RNA-Seq项目常见问题与解答
- 格式:docx
- 大小:61.13 KB
- 文档页数:5


RNA-seq基础知识1.RNA-Seq名词解释2.测序名词解释3.高通量测序常用名词解释4.转录组测序问题集锦RNA-Seq名词解释1.index 测序的标签,用于测定混合样本,通过每个样本添加的不同标签进行数据区分,鉴别测序样品。
2.碱基质量值(Quality Score或Q-score)是碱基识别(Base Calling)出错的概率的整数映射。
碱基质量值越高表明碱基识别越可靠,碱基测错的可能性越小。
3.Q30 碱基质量值为Q30代表碱基的精确度在99.9%。
4.FPKM(Fragments Per Kilobase of transcript per Millionfragments mapped)每1百万个map上的reads中map到外显子的每1K个碱基上的fragment个数。
计算公式为公式中,cDNAFragments 表示比对到某一转录本上的片段数目,即双端Reads数目;Mapped Reads(Millions)表示Mapped Reads总数,以10为单位;Transcript Length(kb):转录本长度,以kb个碱基为单位。
5.FC(Fold Change)即差异表达倍数。
6.FDR(False Discovery Rate)即错误发现率,定义为在多重假设检验过程中,错误拒绝(拒绝真的原(零)假设)的个数占所有被拒绝的原假设个数的比例的期望值。
通过控制FDR来决定P值的阈值。
7.P值(P-value)即概率,反映某一事件发生的可能性大小。
统计学根据显著性检验方法所得到的P 值,一般以P<0.05为显著,P<0.01为非常显著,其含义是样本间的差异由抽样误差所致的概率小于0.05或0.01。
8.可变剪接(Alternative splicing)有些基因的一个mRNA前体通过不同的剪接方式(选择不同的剪接位点)产生不同的mRNA剪接异构体,这一过程称为可变剪接(或选择性剪接,alternative splicing)。

RNAseq汇总篇,一文掌握RNAseqRNA测序(RNA-seq)在过往十年里逐渐成为全转录组水平分析差异基因表达和研究mRNA差异剪接必不可少的工具。
RNA-seq帮助大家对RNA生物学的理解会越来越全面:从转录本在何时何地转录到RNA折叠以及分子互作发挥功能等。
1.RNA-seq相关名词详细介绍了RNA seq的专业词、高通量测序常用词、转录组测序问题等,是入门RNA seq较好的资料。
2.什么是RNA-seq?一文读懂了解了RNA-seq相关基础知识,需要进一步了解RNA seq究竟是什么?能做什么?一文读懂。
RNA-seq是一种集合实验方法和计算机手段的一种技术,它可以确定生物样本中RNA序列的特征性和丰度。
RNA-seq方法的产生源自于测序技术的世代革新。
RNA-seq数据可以让我们知道很多未知的东西,比如,我们可以识别出胚胎干细胞中编码新蛋白质的转录本,可以找到皮肤癌细胞中那些过表达的转录本。
3.RNA-seq测序基本知识一般来说,NGS测序特别是RNA-seq正在迅速改变实验的设计和执行方式。
由于技术的飞速发展,可以公平地说,对于一个特定问题没有单一的正确答案。
而且许多RNA-seq项目有多个目标,例如,可能需要鉴定样本中的新基因融合转录物,对已知基因的丰度进行量化,并鉴定已知基因中的任何SNP。
因此,根据研究设计原则提供指导是更为合理的,用户既可以对预期成果充满信心地计划项目,也可以理解为什么做出某些选择。
在一项研究中所使用的覆盖范围和平台的数量可能需要进行权衡,而且由于实验室资源有限,因此需要进行权衡。
4.RNA-Seq怎么做,又会遇到哪些问题这两年随着测序成本的下降和转录组研究的日渐火热,RNA-seq 俨然已经成为了分子生物学课题组推进项目的首选方向。
5.37个RNA-seq工具大PK,教你数据处理方法如何选择RNA-seq技术的广泛应用为转录组研究迎来了一个新时代。
根据研究内容的方向,精度、速度和成本要求不同,科研人员需要对包括采取何种具体测序方法流程、样品类型、所需的分析结果,以及基因组研究现状和计算数据处理可用资源等内容进行权衡。

RNA-seq从过滤低质量到去接头完整质控步骤3.1 碱基质量碱基的质量反映了碱基识别的好坏,这个在phred的质量评分中得以体现,将碱基出错的比例作为log10的对数,然后乘-10得到的标准值。
举例说明,当100个碱基中有1个错误,质量评分为q = −10*log10(0.01) = 20,质量评分通常在0至40的范围内。
除了数字,在FASTQ文件中质量信息通常用ASCII字符来表示以节约字符空间。
现今的FASTQ的文件最初是由sanger开发,值的范围从0到92,对应的ASCII码从33开始至126。
值得注意的是1.8版之前的Illumina 软件(phred)使用的ASCII字符是由64开始的。
进一步了解不同软件不同的编码方式,参考FASTQ文件格式手册。
如果你不知道你文件的质量编码格式,利用FastQC可以帮你辨别,如果你希望转换FASTQ文件的质量评分,Trimmomatic可以解决这个问题。
一般碱基质量值在之后的循环测序中会下降,通过查看reads的可视化箱形图可以轻松得出结论,在FastQC和PRINSEQ的质量报告中都有这种类型的箱形图。
图2展示了我们以双端测序为样本的FastQC碱基序列质量报告,如图所示,正向(fastq1)reads的质控质量很高,然而反向(fastq2)reads的质量很差,特别是在reads越接近末尾处质量越差。
除了检测每个碱基位点的碱基质量分布,检测reads的平均质量分布也是有价值的,这允许我们查看reads中是否有一部分整体质量不好,FastQC和PRINSEQ都可以绘制reads的平均碱基质量分布图。
理想状态下,大多数的reads的碱基质量均值都应当为25或高于这个值,如图3所示,正向与反向的reads都包含有一段碱基质量普遍不好的2000000个碱基的reads。
可以对包含低碱基质量的reads进行过滤和修剪操作,过滤会去除整段reads,而修剪则会允许仅去除reads末尾低质量的片段,如果你打算过滤或质量矫正双端测序的reads,选择一个能够在输出时保存reads的匹配位置信息的工具,即使某一read或者其对应碱基被删除的情况下。

名词专题RNA-seq常见名词解释前言各位亲们,文献中的很多名字是否困惑过?别怕!我们会用一个专题来解释相关的名词,以期给各位带来一些帮助。
RNA-seq:基于二代测序技术,研究特定细胞在某一功能状态下所有RNA 的功能,主要包括 mRNA 和非编码RNA。
能够全面快速地获得某一物种特定组织或器官在某一状态下的几乎所有转录本序列信息,已广泛应用于基础研究、临床诊断和药物研发等领域。
Q20,Q30:二代测序中,每测一个碱基会给出一个相应的质量值,这个质量值是衡量测序准确度的。
碱基的质量值20的错误率为1%,30的错误率为0.1%。
Q20与Q30表示质量值≧20或30的碱基所占百分比,如碱基质量值为20则表示该碱基的错误率为10^(20/(-10))=0.01=1%(根据Q=-10lgP计算,P为错误率)intron:内含子,是真核生物细胞DNA 中的间插序列。
这些序列被转录在前体RNA 中,经过剪接被去除,最终不存在于成熟RNA 分子中。
术语内含子也指编码相应RNA 内含子的DNA 中的区域。
exon:外显子,是真核生物基因的一部分,它在剪接(Splicing)后仍会被保存下来,并可在蛋白质生物合成过程中被表达为蛋白质。
外显子是最后出现在成熟RNA 中的基因序列,又称表达序列。
既存在于最初的转录产物中,也存在于成熟的RNA 分子中的核苷酸序列。
术语外显子也指编码相应RNA 外显子的DNA 中的区域。
intergenic:基因间区,指基因与基因之间的间隔序列,不属于基因结构,不直接决定氨基酸,可能通过转录后调控影响性状的区域。
UTR:Untranslated Regions, 非翻译区域。
是信使RNA (mRNA)分子两端的非编码片段。
5'-UTR 从mRNA 起点的甲基化鸟嘌呤核苷酸帽延伸至AUG 起始密码子,3'-UTR 从编码区末端的终止密码子延伸至多聚A 尾巴(Poly-A)的前端。
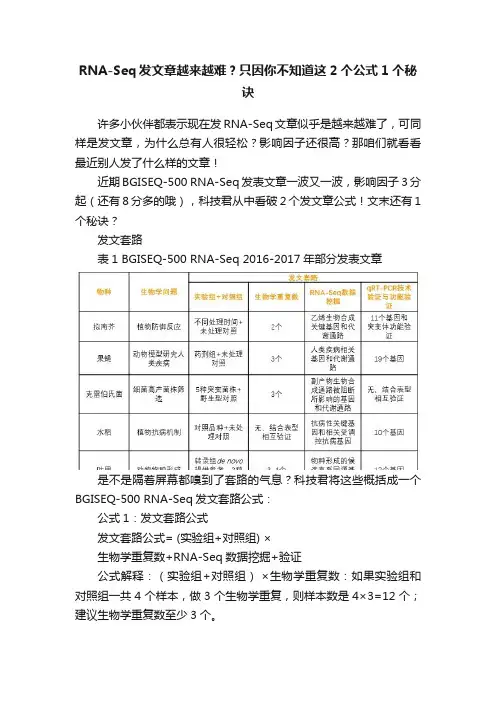
RNA-Seq发文章越来越难?只因你不知道这2个公式1个秘诀许多小伙伴都表示现在发RNA-Seq文章似乎是越来越难了,可同样是发文章,为什么总有人很轻松?影响因子还很高?那咱们就看看最近别人发了什么样的文章!近期BGISEQ-500 RNA-Seq发表文章一波又一波,影响因子3分起(还有8分多的哦),科技君从中看破2个发文章公式!文末还有1个秘诀?发文套路表1 BGISEQ-500 RNA-Seq 2016-2017年部分发表文章是不是隔着屏幕都嗅到了套路的气息?科技君将这些概括成一个BGISEQ-500 RNA-Seq发文套路公式:公式1:发文套路公式发文套路公式= (实验组+对照组) ×生物学重复数+RNA-Seq数据挖掘+验证公式解释:(实验组+对照组)×生物学重复数:如果实验组和对照组一共4个样本,做3个生物学重复,则样本数是4×3=12个;建议生物学重复数至少3个。
RNA-Seq数据挖掘:找到关键基因、功能和通路验证:再做10个左右的qRT-PCR技术验证,推荐继续做功能验证,如果要发表高分文章,尤其是疾病研究文章,还需大样本量的验证。
另外,对于无参考序列的物种,混合所有样本做一个转录组de novo组装后作为参考序列。
公式2:样本选择公式“实验组+对照组”,样本选择要充分考虑基因差异表达来源进行样本选择,科技君也概括了一个样本选择公式:样本选择公式=空间+时间+条件+对照-表型公式解释:空间:不同的组织、器官、性别;时间:不同的发育时期/处理时间/疾病发展时期;条件:不同处理条件,例如剂量梯度;对照:对照品种或样本,例如野生型、感病/抗病品种、正常与疾病样本;对照处理,例如未处理、阴性处理;表型:结合表型是关键,找出关键时间点、最佳处理条件,减少盲目设计大量样本。
接下来科技君为大家一一介绍发表文章☟☟☟文章一生物学问题——发现植物防御反应中的关键基因和调控元件铜离子促进拟南芥乙烯合成,进而引起植物防御反应,RNA-Seq 及后期功能验证阐明关键基因和调控元件在铜离子刺激下能够促进合成乙烯中的分子机制。

rna seq测序原理-回复RNA测序(RNA sequencing)是一种先进的技术,用于研究以及阐明RNA分子在生物体中的表达和调控。
通过RNA测序,科学家可以获得关于基因转录的全面信息,此类信息对于研究基因功能、疾病发展和对药物治疗的应用具有重要作用。
本文将一步一步回答有关RNA测序原理的问题。
Q1:RNA测序是什么?A:RNA测序是一种实验技术,用于测量和记录RNA分子的序列信息。
通过RNA测序,我们可以获得RNA分子构成的“蓝图”,这些蓝图指示了基因在细胞中如何被转录为RNA,并揭示了这些RNA分子如何影响细胞的功能和表型。
Q2:RNA测序的方法有哪些?A:RNA测序有多种方法,其中最常用的是转录组测序(transcriptome sequencing),它可以测量基因在特定生物样本中的转录水平。
其他常见的RNA测序方法包括全长转录组测序(full-length transcriptome sequencing),用于测量全长转录本的序列信息,以及单细胞RNA测序(single-cell RNA sequencing),用于研究单个细胞中的基因表达。
Q3:RNA测序的工作原理是什么?A:RNA测序的工作原理主要包括RNA提取、合成cDNA(互补DNA)、文库构建、测序和数据分析等步骤。
1. RNA提取:从细胞或组织中提取RNA分子,通常使用柱式提取法或磁珠提取法。
2. 合成cDNA:通过逆转录反应,将RNA转录成合成DNA(cDNA)。
这涉及到使用反转录酶将RNA作为模板,合成一条互补的DNA链。
3. 文库构建:将合成的cDNA进行文库构建,这包括将DNA片段连接到测序适配体上,以便在测序前进行扩增。
4. 测序:使用高通量测序平台(如Illumina)对文库中的DNA片段进行测序。
这一步会生成大量的短序列读数,其中包含着DNA片段的序列信息。
5. 数据分析:对测得的序列数据进行处理、比对和解读,以获得准确的转录组信息,包括转录本的丰度、剪接变异以及基因表达的变化等。

RNA-seq,你必须知道的事1转录组测序能解决哪些问题?(1)差异基因分析:选用来自不同生理或病理状态组织/细胞的样本为研究对象,通过分析各样本基因表达情况,进行表达差异分析,从而筛选出与其状态相关的候选基因。
(2)差异基因富集分析:对筛选到的差异基因进行功能富集分析(GO,KEGG 富集分析),进一步全面地挖掘与样本性状相关的分子机制。
(3)分子标记开发:通过转录组测序进行SNP、SSR 等分析,能够更加全面高效地挖掘样本的分子标记。
该分析广泛应用于遗传育种、基因定位、物种亲缘关系鉴别、基因库构建、基因克隆等方面。
(4)模式生物转录本深入研究:对已有参考基因组的模式生物,转录组测序可以更加深入地分析其基因结构相关信息,包括分析可变剪切、基因融合、新转录本的预测和注释、lncRNA 的预测等。
2送样需要注意哪些?(1)组织样品动物组织:> 2g;植物组织:> 4g;培养细胞:>1×107 个;血液样品:≥ 2ml(最好是全血)(2)真核生物RNA请提供浓度≥ 200ng/μL,总量≥10μg 的RNA(单次建库用量为5μg);OD260/280介于1.8~2.2 之间,OD260/230≥2.0,RIN≥6.5,28S:18S ≥1.0,确保RNA 无降解;送样时请标记清楚样品编号,管口使用Parafilm 膜密封;样品保存期间切忌反复冻融;送样时请使用干冰运输。
(3)原核生物RNA请提供浓度≥200ng/μL,总量≥10μg 的RNA(单次建库用量为5μg);OD260/280 介于1.8~2.2 之间,OD260/230≥2.0,RIN≥6.5,23S:16S ≥1.0,确保RNA 无降解;送样时请标记清楚样品编号,管口使用Parafilm 膜密封;样品保存期间切忌反复冻融;送样时请使用干冰运输。
3转录组测序需要多大测序量?转录组测序所需的测序量随物种转录组大小的不同而有所差异。

转录组测序结题报告怎么看?点进来就知道啦!转录组测序(RNA-Seq)作为研究基因表达的利器,是发掘基因功能的重要途径。
随着RNA-Seq技术的普及,那么问题来了,很多不了解RNA-Seq的小伙伴,在点开结题报告的一瞬间,是不是满脑子的问号,不知所措呢?没关系!我们懂你!不了解RNA-Seq?不会看结题报告?莫慌,我们来给大家理头绪、划重点!首先,可将整个结题报告分成四个主要模块。
图 1 转录组测序结题报告主要模块差异基因的鉴定与功能富集分析是构成转录组文章的主体,数据挖掘与分析也是基于这两个模块进行,是结题报告的重心。
接下来详细告诉大家每个模块需要关注的重点内容。
原始数据整理与质量评估数据量的大小与测序质量的好坏是评判测序数据可靠性的重要标准。
▶数据量一般用Bases或Raw data表示,对于绝大部分物种来说,转录组测序6G数据量即可,若想获得更多低丰度基因的信息,可适当增加测序数据量。
▶数据质量主要包括碱基质量与碱基含量。
Illumina官方的碱基质量评价标准一般为Q30(即碱基错误识别率为0.1%),Q30的值越大越好,一般不能低于80%。
碱基含量即ATGC四种碱基所占的比例,除了前几个碱基位置之外,4种碱基的含量线条应平行且接近。
图 2 测序质量评估差异基因表达鉴定看基因的表达量与鉴定差异基因是做转录组测序的主要目的,生物学重复之间的相关性高低与差异基因鉴定的准确性息息相关。
▶样品相关性检验一般以矩阵图与PCA分析图展示。
在矩阵图中基因表达相近的样品会被聚到一起,生物学重复间相关系数越高越好,低于0.8表示相关性较差。
PCA分析图更加直观,可以把基因表达相关性好的样品展示到一起。
图3 样品相关性检验▶差异表达基因的鉴定在这里可以看到各个处理组与对照组之间基因的上、下调表达的信息。
从中查找所关注基因的表达情况。
显著差异基因判定标准:|log2 Foldchange|>1;P value < 0.05。

RNA-seq结果怎么才能看懂?答案全在这些图里---(2)基础分析结果篇上一期的RNA-seq结果怎么才能看懂?答案全在这些图里---(1)测序质量篇,(点这里查看这一期微信),我们介绍了评估RNA测序质量的一些常见图示。
本期,小编继续“看图说话”,一起看看RNA-seq基础分析里的图示都反映了哪些内容吧。
1主成分分析图(PCA图)----用RNA测序结果体现样本聚类主成分分析图是生信分析中最朴实无华的,因为谁都能看的懂。
我们不需要操心X,Y轴的主成分到底是什么,只要明白每个样本都被一个2维坐标(X,Y)定位到了这张图上。
对于基于转录组的PCA图中,如果两个样本距离越远,则说明两个样本转录组差异越大。
我们最想看到的情况就是,相同表型的个体(比如疾病组)会在图中聚类在一起。
2差异基因表达散点图----体现重复样本的重复性好不好我们可以简单的把这张图理解为2个样本的RNAseq结果关联度散点图。
X,Y轴分别是两个样本,每个点代表一个基因在两个样品中FPKM 的对数值(FPKM是RNAseq中衡量基因表达高低的常用数值)。
从这张图可以观察,偏离对角线的点越多,说明样品表达量的相关性越低,重复性越差;偏离对角线的点越少,则说明样品间表达量的相关性越高,重复样品的重复性越好。
3差异基因表达火山图---直观展示上调表达和下调表达基因数量对于常规的2组样本RNAseq研究,我们关心的是组1和组2到底哪些基因有显著的差异表达(T检验获得P值,p值反映显著性),差异表达基因在组1和组2之间到底差了多少倍。
这些信息都是通过火山图展示了出来的。
火山图是以log2(差异倍数)为横坐标,以T检-log10(P值)为纵坐标。
所以,我们最关心的基因就是图中左上角和右上角的点,分别表示表达水平差异非常显著的下调基因和上调基因。
4差异基因聚类热图---体现样本聚类和基因聚类聚类热图体现了2个层次的聚类,一般会在横轴和纵轴的位置展示。

RNA提取常见问题、原因分析及其对策RNA提取常见问题、原因分析及其对策一、RNA提取的通用方法异硫氰酸胍/苯酚法(即TriZol类试剂)细胞在变性剂异硫氰酸胍的作用下被裂解,同时核蛋白体上的蛋白变性,核酸释放;释放出来的DNA和RNA由于在特定pH下溶解度的不同而分别位于整个体系中的中间相和水相,从而得以分离;有机溶剂抽提,沉淀,得到纯净RNA。
步骤:材料准备:尽量新鲜。
裂解变性:异硫氰酸胍(亚硫氢胍,巯基乙醇,N-月桂肌氨酸等)。
使细胞及核蛋白复合物变性,释放RNA,有效抑制核酸酶。
纯化分离:苯酚,氯仿,异戊醇。
苯酚/氯仿可抽提去除杂物。
洗涤:70%乙醇。
沉淀:异丙醇、无水乙醇。
乙酸钠(pH4.0):维持变性的细胞裂解液的pH值,沉淀RNA。
此外还常用氯化锂选择沉淀RNA。
二、影响RNA提取的因素由于RNA样品易受环境因素特别是RNA酶的影响而降解,提取高质量的RNA样品在生命科研中具有相当的挑战性。
RNA提取对样品的新鲜性要求非常高,获取样品后最好立即提取RNA,若无条件立即实验,应于-80℃或液氮中保存样品,提取时取出样品后立即在低温下研磨裂解细胞,以防RNA降解。
1.材料:新鲜,切忌使用反复冻融的材料,如若材料来源困难,且实验需要一定的时间间隔。
可以先将材料贮存在TRIzol或样品贮存液中,于-70℃或-20℃保存如要多次提取,请分成多份保存,液氮长期保存,-70℃短期保存。
2.样品破碎及裂解:根据不同材料选择不同的处理方法:培养细胞:通常可直接加裂解液裂解酵母和细菌:一般TRIzol可直接裂解,对一些特殊的材料可先用酶或者机械方法破壁动植物组织:先液氮研磨和匀浆,后加裂解液裂解。
期间动作快速,样品保持冷冻,样品量适当,保证充分裂解为减少DNA污染,可适当加大裂解液的用量3.纯化:在使用氯仿抽提纯化时,一定要充分混匀,且动作快速;经典的纯化方法,如LiCl 沉淀等,虽然经济,但操作时间长,易造成RNA 降解;柱离心式纯化方法:抽提速度快,能有效去除影响RNA 后续酶反应的杂质,是目前较为理想的选择。
RNA 抽提比基因组DNA 抽提要困难得多。
事实上,现有的RNA 抽提方法/试剂,如果用于从培养细胞中抽提RNA,比抽提基因组DNA 更方便,成功率也更高。
那为什么同样的方法用于组织RNA 的抽提,总会碰到问题呢?组织RNA 抽提失败的两大现象是:RNA 降解和组织内杂质的残留。
关于降解问题,首先看一下为什么从培养细胞中抽提RNA 不容易降解。
现有的RNA 抽提试剂,都含有快速抑制Rnase 的成分。
在培养细胞中加入裂解液,简单的混匀,即可使所有的细胞与裂解液充分混匀,细胞被彻底裂解。
细胞被裂解后,裂解液中的有效成分立即抑制住细胞内的Rnase,所以RNA 得以保持完整。
也就是说,培养细胞由于很容易迅速与裂解液充分接触,所以其RNA 不容易被降解;反过来讲,组织中的RNA 之所以容易被降解,是因为组织中的细胞不容易迅速与裂解液充分接触所致。
因此,假定有一种办法,在抑制RNA 活性的同时能使组织变成单个细胞,降解问题也就可以彻底解决了。
液氮碾磨就是最有效的这样一种办法。
但是,液氮碾磨方法非常麻烦,如果碰到样品数比较多的时候更加会有此感觉。
这样就产生了退而求其次的方法:匀浆器。
匀浆器方法没有考虑细胞与裂解液接触前如何抑制Rnase 活性这个问题,而是祈祷破碎组织的速度比细胞内的Rnase 降解RNA 的速度快。
电动匀浆器效果较好,玻璃匀浆器效果较差,但总的来说,匀浆器方法是不能杜绝降解现象的。
因此,如果抽提出现降解,原来用电动匀浆器的,改用液氮碾磨;原来用玻璃匀浆器的,改用电动匀浆器或者直接用液氮碾磨,问题几乎100% 能获得解决。
影响后续实验的杂质残留问题,其原因比降解更多样,解决方法响应也不同。
总之,如果出现降解现象或者组织内杂质残留现象,则必须对具体实验材料的抽提方法/试剂进行优化。
优化大可不必使用您的宝贵样品:可以从市场上购买一些鱼/鸡之类的小动物,取相应部分的材料用于RNA 抽提,其它部分用于抽提蛋白质–用嘴碾磨,肠胃抽提。
第6章-思考题解析第6章思考题解析1.写出用RNA-seq技术进行转录组学分析的原理。
答:RNA-Seq是利用高通量测序技术对转录组进行测序分析,对测序得到的大量原始读长(reads)进行过滤、组装及生物信息学分析的过程。
对于有参考基因组序列的物种,需要根据参考序列进行组装;对于没有参考序列的,需要进行从头组装,利用大量读长之间重叠覆盖和成对读长的相对位置关系,组装得到尽可能完整的转录本,并以单位长度转录本上覆盖的读长数目,作为衡量基因表达水平的标准。
2.写出RNA原位杂交的主要实验过程及应用。
答:RNA原位杂交用放射性或非放射性(如地高辛、生物素等)标记的特异性探针与被固定的组织切片反应,若细胞中存在与探针互补的mRNA分子,两者杂交产生双链RNA,就可通过检测放射性标记或经酶促免疫显色,对该基因的表达产物在细胞水平上做出定性定量分析。
应用:RNA原位杂交常被用于检测动植物组织中某种特定基因的mRNA的表达情况;还可用在基因分析和诊断方面能作定性、定位和定量分析。
3.说出免疫共沉淀实验的原理与过程,比较酵母双杂交技术和免疫共沉淀技术在研究蛋白质相互作用方面的优缺点。
答:免疫共沉淀实验(Co-Immunoprecipitation)的原理与过程:免疫共沉淀技术的核心是通过抗体来特异性识别候选蛋白。
首先,将靶蛋白的抗体通过亲和反应连接到固体基质上,再将可能与靶蛋白发生相互作用的待筛选蛋白加入反应体系中,用低离心力沉淀或微膜过滤法在固体基质和抗体的共同作用下将蛋白复合物沉淀到试管的底部或微膜上。
如果靶蛋白与待筛选蛋白质发生了相互作用,那么这个待筛选蛋白质就通过靶蛋白与抗体和固体基质相互作用而被分离出来。
免疫共沉淀技术在研究蛋白质相互作用方面的优点:(1)相互作用的蛋白质都是经翻译后修饰的,处于天然状态;(2)蛋白质的相互作用是在自然状态下进行的,可以避免人为的影响;(3)可以分离得到天然状态的相互作用蛋白复合物。
转录组测序问题集锦转录组测序问题集锦转录组是某个物种或者特定细胞类型产生的所有转录本的集合,转录组测序(RNA-seq)是最近发展起来的利用深度测序技术进行转录分析的方法,可以对全转录组进行系统的研究。
Roche GS FLX Titanium 、Illumina Solexa GA IIx和AB SOLID 4均可以对转录组进行测序,Roche GS FLX Titanium与Illumina Solexa GA IIx和AB SOLID 4相比,拥有更长的读长和较小的数据量,适用于表达量较高基因的RNA全长测序。
但是对低表达丰度的基因,可能需要多次测序才能得到足够的数据,成本比较高,而Illumina Solexa GA IIx和AB SOLID 4数据读取量大,能够得到较高的覆盖率,可以较好的降低成本。
若是位置基因组序列的物种,则Roche GS FLX Titanium测序更有优势,其较长的读长便于拼接,获得更好的转录本数据。
转录组测序可以供研究者在转录本结构研究(基因边界鉴定、可变剪切研究等),转录本变异研究(如基因融合、编码区SNP研究),非编码区域功能研究(Non-coding RNA研究、miRNA前体研究等),基因表达水平研究以及全新转录本发现等方面进行深入研究。
1.研究转录组的方法有哪些?答:目前研究转录组的方法主要三种,基于杂交技术的cDNA芯片和寡聚核苷酸芯片,基于sanger测序法的SAGE (serial analysis of gene expression)、LongSAGE和MPSS(massively parallel signature sequencing),基于第二代测序技术的转录组测序,又称为RNA-Seq。
2.转录组测序比其他研究方法有哪些优势?答:转录组测序具有以下优势:(1)可以直接测定每个转录本片段序列、单核苷酸分辨率的准确度,同时不存在传统微阵列杂交的荧光模拟信号带来的交叉反应和背景噪音问题;(2)灵敏度高,可以检测细胞中少至几个拷贝的稀有转录本;(3)可以对任意物种进行全基因组分析,无需预先设计特异性探针,因此无需了解物种基因信息,能够直接对任何物种进行转录组分析,同时能够检测未知基因,发现新的转录本,并准确地识别可变剪切位点及cSNP,UTR区域。
RNA-Seq基因表达水平衡量方法近年来RNA-Seq被广泛应用,报告结果中衡量基因表达水平的方法也变得多种多样,如RPK、RPKM、FPKM、TPM等。
然而大家对于这些单位还存在着很多困惑和错误的理解。
今天小宇就与大家分享一下他们之间的区别与换算。
首先要明确的是实验之间基因表达水平单位不具有可比性。
RNA-Seq的结果是一个相对度量,不是绝对的。
在解读FPKM、RPKM、TPM之前,先明确几个概念:本文中read指的是单末端或双末端reads。
计数的概念在两种reads中是一样的,每个read都是指被测序的一个片段。
本文中feature指的是一个表达特征,就是说一个基因组区域包含一段可以正常出现在RNA-Seq实验中的序列,如基因、亚型、外显子等。
用随机变数X i表示观察到的感兴趣的特征i的数目。
然而由于可变剪切的存在,我们不能直接观察到X i,所以我们用,这是用eXpress,RSEM,Sailfish,Cufflinks或其他算法估计出来的一个值。
下面介绍几个样品未均一化的基因表达水平单位:CountCount数目通常指比对到某个特殊的特征的reads数目,用随机变量X i表示。
这些数目主要依赖于两个方面:(1)测得的片段数目(与相对丰度有关);(2)特征的长度,或者更适合的有效长度。
有效长度指一个特征可能的起始位点数目可以生成特定长度的片段,计算公式如下:从比对read得到的片段长度分布的平均值。
如果丰度估算方法用包含序列偏差建模(如express或Cufflinks),偏差通常并入到有效长度,从而特征的长短受偏差的影响。
由于counts不是由ferture的长度来衡量,一个样本里没有调整feature长度,那么这个范畴里的所有单位都没有可比性。
这意味着不能说一组features的counts相加之和就代表这组feature的表达。
(如,不能说亚型的数目总和就是得到的基因数目)Effective counts使用eXpress方法计算得到的是有效数目。
RNA-Seq技术的优缺点RNA-Seq技术的优缺点一直以来,研究人员都很有兴趣了解细胞在各种不同状态下的基因表达差异,并开发出多种方法,来不断提高灵敏度和增加通量。
基因表达芯片是近年来较多采用的方法,但它如今却碰上了一个强劲对手——RNA-Seq.RNA-Seq可进行全基因组水平的基因表达差异研究,具有定量更准确、可重复性更高、检测范围更广、分析更可靠等特点。
除了分析基因表达水平,RNA-Seq还能发现新的转录本、SNP、剪接变体,并提供等位基因特异的基因表达。
RNA-Seq的动态范围更广,且假阳性可能更小,这意味着RNA-Seq的数据重复性应当比芯片要高。
RNA-Seq能够检测样品中的所有RNA,这对于鉴定细胞的新颖转录本来说是个优点,但同时缺点在于,它检测了总的RNA,而细胞中很大一部分RNA都来自核糖体和线粒体。
这限制了其他RNA的读取数量以及这些RNA表达水平的准确性。
因此,polyA RNA选择和核糖体RNA去除等方法被开发出来,以便解决这个问题。
然而,这些分离方法有可能会引入潜在误差,影响实验结果。
因此,第三代测序公司Helicos BioSciences的研究人员对操作方法修改后可能发生哪些差异进行了研究,文章发表在最近一期的PloS ONE上。
他们认为,对于研究人员来说,必须了解以下几点:技术差异如何影响结果的质量和可解释性,操作方法如何引入潜在误差,RNA的来源如何影响转录检测,以及所有这些差异如何影响得到的结论。
Helicos BioSciences公司的研究人员使用了多个人RNA样品,来评估RNA片段化、RNA分离、cDNA合成,单个以及多个标签计算。
尽管采用polyA RNA选择的操作方法得到了最多的非核糖体读取,并能够最精确测定编码转录本,但研究人员发现这种方法只能检测人细胞中的一部分非核糖体RNA.polyA RNA排除了数千个注释的转录本以及更多未注释的转录本,使得转录组查看不完全。
RNA-seq结果怎么才能看懂?答案全在这些图⾥---(1)测序质量篇 2016-10-17测序结束,满怀期待的拿到了沉甸甸的数据。
好长的实验报告,好多⽂件夹,好多excel表格,好多图。
于是,很多医⽣默默选择关闭笔记本电脑,暂且放下这天书⼀般的数据……其实,RNA-seq数据解读并不难,最核⼼的内容就是要解读各种数据展⽰图形。
实验报告⾥的图,都是把测序获得的⼤数据,经过⽣物信息学⽅法分析,最终以最直观的图形展⽰出来。
所以,只要理解了RNA-seq结果中的所有图⽰,基本上就对RNA-seq的结果有了充分的掌握。
今天⼩编先为⼤家介绍RNA-seq结果第⼀部分常见的图⽰,这些图反映了测序的质量。
有了质量的保证,后续的数据分析才有价值。
接下来,便是”看图说话“时间!Pat1⽤于展⽰RNA-seq测序原始数据质量的图⽰当⼆代测序的原始数据拿到⼿之后,第⼀步要做的就是看⼀看原始reads的质量。
如果⼀开始质量就不⾏,后⾯什么分析都是在浪费时间啊!这⼀步常⽤的⼯具是Fastqc。
通常,会以单碱基质量分布图,ATCG含量分布图去展⽰原始数据的质量。
01单碱基质量分布图(体现了测序错误率⾼不⾼)为什么⼀个样本会有2张这个图?答: 测序的时候,所有上机⽚段都是约300bp的⽂库。
测序采⽤2*150bp的测序模式,即从最左端测150bp,再从最右端测150bp。
所以每个⽚段都会得到2个序列,这两个序列就是我们常说的read。
所以,碱基质量分布图会有2个,分别与read1和read2对应。
X和Y轴都是什么意思?答:X轴是⼀条read中,每⼀个碱基的位置(因为read1⼀共就150bp长度,所以X轴⼀般都是1到150左右);Y轴是每⼀个碱基的碱基质量值,这个质量计算公式为-10*log10(p),p为测错的概率。
所以如果⼀条read 1第⼀个碱基出错概率为0.01,其quality就是20。
最上⾯的竖线,黄框,蓝线是什么意思?答:对于⼀个样本,在RNA测序完成后会获得⼏千万条read1. 对于read1的第⼀个碱基,也就会有⼏千万个碱基质量值。
RNA-Seq项目常见问题与解答这两年随着测序成本的下降和转录组研究的日渐火热,RNA-seq俨然已经成为了分子生物学课题组推进项目的首选方向。
在我们接触的转录组项目中,有些老师对项目分析结果存在或多或少不清楚或有疑惑的地方。
那么春天来了,花儿开了,今天福利也到了,我们特意将转录组项目中常见的一些问题进行了汇总,各位老师可以按需自取哈。
1.如何判定生物学重复一致性的高低?生物学重复统计方法及公式答:(1)皮尔逊相关系数r可以作为生物学重复相关性的评估指标,理想的生物学重复试验r2≧0.92。
考虑到个体差异、取材环境、时间以及人员操作熟练程度等因素对测序数据的影响,一般r2≧0.8为可接受范围。
(2)Pearson(皮尔逊)相关系数:皮尔逊相关也称为积差相关(或积矩相关)是英国统计学家皮尔逊于20世纪提出的一种计算直线相关的方法。
2.DEG基因用Transcripts还是Unigenes?答:DEG基因用的是Unigene。
3.transcript-id代表什么意思?为什么有的基因有多个transcript-id?答:基因转录本id;因为可变剪切的缘故,一个基因可能有多个转录本。
4.在miRNA鉴定中,可能成为miRNA的reads是怎样计算的?哪些条件会影响到mrd值?micro RNA在不同组织有异构体的存在,是如何处理的?答:与 Rfam, miRbase, RepBase和 Exon\Intro 序列库进行比对,获得 sRNA 注释信息,以此作为预测新的 miRNA 的基础。
miRNA的鉴定是利用miRDeep2软件进行已知及新(保守及非保守)的miRNA鉴定。
miDeep2会在reads比对到基因组上的位置两端分别延伸75、15bp进行结构预测,此软件认为极可能与可能是miRNA的根据是通过mrd值来区分的,mrd>-10为可能,mrd>0为极可能;影响mrd值的有reads在基因组上的分布和碱基结合的自由能等;5.对于有生物学重复的项目,怎样计算差异基因?答:两两比对使用的是R的EBseq包, 是基于负二项分布检验的方式对reads数进行差异显著性检验,重复间的比对使用的是R的DEseq包,是基于分层贝叶斯模型的原理对组合内样品进行分析。
6.外显子,内含子及基因间区各自的比例如何评估建库情况?答:理论上,来自成熟mRNA的reads应该比对到外显子区。
但是,由于基因组注释水平、可变剪切导致的内含子序列保存,以及很多RNA(比如lncRNA)就来自基因间区和内含子,因此有比对到内含子和基因间区的reads。
受物种等的影响外显子所占比例不同,一般情况下外显子区域所占比例超过70%即比较理想。
7.影响组装Contig结果的因素?答:a.物种的特异性;b.测序质量;c.测序的数据量;d.SNP的杂合率;e.组装参数的选择。
(1)、在不考虑物种特异性和测序质量的情况下,测序的数据量越大,SNP的杂合率越高,得到的短片段Contig的数目就越多。
根据Trinity组装Contig的策略,将Reads构建K-mer 库,选取频数最高的K-mer,按照k-1的overlap进行延伸,用于延伸的K-mer全部从库中清掉,因此测到的reads越多,SNP的杂合率越高,延伸完后的短片段就越多。
(2)、对于组装参数的选择,是用于过滤低频数K-mer,选择的参数不同,过滤掉的K-mer 数目不同,如果过滤掉的越多,那么留下的短片段的Contig就会少。
所以即使用同一个软件(Trinity)进行组装,如果不知道组装参数的时候,对于组装结果没有很大的可比性。
(3)、组装结果的好坏最主要的还是看Unigene的组装数据,包括组装出的数目和N50。
一般来说,组装出的Unigene的数目在一个合理范围内(比如10W以内),N50越大,组装的结果越好。
8.转录组测序Contig 与transcript的区别?答:转录组测序的原始数据包含了很多的reads,通过序列的拼接,具有重叠区的reads会被组装成更大的片段,称之为contig。
将reads比对回contig,通过paired-end reads 能确定来自同一转录本的不同contig 以及这些contig之间的距离,将这些contig连在一起,最后得到两端不能再延长的序列,称之为Unigene。
Transcript即转录本。
9.不同ID号代表的基因相同吗?不同ID号功能注释相同的,为什么?答:不同的ID可以认为是代表不同的基因。
不同的基因注释的功能相同,原因有:一是有些长的基因没有组装出完整的序列,而是分成了多个小片段,这种情况去进行注释的话会注释到同一个功能蛋白;二是基因的核酸序列不同,但是蛋白序列具有一定的相似性或者具有相似的功能区域,这些基因在比对注释用的蛋白序列时,会注释到相同的功能。
10.多个Unigene注释一样,序列长度不同,相似性较低,为什么?答:1)首先某一基因可能比较长,但无参考基因组装出的片段即Unigene很难组装得到全长,得到的是这个基因上的大小不等的片段,在进行比对的时候就会比对到同一个基因上,因此他们的注释信息一致;2)从序列来看Unigene基因的序列相似度不高,但是因为比对的是蛋白,所以可能他们的蛋白相似度会比较高,因此会注释到同一基因上。
11.transcript_id、gene_id、length、effective_length、expected_count、TPM、FPKM、IsoPct这几个字段的意思?答:一个Unigene可能对应多个转录本。
Transcript id:为组装转录本编号;gene_id:Unigene 编号;length:Unigene的长度;effective_length:各个转录本的平均长度;TPM:Transcripts per million,公式为:Unigene 的reads数×10^6/总reads数;FPKM即RPKM (双端Reads数目/(比对到转录本上的片段总数*转录本长度));IsoPct:某一个转录本的表达量占相应的组装原件表达量的百分比。
12.同一ID下有多条序列,想得到此序列的核苷酸信息应选哪一条?答:同一个ID号下面好几条序列,这个应该是组装过程中装出来的转录本序列,来自同一个Component(具体见Trinity组装的第二步),其ID前缀相同,后面跟着seq+数字的编号。
Trinity软件认为这些转录本来源于同一个基因,因此,选取其中最长的那个转录本的序列作为该基因的序列。
13.生物云转录组APP上的差异筛选阈值采用的是哪种方法?p值与FDR值的区别是?答:生物云转录组APP在差异表达分析过程中采用了公认有效的Benjamini-Hochberg方法对原有假设检验得到的显著性p值(p-value)进行校正,并最终采用校正后的p值,即FDR (False Discovery Rate)作为差异表达基因筛选的关键指标,以降低对大量基因的表达值进行独立的统计假设检验带来的假阳性。
p值与FDR之间没有单纯的换算公式,是在linux 操作系统下,运用R语言编写的程序完成的fisher精确检验,在筛选过程中,默认将FDR<0.01且差异倍数(Fold Change)≥2作为筛选标准。
14.生物云转录组在分析差异基因时,对于表达量为0的,如何计算差异倍数?答:差异基因分析软件EBseq在分析表达量为0的基因的差异倍数时,会采用贝叶斯估计给出一个估计值,然后使用这个估计值计算差异倍数。
由于计算估计值时综合考虑多项因素,因此不同基因间FPKM和FC不具有一致性。
15.如何定义的已知micRNA、保守的micRNA以及新预测的micRNA?答:已知micRNA指的是序列在miRBase数据库中百分百的比对到该物种的序列上,如果在该物种上没有比对上但比对上了数据库中的其他物种上我们称之为保守的micRNA;新预测的micRNA:通过miRDeep2软件进行预测,有一定的read能够比对到基因组上,并且比对位置的序列可以形成发卡结构,那么就会作为新预测的miRNA。
16.分析时发现不同的名,但是他们的前体序列和成熟序列都一样,表达量在各个样品中也相同,为什么?答:这个是由于在染色体上的位置不同导致的,可以参考miRBase数据库中的hsa-mir-1233-1 和 hsa-mir-1233-2 这两个 ID,它们对应的前体序列,3' 和 5' 成熟序列均相同,但在基因组上的位置不同,软件将它们区别成两个不同的小RNA,又因为它们的序列一致,所以比对上的reads是一样的,表达量因此一样。
具体见下:17.测序得到的lncRNA,如何知道哪些是已知的?哪些是未知的?答:目前长链分析结果中如果分析的物种是比较常见的物种比如人、大鼠、小鼠,这些物种具有比较完整的已知lncRNA数据库,这种情况:(1)通过确切的位置关系(位置相交则认为相同)对预测出来的那些lncRNA鉴定其是否为已知;(2)根据fa序列进行比对,对预测出的lncRNA序列与数据库中已知的lncRNA序列比对,达到一定比对值的会认为该预测长链是已知的长链。
注:NONCODE DB中包含的物种主要是动物方面的,包括:人、小鼠、大鼠、奶牛、鸡、果蝇、斑马鱼、线虫、酵母、拟南芥、黑猩猩、大猩猩、恒河猴、复鼠、鸭嘴兽、猩猩18.转录组测序之后,用QPCR进行验证,但验证的基因表达趋势与测序结果中不一致,这是什么原因?如何解决呢?答:首先,我们需要确定检验的样品是否是同一批次,验证样品的上下调关系是否与测序结果中的一致(这个需要根据测序公司具体的分析结果,比如某个基因的FC值对应的样品写的是T01 vs T02 ,那么T01就是对照组、T02是实验组),若样品不为同一批次或其上下调关系颠倒了,则势必会导致验证基因表达趋势不一致的情况。
其次,我们需要查看验证基因的表达量、样品和实验用的引物是否被污染,若验证基因表达量过低,则有可能导致差异不显著,若样品或实验用的引物被污染则后续结果可能也不会准确,所以我们尽量不要挑选表达量太低的基因,同时,需要保证样品和实验引物没有被污染。
当以上所有情况都不存在,且结果依然不一致,这时我们需要检查QPCR结果是否正确。
如果仅一个基因验证结果不一致,则不足以说明测序或者验证有问题,但当我们选择了15个基因甚至更多时,结果依然不一致时,那么我们可能需要分析测序数据的结果是否正确,同时检查结果预期是否正确。
19.从NCBI上下载的数据都是SAR格式的,如何转化成FASTQ格式?答使用软件sra2fastq进行转换。