BP 非无菌制剂的微生物限度
- 格式:doc
- 大小:171.00 KB
- 文档页数:4

【2020版中国药典】通则-非无菌微生物限度检查1105非无菌产品微生物限度检查:微生物计数法微生物计数法系用于能在有氧条件下生长的嗜温细菌和真菌的计数。
当本法用于检查非无菌制剂及其原、辅料等是否符合规定的微生物限度标准时,应按下述规定进行检验,包括样品的取样量和结果的判断等。
除另有规定外,本法不适用于活菌制剂的检查。
研究:将旧版的“相应”更换为“规定”,更便于按照1107进行判定执行。
微生物计数试验环境应符合微生物限度检查的要求。
检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。
洁净空气区域、工作台面及环境应定期进行监测。
研究:将旧版的“单向流空气区域”更换为“洁净空气区域”。
计数方法……供试品检查时,应根据供试品理化特性和微生物限度标准等因素选择计数方法,检测的样品量应能保证所获得的试验结果能够判断供试品是否符合规定。
所选方法的适用性须经确认。
……提醒:后文增加了关于“贵重药品、微量包装药品”的检验量的更全面的表述,因留意结合此处的请求。
计数培养基适用性检查和供试品计数方法适用性试验……菌液制备……取黑曲霉的新鲜培养物加人适量含0.05%(ml/ml)聚山梨酯80的pH7.0无菌氯化钠-蛋白胨缓冲液或……研究:此处改动同无菌检查法,将“3~5ml”的具体量调整为“适量”,便于根据孢子的量灵活掌握菌液制备方法。
培养基适用性检查微生物计数用的商品化的预制培养基、由脱水培养基或按处方配制的培养基均应进行培养基适用性检查。
研究:类似于无菌检查法,此处将“成品培养基”修改为“商品化的预制培养基”,表述更准确,下文还有,不再赘述。
……计数方法适用性试验1.供试液制备根据供试品的理化特性与生物学特性,采取适宜的方法制备供试液。
供试液制备若需加温时,应均匀加热,且温度不应超过45°C。
供试液从制备至加人检验用培养基,不得超过1小时。
……研究:此处应注意同时进行数个品种计数方法适用性试验时的时效问题。

非无菌制剂微生物限度检查操作标准程序起草人日期年月日执行日期2015年12月01日审核人日期年月日颁发部门质保部批准人日期年月日分发部门质保部()生产部()采供部()销售部()设备部()物资部()销售部()行政部()财务部()变更记载:修订号执行日期00 2012年06月01日01 2014年05月01日02 2015年12月01日1. 目的:建立非无菌药品微生物限度检查检验标准操作规程,规范检验操作,确保检验结果准确。
2. 适用范围:适用于本公司所有采用非无菌药品微生物限度检查法测定的供试品。
3. 责任者:QC检验员、QC经理。
4. 正文:4.1 非无菌产品微生物限度检查:微生物计数法4.1.1 简述微生物计数法系用于能在有氧条件下生长的嗜温细菌和真菌的计数。
当本法用于检查非无菌制剂及其原、辅料等是否符合规定的微生物限度标准时,应按下述规定进行检验,包括样品的取样量和结果的判断等。
除另有规定外,本法不适用于活菌制剂的检查。
本检查法可采用替代的微生物检查法,包括自动检测方法,但必须证明替代方法等效于药典规定的检查方法。
微生物计数试验应在受控洁净环境下的局部洁净度不低于B 级的单向流空气区域内进行。
检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。
单向流空气区域、工作台面及环境应定期进行监测。
如供试品有抗菌活性,应尽可能去除或中和。
供试品检查时, 若使用了中和剂或灭活剂,应确认其有效性及对微生物无毒性。
供试液制备时如果使用了表面活性剂,应确认其对微生物无毒性以及与所使用中和剂或灭活剂的相容性。
4.1.2 计数方法计数方法包括平皿法、薄膜过滤法和最可能数法(Most-Probable-NumberMethod,简称MPN 法)。
MPN 法用于微生物计数时精确度较差,但对于某些微生物污染量很小的供试品,MPN 法可能是更适合的方法。
供试品检查时, 应根据供试品理化特性和微生物限度标准等因素选择计数方法,所选的方法必须具备检测充足样品量的能力,以保证所获得的试验结果能够判断供试品是否符合规定。



對非無菌劑型的註冊藥劑製品微生物品質的要求背景根據《藥劑業及毒藥條例》(第138 章)及其附屬規例(第138A 章),藥劑製品應符合安全、效能及素質方面的標準,並獲藥劑業及毒藥管理局註冊,方可於本港銷售。
2. 在非無菌製劑中,某些微生物的存在可使產品減低甚至失去治療作用,亦對病人的健康構成潛在風險。
因此,非無菌劑型的藥劑製品的微生物限量應受監控。
3. 非無菌劑型的藥劑製品的微生物檢查已被編入不同的藥典,如美國藥典、英國藥典、歐洲藥典及日本藥典等。
在2009年5月,上述藥典中的微生物品質要求均已訂立相同的標準。
註冊要求4. 為確保註冊藥劑製品在微生物品質方面的安全及素質,藥劑業及毒藥(藥劑製品及物質註冊:臨牀試驗及藥物測試證明書)委員會(委員會)決定從2019年10月1日起,所有非無菌劑型的註冊藥劑製品(包括正在申請註冊的新藥和已註冊藥劑製品),在整個保質期內必須符合上述第3段的主要藥典訂明的微生物限度標準(節錄於表一)。
表一: 非無菌劑型的微生物限度標準給藥途徑需氧菌總數(cfu/g 或cfu/ml)霉菌和酵母菌總數(cfu/g 或cfu/ml)控制菌口服給藥固體製劑103102大腸埃希菌陰性(1 g or 1 mL) 口服給藥液體製劑102101大腸埃希菌陰性(1 g or 1 mL) 直腸給藥製劑103102-口腔黏膜給藥製劑齒齦給藥製劑皮膚給藥製劑鼻用製劑耳用製劑102101金黃色葡萄球菌陰性(1 g or 1 mL)銅綠假單胞菌陰性(1 g or 1 mL)陰道給藥製劑102101銅綠假單胞菌陰性(1 g or 1 mL)金黃色葡萄球菌陰性(1 g or 1 mL)白色念珠菌陰性(1 g or 1 mL)透皮貼劑(對一個含有黏著層與背襯的貼劑限量)102101金黃色葡萄球菌陰性(1 patch)銅綠假單胞菌陰性(1 patch)呼吸道吸入給藥製劑(適用於霧化用液體製劑的特別要求)102101金黃色葡萄球菌陰性(1 g or 1 mL)銅綠假單胞菌陰性(1 g or 1 mL)耐膽鹽革蘭陰性菌陰性(1 g or 1 mL)備註:非無菌產品的微生物檢驗方法應參考美國藥典、英國藥典、歐洲藥典或日本藥典。


非无菌药品微生物限度检查指导原则微生物限度检查是药品质量控制中的重要环节,尤其对于非无菌药品而言更为重要。
本文将对非无菌药品微生物限度检查的指导原则进行详细介绍,旨在提高药品的质量安全性。
一、检验项目及标准非无菌药品微生物限度检查的主要检验项目包括总生菌数、大肠菌群、霉菌和酵母菌等。
每一项检验项目都有相应的标准来衡量合格与否。
以下是常用的标准:1. 总生菌数:根据药典要求,大部分非无菌药品每克不得超过1000 CFU(菌落形成单位)。
2. 大肠菌群:大肠菌群是肠道中的常见菌种,其存在可能暗示有肠源性污染。
检验结果一般要求不得检出大肠菌群。
3. 霉菌和酵母菌:霉菌和酵母菌是环境中广泛存在的微生物,在非无菌药品中的存在可能引发变质,甚至导致严重的药品质量问题。
一般情况下,每克药品中不得检出霉菌和酵母菌。
二、样品的选择和采集在进行微生物限度检查前,需选择合适的样品,并采取正确的样品采集方式。
以下是一些常用的样品选择和采集方法:1. 样品选择:根据药品的特性,选择代表性的样品进行检测。
选取多个批次的不同规格的样品进行检验更有利于全面评估该药品的微生物污染水平。
2. 样品采集:在采集样品前,先进行适当的表面消毒,以避免外源性污染。
采集时应遵循严格的无菌操作,确保样品的真实性和可靠性。
常用的样品采集方法包括划线法、切割法、稀释法等。
三、检验方法和操作流程微生物限度检查需要使用一系列严格的操作流程和检验方法,以保证结果的准确性和可比性。
以下是一般的操作流程:1. 样品预处理:根据药品的特性,选择适当的预处理方法,如溶解、稀释、震荡等,以提高微生物的检出率。
2. 培养基选择:根据不同的菌种需求,选择适宜的培养基进行菌落的培养。
常用的培养基有营养琼脂平板、大肠埃希菌选择平板、马铃薯葡萄糖琼脂平板等。
3. 培养条件:根据菌种的生长特性和检验项目的要求,设定适当的温度、时间和培养条件,以促进菌落的生长。
4. 菌落计数:通过目视或自动计数法,对培养基上的菌落进行计数。


2.6.12非无菌产品的微生物限度检查:微生物计数试验1.简介:该法所描述的是在需氧条件下生长的嗜温性细菌和真菌类微生物的定量检测。
设计该试验的最初目的是为了确定一种物质或制剂是否符合微生物质量的既定标准。
如果是这种目的,则需要遵从下面的说明,包括所需样品数,从而根据下面所提到的方法来进行结果解释。
该法不适用于包含微生物作为活性成分的产品。
如果与药典的等效性作了相关说明,可以使用自动收集法作为替代法。
2.一般步骤设计方法时所用条件要避免外在微生物对待测产品的污染。
必须釆取措施,使其不影响任何待测微生物。
如果待测产品有抗菌活性,必须将这种活性去除或中和掉。
如果因此使用了灭活剂,必须证明它们对微生物的高效性和无毒性。
如果在样品制备中加了表面活性剂,必须证明它们对微生物的无毒性和与灭活剂的兼容性。
3.计数方法按规定用微孔滤膜法或平板计数法。
MPN法对微生物测定来说是最不精确的方法,但是,如果某些产品所含极少的微生物量,该法是最合适的。
方法选择基于下列因素:如产品的特性和所要求的微生物量。
所选方法必须能够测定充足的样品从而来判断其是否符合标准。
所选方法的适用性必须建立。
4.促生长实验、计数法的适用性和阴性对照4-1 总则在不加产品的条件下该试验检测微生物的能力必须已确定。
如果测试条件改变或产品改变,必须证实方法的适适用性,因为这些改变可能影响试验的最终结果。
4-2 试验菌株的准备使用测试菌株标准稳定的悬浮液或按下面方法制备。
使用批种子培养技术(seed-lot systems)使得用于接种的微生物从最初的主批种子传代不多于5次。
细菌和真菌试验株的生长分别见表2.6.12-2。
使用氯化钠蛋白胨(pH 7.0)缓冲液或pH 7.2的磷酸缓冲液制作试验用悬浮液。
对于黑曲霉菌需要加入0.05%聚山梨醇酯80到缓冲液中。
在两小时内使用该菌悬液或2-8℃下可保存24小时。
作为一种可替代的制备方法,稀释含有黑曲霉菌或枯草芽孢杆菌的新鲜悬浮液,该悬浮液中加有营养细胞,从而可以制得一种稳定的孢子悬浮液,对于接种试验来说需要将该孢子悬浮液调整到一个合适的体积下。
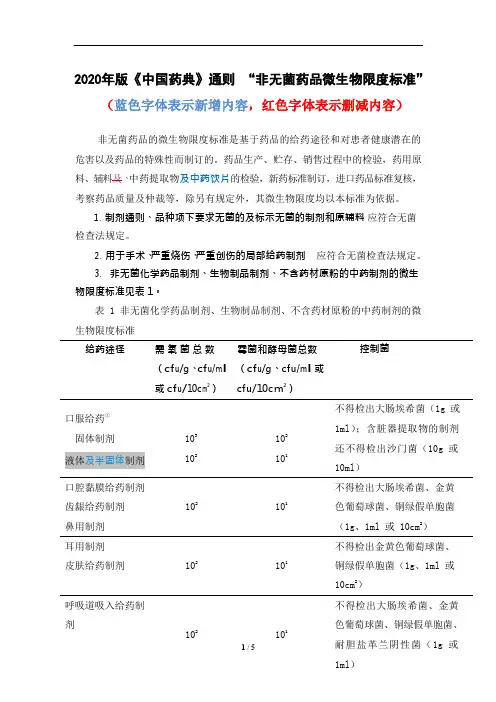
2020年版《中国药典》通则“非无菌药品微生物限度标准”(蓝色字体表示新增内容,红色字体表示删减内容)非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,药用原料、辅料及、中药提取物及中药饮片的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的及标示无菌的制剂和原辅料应符合无菌检查法规定。
2.用于手术、严重烧伤、严重创伤的局部给药制剂应符合无菌检查法规定。
3.非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准见表 1。
表 1 非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准给药途径需氧菌总数(cf u/g、cf u/m l或cf u/10c m2)霉菌和酵母菌总数(cf u/g、c fu/m l或cfu/10cm2)控制菌口服给药①固体制剂液体及半固体制剂103102102101不得检出大肠埃希菌(1g 或1ml);含脏器提取物的制剂还不得检出沙门菌(10g 或10ml)口腔黏膜给药制剂齿龈给药制剂鼻用制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌(1g、1ml 或 10cm2)耳用制剂皮肤给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml 或10cm2)呼吸道吸入给药制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌、或 10ml ) 阴道、尿道给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌(1g 、1ml 或 10cm 2);中药制剂还不得检出梭菌(1g 、 1ml 或 10cm 2)直肠给药 固体制剂103102 不得检出金黄色葡萄球菌、铜绿假单胞菌(1g 或 1ml )其他局部给药制剂102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g 、1ml 或 10cm 2)注 ①化学药品制剂和生物制品制剂若含有未经提取的动植物来源的成份及矿物质还不得检出沙门菌(10g 或 10ml )。

2020年版《中国药典》通则—“非无菌药品微生物限度标准”2020年版《中国药典》通则“非无菌药品微生物限度标准”(蓝色字体表示新增内容,红色字体表示删减内容)非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,药用原料、辅料及、中药提取物及中药饮片的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的及标示无菌的制剂和原辅料应符合无菌检查法规定。
2.用于手术、严重烧伤、严重创伤的局部给药制剂应符合无菌检查法规定。
3.非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准见表 1。
表1 非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准给药途径需氧菌总数(cf u/g、cf u/m l或cf u/10c m2)霉菌和酵母菌总数(cf u/g、c fu/m l或cfu/10cm2)控制菌口服给药①固体制剂液体及半固体制剂103102102101不得检出大肠埃希菌(1g 或1ml);含脏器提取物的制剂还不得检出沙门菌(10g 或10ml)口腔黏膜给药制剂齿龈给药制剂鼻用制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌(1g、1ml 或 10cm2)耳用制剂皮肤给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml 或10cm2)呼吸道吸入给药制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌、或 10ml )阴道、尿道给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌(1g 、1ml 或 10cm 2);中药制剂还不得检出梭菌(1g 、 1ml 或 10cm 2)直肠给药固体制剂103102 不得检出金黄色葡萄球菌、铜绿假单胞菌(1g 或 1ml )其他局部给药制剂102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g 、1ml 或 10cm 2)注①化学药品制剂和生物制品制剂若含有未经提取的动植物来源的成份及矿物质还不得检出沙门菌(10g 或 10ml )。
药品微生物限度标准非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,化学药品原料药、中药提取物及辅料的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
非无菌制剂的总需氧菌数、总霉菌及酵母菌数测定照附录×××检查;非无菌制剂的控制菌检查照附录×××检查。
本限度标准解释如下:101CFU:最大可接受限值=20;102CFU:最大可接受限值=200;103CFU:最大可接受限值=2000。
以此类推。
1.制剂通则、品种项下要求无菌的制剂及标示无菌的制剂应符合无菌检查法规定。
2.口服给药制剂2.1 不含药材原粉的口服给药制剂需氧菌总数每1g不得过103cfu。
每1ml不得过102cfu。
霉菌及酵母菌总数每1g不得过102cfu。
每1ml不得过101cfu。
大肠埃希菌每1g或1ml不得检出。
沙门菌含脏器提取物的口服给药制剂每10g或10ml不得检出。
2.2含药材原粉的口服制剂2.2.1不含豆豉、神曲等发酵原粉的口服给药制剂需氧菌总数每1g不得过10000cfu。
每1ml不得过100cfu。
霉菌及酵母菌总数每1g或1ml不得过100cfu。
大肠埃希菌每1g或1ml不得检出。
沙门菌每10g或10ml不得检出。
耐胆盐革兰阴性菌每1g应小于102个。
每1ml应小于101个。
2.2.2 含豆豉、神曲等发酵原粉的口服制剂需氧菌总数每1g不得过100000cfu。
每1ml不得过1000cfu。
霉菌和酵母菌总数每1g不得过500cfu。
每1ml不得过100cfu。
大肠埃希菌每1g或1ml不得检出。
沙门菌每10g或10ml不得检出。
耐胆盐革兰阴性菌每1g应小于102个。
每1ml应小于101个。
3.局部给药制剂3.1 用于手术、烧伤或严重创伤的局部给药制剂应符合无菌检查法规定。
01/2011:50104 5.1.4. MICROBIOLOGICAL QUALITY OF NON-STERILE PHARMACEUTICAL PREPARATIONS AND SUBSTANCES FOR PHARMACEUTICAL USE (1) 非无菌制剂和药用物质的微生物质量The presence of certain micro-organisms in non-sterile preparations may have the potential to reduce or even inactivate the therapeutic activity of the product and has a potential to adversely affect the health of the patient.在非无菌制剂中如果存在某些微生物,可能会降低或抑制药物的治疗活性,可能会对病人健康有潜在不良影响。
Manufacturers therefore have to ensure a low bioburden of finished dosage forms by implementing current guidelines on Good Manufacturing Practice during the manufacture, storage and distribution of pharmaceutical preparations.因此,生产商应在药品生产、存贮和销售过程中遵守现行GMP指南,来保证制剂的微生物在一个较低的水平。
Microbial examination of non-sterile products is performed according to the methods given in general chapters 2.6.12 and 2.6.13. Acceptance criteria for non-sterile pharmaceutical products based upon the total aerobic microbial count (TAMC) and the total combined yeasts/moulds count (TYMC) are given in Tables 5.1.4-1 and 5.1.4-2. Acceptance criteria are based on individual results or on the average of replicate counts when replicate counts are performed (e.g. direct plating methods).非无菌产品微生物检查应依据通则2.6.12和2.6.13中指定的方法进行。
附录XVI D的微生物质量非无菌制剂和药品物质应用1(欧洲药典正文5.1.4)无菌制剂中存在某种微生物可能会减弱甚至阻碍此产品发挥治疗作用和危害病人健康。
因此制造商必须在药物制剂的生产、贮存和配伍过程中严格遵守现行的《优良制造规范》(GMP),以确保成品剂型低生物负荷。
无菌产品制剂的细菌检测依照总章2.6.12 和 2.6.13中给出的方法进行。
无菌产品制剂的验收标准见表 5.1.4.-1 和表 5.1.4.-2列出的总需氧微生物计数(TAMC)和总酵母菌/霉菌计数(TYMC)。
验收标准依据单次结果,若进行了重复计数,则取其平均值(例如直接培养法)。
微生物检测标准一旦制定,应按如下方式进行标注:— 101 CFU: 最多不超过20;— 102 CFU: 最多不超过200;— 103 CFU:最多不超过2000, 以此类推。
表 5.1.4.-1 列出一些已有验收标准的特定微生物,该目录不一定完全,对于确定的药剂还要根据最初材料的性质和生产过程确定是否应检测其他种类微生物。
如果已证明任何规定的微生物检测结果均未达到微生物验收标准中规定的有效的量,则可以采用有限稀释法,稀释到接近验收标准规定的量。
除了微表5.1.4.-1所列的生物,其他微生物的意义在以下方面进行评估:- 产品用途:不同的使用途径所带来的危险(眼睛,鼻子,呼吸道)-产品性质:它有助于成长,能够体现抗菌效力;-使用方法-接受者:风险可能会因新生儿,婴幼儿,衰弱而有所不同-免疫抑制物:皮质甾体类-表现为疾病、伤、器官损伤表5.1.4.-1无菌剂型的微生物检测验收标准适用范围TAMC(CFU/g 或CFU/ml)TYMC(CFU/g 或CFU/ml)细菌种类内服非水药物103102不含大肠杆菌(1克或1毫升)内服水类药物102101不含大肠杆菌(1克或1毫升)直肠类药物103102_粘膜、牙龈、皮肤、鼻、耳用药物102101不含葡萄球菌(1克或1毫升)不含绿脓杆菌(1克或1毫升)阴道用药物102101不含绿脓杆菌(1克或1毫升)不含葡萄球菌(1克或1毫升)不含白色念珠菌(1克或1毫升)贴剂(限一片的粘结102101不含葡萄球菌(1克或1毫升)如有需要,对相关因素基于风险的评估工作由经过专门培训的微生物学人员和微生物资料库解释。
附录×××非无菌药品微生物限度标准非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,药用原料、辅料及中药提取物的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的制剂及标示无菌的制剂和原辅料应符合无菌检查法规定。
2.用于手术、烧伤或严重创伤的局部给药制剂应符合无菌检查法规定。
3. 非无菌不含药材原粉的中药制剂微生物限度标准(见表1)表1 非无菌不含药材原粉的中药制剂微生物限度标准给药途径需氧菌总数(CFU/g、 CFU/m或CFU/10cm2)霉菌及和酵母菌总数(CFU/g、 CFU/m或CFU/10cm2)控制菌口服固体给药制剂103102不得检出大肠埃希菌(1g);含脏器提取物的制剂还不得检出沙门菌(10g)口服液体给药制剂102101不得检出大肠埃希菌(1ml);含脏器提取物的制剂还不得检出沙门菌(10ml)口腔黏膜给药制剂齿龈给药制剂鼻用制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)耳用制剂皮肤给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)呼吸道吸入给药制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌、耐胆盐革兰阴性菌(1g或1ml)阴道、尿道给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌、梭菌(1g、1ml或10cm2)直肠给药固体制剂液体制剂103102102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g或1ml)其他局部给药制剂102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)4. 非无菌含药材原粉的中药制剂微生物限度标准(见表2)表2非无菌含药材原粉的中药制剂微生物限度标准给药途径需氧菌总数(CFU/g、 CFU/mL或CFU/10cm2)霉菌和酵母菌总数(CFU/g、 CFU/mL或CFU/10cm2)控制菌固体口服给药制剂不含豆豉、神曲等发酵原粉含豆豉、神曲等发酵原粉104(丸剂3×104)1051025×102不得检出大肠埃希菌(1g);不得检出沙门菌(10g);耐胆盐革兰阴性菌应小于102(1g)。
湖南湘大兽药有限公司工作标准文件题目非无菌产品微生物限度检查标准操作规程编号ZLJG F-048-00 修定质检部审核批准修定日期2016.12 审核日期批准日期颁发部门GMP办公室颁发数量 2 份生效日期分发部门质检部、档案室文件页数共13页一、目的:为规定非无菌产品微生物限度检查标准的检测方法和操作要求,特制定本操作规程。
二、引用标准:中华人民共和国兽药典(2015年版一部附录P195)。
三、适用范围:适用于本公司检品采用非无菌产品微生物限度检查标准的质量检测。
四、责任者:理化分析员对本标准的实施负责。
五、正文:1 简介非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,药用原料、辅料及中药提取物的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
制剂通则、品种项下要求无菌的制剂及标示无菌的制剂和原辅料应符合无菌检查法规定。
用于手术、烧伤或严重创伤的局部给药制剂应符合无菌检查法规定。
非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准(见表1)表1 非无菌兽药制剂的微生物限度标准给药途径需氧菌总数(cfu/g、cfu/ml 或cfu/10cm2)霉菌和酵母菌总数(cfu/g、cfu/ml或cfu/10cm2)控制菌口服给药①固体制剂液体制剂103102102101不得检出大肠埃希菌(1g或1ml);含脏器提取物的制剂还不得检出沙门菌(10g或10ml)耳用制剂皮肤给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)尿道给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌(1g、1ml或10cm2)直肠给药固体制剂液体制剂103102102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g或1ml)其他局部给药制剂102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml或10cm2)注:①兽药制剂若含有未经提取的动植物来源的成分及矿物质,还不得检出沙门菌(10g或10ml)。
非无菌产品微生物限度检查:微生物计数法1.导言微生物计数法系用于能在有氧条件下生长的嗜温细菌和霉菌的定量计数当本法用于检查非无菌制剂及其原、辅料等是否符合相应的微生物限度标准时,应按照下述规定进行检验,包括样品的取样量,结果的判断。
本法不适用于活菌制剂的检查,可使用包括自动化法在内的替代方法,需确认其与药典方法的等同性。
2. 通用规程微生物计数环境应能防止外来微生物对供试品的污染。
防止污染的措施不能影响供试品中微生物的检出。
备注:检验全过程必须严格遵守无菌操作,防止再污染。
单向气流区域、工作台面及环境应定期进行监测。
如果供试品有抗菌活性,应尽可能去除或中和。
若使用了中和剂或灭活剂,需确认其有效性及对微生物无毒性。
供试品制备过程中若使用了表面活性剂,需确认其对微生物无毒性以及与所使用的中和剂或灭活剂的相容性。
3.计数方法计数方法包括薄膜过滤法、平皿法和最可能数法(Most-Probable-Number 简称 MPN)。
MPN 法用于微生物计数时精确度最差,但用于微生物污染量小的供试品,MPN 法可能是最合适的方法。
(MPN法不适用于霉菌的检测,仅在供试品总需氧菌数没有适应计数方法的情况下使用。
)供试品检查时,应根据供试品理化特性和微生物限度标准等因素选择计数方法。
所选择的计数方法应能够通过检测足量的供试品,判断与质量标准的符合性。
且该计数方法的适用性须经确认。
4. 促生长实验,计数法适用性试验4.1一般要求在有供试品存在的情况下,所选用的计数方法需能够检测微生物。
若检测程序或产品发生变化可能影响检测结果时,计数方法需重新进行适用性确认。
4.2试验菌液的制备试验菌液:使用试验菌株的标准化稳定悬浮液或按下述规定制备。
按表 1 规定程序分别培养各试验菌液。
菌株:采用适宜的菌种保藏技术(种子批系统),试验用菌株传代次数自主种子批(第 0 代)算起不得超过 5代。
菌液制备:取表 1各试验菌株的新鲜培养物,用 PH7.0的氯化钠-蛋白胨缓冲液或 PH7.2的磷酸缓冲液制备混悬液。
Appendix XVI D. Microbiological Quality of Non- sterile Pharmaceutical Preparations and Substances
非无菌制剂的微生物限度
(Ph. Eur. general text 5.1.4)
The presence of certain micro-organisms in non-sterile preparations may have the potential to reduce or even inactivate the therapeutic activity of the product and has a potential to adversely affect the health of the patient. Manufacturers therefore have to ensure a low bioburden of finished dosage forms by implementing current guidelines on Good Manufacturing Practice during the manufacture, storage and distribution of pharmaceutical preparations.
非无菌制剂中存在一定量的微生物可能导致药物的治疗活性降低或消失,并对患者健康有着潜在的危害。
因此生产商应在药品生产、储存、运输过程中实施GMP管理,使制剂的微生物负荷保持在低水平。
Microbial examination of non-sterile products is performed according to the methods given in general chapters 2.6.12 and 2.6.13. Acceptance criteria for non-sterile pharmaceutical products based upon the total aerobic microbial count (TAMC) and the total combined yeasts/moulds count (TYMC) are given in Tables 5.1.4.-1 and 5.1.4.-2. Acceptance criteria are based on individual results or on the average of replicate counts when replicate counts are performed (e.g. direct plating methods).
非无菌制剂的微生物检测按照通则2.6.12和2.6.13的方法执行。
非无菌制剂的微生物限度基于表5.1.4-1和5.1.4-2的需氧微生物总数(TAMC)及酵母菌/霉菌总数(TYMC)。
接受标准适用于单个检出结果或者做重复样品时结果的平均数(例如,直接平板法)。
When an acceptance criterion for microbiological quality is prescribed it is interpreted as follows:
给出的微生物质量接受标准解读如下:
— 101 CFU: maximum acceptable count = 20;
— 101 CFU: 最大可接受数= 20;
— 102 CFU: maximum acceptable count = 200;
— 102 CFU: 最大可接受数= 200;
— 103 CFU: maximum acceptable count = 2000;
— 103 CFU: 最大可接受数= 2000;
Table 5.1.4.-1 includes a list of specified micro-organisms for which acceptance criteria are set. The list is not necessarily exhaustive and for a given preparation it may be necessary to test for other micro-organisms depending on the nature of the starting materials and the manufacturing process.
表5.1.4-1给出了各种剂型的具体微生物接受标准。
此表格并不完全,根据起始物料和生产工艺的性质可能需要其他的微生物检查。
If it has been shown that none of the prescribed tests will allow valid enumeration of micro-organisms at the level prescribed, a validated method with a limit of detection as close as possible to the indicated acceptance criterion is used.
若所有给出的测试方法都不能有效的测出描述微生物限度,需要使用其检测限尽可能接近接受标准的经过验证的方法。
In addition to the micro-organisms listed in Table 5.1.4.-1, the significance of other micro-organisms recovered is evaluated in terms of:
除了表5.1.4-1给出的微生物外,评估其他发现微生物的重要性要根据以下项目的评估:— use of the product: hazard varies according to the route of administration (eye, nose, respiratory tract);
— nature of the product: its ability to support growth, the presence of adequate antimicrobial preservation;
— method of application;
— intended recipient: risk may differ for neonates, infants, the debilitated;
— use of immunosuppressive agents, corticosteroids;
— presence of disease, wounds, organ damage.
Where warranted, a risk-based assessment of the relevant factors is conducted by personnel with specialised training in microbiology and the interpretation of microbiological data. For raw materials, the assessment takes account of processing to which the product is subjected, the current technology of testing and the availability of materials of the desired quality.
--产品使用:根据使用途径不同危害不同(眼、鼻、呼吸道);
--产品的性质:促生长的能力,是否添加足量的防腐剂;
--使用方法:
--使用人群:新生儿、婴儿、体弱者;
--免疫抑制剂、类固醇的使用;
--有疾病、伤口、器官衰竭者;
如果必要,应有受过专业微生物培训的人员对相关因素进行风险评估,并对微生物数据进行解读,对于原料药,评估要考虑其加工成制剂的工艺,目前的检验技术及能达到质量要求物料的可获得性。