on the A1surface(0.43eV),plus the energy cost of the self-trapping distortion on the ground-state energy surface(1.29eV).The data reported in Ref.[8]show a shift of about1eV in the positions of the peaks associated to the1s core exciton in X-ray absorption and emission spectra.The emission peak is very broad,with a large sideband that corresponds to Stokes shifts of up to5eV.As pointed out in Ref.[8],this large sideband is likely to be the effect of incomplete relaxation. This is to be expected since the core exciton lifetime should be comparable to the phonon period[8].As a consequence,the atomic lattice would be able to perform only a few damped oscillations around the distorted minimum structure during the lifetime of the core exciton.We now present our results for the valence excitations.While in the case of a single exciton the energy is minimum for the undistorted crystalline lattice,in the case of a biex-citon wefind that the energy is minimized in correspondence of a localized distortion of theatomic lattice.This is characterized by a large outward symmetric displacement along the (111)direction of the atoms a and b in Fig.1.As a result the(a,b)-bond is broken since the distance between the atoms a and b is increased by51.2%compared to the crystalline bondlength.This distortion can be viewed as a kind of local graphitization in which the atoms a and b change from fourfold to threefold coordination and the corresponding hy-bridized orbitals change from sp3to sp2character.Again,in agreement with the model based on simple scaling arguments,the distortion is strongly localized on a single bond.As a matter of fact and with reference to the Fig.1,the atoms c and d move by1.2%of the bondlength,the atoms e and f move by2.3%,and the atoms not shown in thefigure by less than0.9%.The self-trapping distortion of the biexciton gives rise to two deep levels in the gap: a doubly occupied antibonding level,at1.7eV below the conduction band edge,and an empty bonding level,at1.6eV above the valence band edge.Both states are localized on the broken bond.In Fig.3we show how different BO potential energy surfaces behave as a function of the self-trapping distortion of the valence biexciton.In particular,from thisfigure we see that,while for the biexciton there is an energy gain of1.74eV in correspondence with the self-trapping distortion,the same distortion has an energy cost of1.49eV for the single exciton,and of4.85eV for the unexcited crystal.We notice that,while DFT-LDA predicts self-trapping for the valence biexciton,it does not do so for the single exciton,in agreement with experiment.Similarly to the case of the core exciton the major experimental consequence of the self-trapping of the valence biexciton is a large Stokes shift in the stimulated-absorption spontaneous-emission cycle between the exciton and the biexciton BO surfaces.As it can be seen from Fig.3,this Stokes shift should be equal to3.23eV,i.e.to the sum of the energy gain of the biexciton(1.74eV)and of the energy cost of the exciton(1.49eV) for the self-trapping relaxation.The fundamental gap of diamond is indirect.Thus the spontaneous decay of a Wannier exciton in an ideal diamond crystal is phonon assistedand the radiative lifetime of the exciton is much longer than in direct gap semiconductors. However,after self-trapping of the biexciton,the translational symmetry is broken and direct spontaneous emission becomes allowed.As a consequence the radiative life time of the self-trapped biexciton is much smaller than that of the Wannier ing the DFT-LDA wavefunctions,we obtained a value of∼7ns for the radiative lifetime of the biexciton within the dipole approximation.This is several orders of magnitude larger than the typical phonon period.Therefore the self-trapping relaxation of the valence biexciton should be completed before the radiative decay.A self-trapped biexciton is a bound state of two excitons strongly localized on a single bond.Thus the formation of self-trapped biexcitons requires a high excitonic density.To realize this condition it is possible either to excite directly bound states of Wannier excitons, or to create a high density electron-hole plasma,e.g.by strong laser irradiation.In the second case many self-trapped biexcitons could be produced.This raises some interesting implications.If many self-trapped biexcitons are created,they could cluster producing a macroscopic graphitization.Moreover,since the process of self-trapping is associated with a relevant energy transfer from the electronic to the ionic degrees of freedom,in a high density electron hole plasma biexcitonic self-trapping could heat the crystal up to the melting point in fractions of a ps,i.e in the characteristic time of ionic relaxation.Interestingly,melting ofa GaAs crystal under high laser irradiation has been observed to occur in fractions of a ps[19].In Ref.[19]this phenomenon has been ascribed to the change in the binding properties due to the electronic excitations.Our study on diamond leads one to speculate that in a sub-picosecond melting experiment self-trapping phenomena could play an important role.In conclusion,we have studied excited-state BO potential energy surfaces of crystalline diamond within DFT-LDA.Our calculation predicts self-trapping of the core exciton and provides a coherent description of the X-ray absorption and emission processes,which com-pares well with the experimental data.Moreover,we also predict self-trapping of the valence biexciton,a process characterized by a large local lattice relaxation.This implies a strong Stokes shift in the stimulated absorption-spontaneous emission cycle of about3eV,whichcould be observed experimentally.It is a pleasure to thank F.Tassone for many useful discussions.We acknowledge support from the Swiss National Science Foundation under grant No.20-39528.93REFERENCES∗Present address:Departement of Physics,University of California,Berkeley CA94720, USA.[1]C.A.J.Ammerlaan,Inst.Phys.Conf.Ser.59,81(1981).[2]R.J.Cook and D.H.Whiffen,Proc.Roy.Soc.London A295,99(1966).[3]S.A.Kajihara et al,Phys.Rev.Lett.66,2010(1991).[4]J.F.Morar et al,Phys.Rev.Lett.54,1960(1985).[5]K.A.Jackson and M.R.Pederson,Phys.Rev.Lett.67,2521(1991).[6]J.Nithianandam,Phys.Rev.Lett.69,3108(1992).[7]P.E.Batson,Phys.Rev.Lett.70,1822(1993).[8]Y.Ma et al,Phys.Rev.Lett.71,3725(1993).[9]A.Mainwood and A.M.Stoneham,J.Phys.:Condens.Matter6,4917(1994).[10]R.G.Farrer,Solid State Commun.7,685(1969).[11]W.Hayes and A.M.Stoneham,Defects and defect processes in nonmetallic solids,(Wiley&Sons,New York,1985)pags.29-38.[12]F.Mauri,R.Car,(to be published).[13]The number of equal particles that can be accommodated on one bond of the crystal inthe same quantum state is limited by the Pauli principle.Thus no more than two holes or/and two electrons with opposite spins can be localized on one bond of a sp3bonded semiconductor.[14]G.Bachelet,D.Hamann,and M.Schl¨u ter,Phys.Rev.B26,4199(1982).[15]E.Pehlke and M.Scheffler,Phys.Rev.B47,3588(1993).[16]R.Car and M.Parrinello,Phys.Rev.Lett.55,2471(1985).[17]F.Mauri,R.Car and E.Tosatti,Europhys.Lett.24,431(1993).[18]F.Tassone,F.Mauri,and R.Car,Phys.Rev.B50,10561(1994).[19]orkov,I.L.Shumay,W.Rudolph,and T.Schroder,Opt.Lett.16,1013(1991);P.Saeta,J.-K.Wang,Y.Siegal,N.Bloembergen,and E.Mazur,Phys.Rev.Lett.67, 1023(1991);K.Sokolowski-Tinten,H.Schulz,J.Bialkowski,and D.von der Linde, Applied Phys.A53,227(1991).FIGURESFIG.1.Atoms and bonds in the ideal diamond crystal(left panel).Atoms and bonds after the self-trapping distortion associated with the valence biexciton(right panel).In this case the distance between the atoms a and b increases by51.2%.A similar but smaller distortion is associated with the core exciton:in this case the(a,b)distance is increased by21.9%.FIG.2.Total energy vs self-trapping distortion of the core-exciton.Thefigure displays the BO potential energy surfaces correponding to the ground-state,the A1,and the T2core exciton states.FIG.3.Total energy as a function of the self-trapping distortion of the biexciton.The BO energy surfaces correponding to the ground state,the valence exciton,and the valence biexciton are shown in thefigure.a b ce df(111)ground stateA 1−core excitonT 2−core excitonconduction ideal lattice distorted latticeground statebi−excitonexcitondistorted lattice ideal lattice。