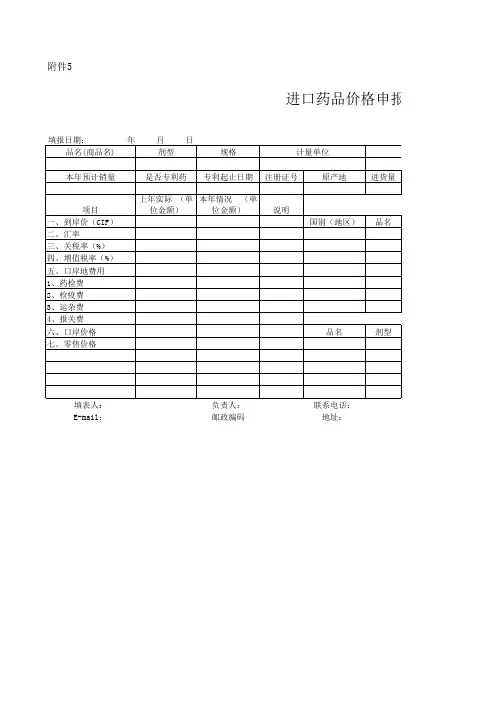
进口药材补充申请表
- 格式:docx
- 大小:14.63 KB
- 文档页数:2

进口药材管理方法(修订稿〕全文进口药材管理方法(修订稿〕全文为进一步加强进口药材监视管理,国家食品药品监视管理总局组织对《进口药材管理方法〔试行〕》进展了修订,起草了《进口药材管理方法〔修订稿〕》,现向社会公开征求意见,进口药材管理方法(修订稿〕全文。
进口药材管理方法(修订稿)第一章总那么第一条为加强进口药材监视管理,保证进口药材质量,根据《中华人民共和国药品管理法》《中华人民共和国海关法》《中华人民共和国行政答应法》《中华人民共和国药品管理法施行条例》及相关法律法规的规定,制定本方法。
第二条满足药用需求的进口药材申请、审批、登记备案、报关、口岸检验及监视管理,适用本方法。
第三条增设允许药材进口的边境口岸应当由国家食品药品监视管理总局会同海关总署提出,报国务院批准。
第四条药材必须从允许药品进口的口岸或者允许药材进口的边境口岸进口。
允许药材进口的边境口岸,只能进口该口岸周边国家或者地区所产药材。
第五条药材进口应当符合国家有关法规的要求。
第二章药材进口申请第六条进口药材申请包括首次进口药材申请和非首次进口药材申请。
首次进口药材,是指从境外某产地首次进口的药材。
第七条进口药材申请人,应当是中国境内获得《药品消费答应证》或者《药品经营答应证》的中药消费企业或者药品经营企业。
药品经营企业经营范围应含有中药材或中药饮片。
第八条受理药材进口申请的食品药品监视管理部门应当在受理场所公示申报资料的工程和有关申请书示范文本。
申请人应当按照规定如实提交标准完好的资料,反映真实情况,并对其申报资料全部内容的真实性负责。
第九条首次进口药材申请,申请人应当按照规定填写《进口药材申请表》,并向允许药品进口的口岸或者允许药材进口的边境口岸所在地省、自治区、直辖市食品药品监视管理部门(以下简称口岸或者边境口岸省级食品药品监视管理部门)报送以下申报资料:(一)《进口药材申请表》;(二)申请人《药品经营答应证》或《药品消费答应证》、《营业执照》复印件;(三)供货方合法登记证明文件(如《营业执照》等)复印件;(四)购货合同及其公证文书复印件;(五)药材标准及其来;(六)申请进口药材的基原腊叶标本及由中国境内具有动、植物基原鉴定资质的机构提供的药材基原研究证明资料。


国家市场监督管理总局令第9号——进口药材管理办法文章属性•【制定机关】国家市场监督管理总局•【公布日期】2019.05.16•【文号】国家市场监督管理总局令第9号•【施行日期】2020.01.01•【效力等级】部门规章•【时效性】现行有效•【主题分类】药政管理正文国家市场监督管理总局令第9号《进口药材管理办法》已于2019年4月28日经国家市场监督管理总局2019年第8次局务会议审议通过,现予公布,自2020年1月1日起施行。
局长张茅2019年5月16日进口药材管理办法(2019年5月16日国家市场监督管理总局令第9号公布)第一章总则第一条为加强进口药材监督管理,保证进口药材质量,根据《中华人民共和国药品管理法》《中华人民共和国药品管理法实施条例》等法律、行政法规,制定本办法。
第二条进口药材申请、审批、备案、口岸检验以及监督管理,适用本办法。
第三条药材应当从国务院批准的允许药品进口的口岸或者允许药材进口的边境口岸进口。
第四条国家药品监督管理局主管全国进口药材监督管理工作。
国家药品监督管理局委托省、自治区、直辖市药品监督管理部门(以下简称省级药品监督管理部门)实施首次进口药材审批,并对委托实施首次进口药材审批的行为进行监督指导。
省级药品监督管理部门依法对进口药材进行监督管理,并在委托范围内以国家药品监督管理局的名义实施首次进口药材审批。
允许药品进口的口岸或者允许药材进口的边境口岸所在地负责药品监督管理的部门(以下简称口岸药品监督管理部门)负责进口药材的备案,组织口岸检验并进行监督管理。
第五条本办法所称药材进口单位是指办理首次进口药材审批的申请人或者办理进口药材备案的单位。
药材进口单位,应当是中国境内的中成药上市许可持有人、中药生产企业,以及具有中药材或者中药饮片经营范围的药品经营企业。
第六条首次进口药材,应当按照本办法规定取得进口药材批件后,向口岸药品监督管理部门办理备案。
首次进口药材,是指非同一国家(地区)、非同一申请人、非同一药材基原的进口药材。

《进口药材管理办法(试行)》(局令第22号)2005年11月24日发布国家食品药品监督管理局令第22号《进口药材管理办法(试行)》于2005年10月21日经国家食品药品监督管理局局务会审议通过,现予公布,自2006年2月1日起施行。
局长:邵明立二○○五年十一月二十四日进口药材管理办法(试行)第一章总则第一条为加强进口药材监督管理,保证进口药材质量,根据《中华人民共和国药品管理法》、《中华人民共和国药品管理法实施条例》(以下简称《药品管理法》、《实施条例》)及相关法律法规的规定,制定本办法。
第二条进口药材申请与审批、登记备案、口岸检验及监督管理,适用本办法。
进口药材申请与审批,是指国家食品药品监督管理局根据申请人的申请,依照法定程序和要求,对境外生产拟在中国境内销售使用的药材进行技术审评和行政审查,并作出是否同意其进口的决定。
进口药材申请人,应当是中国境内取得《药品生产许可证》或者《药品经营许可证》的药品生产企业或者药品经营企业。
第三条国家食品药品监督管理局负责药材进口的审批,并对登记备案、口岸检验等工作进行监督管理。
省、自治区、直辖市(食品)药品监督管理局依法对进口药材进行监督管理。
允许药品进口的口岸或者允许药材进口的边境口岸所在地(食品)药品监督管理局(以下简称口岸或者边境口岸(食品)药品监督管理局)负责进口药材的登记备案,组织口岸检验并进行监督管理。
中国药品生物制品检定所负责首次进口药材的样品检验、质量标准复核等工作。
国家食品药品监督管理局确定的药品检验机构负责进口药材的口岸检验工作。
第四条药材必须从国务院批准的允许药品进口的口岸或者允许药材进口的边境口岸进口。
允许药材进口的边境口岸,只能进口该口岸周边国家或者地区所产药材。
第二章申请与审批第一节一般规定第五条国家食品药品监督管理局应当在药材进口申请受理场所公示申报资料的项目和有关申请书示范文本。
第六条申请人申请药材进口时应当按照规定如实提交规范完整的材料,反映真实情况,并对其申报资料实质内容的真实性负责。




国家药品监督管理局药品补充申请表原始编号:XXXXXXXXX填表说明1.申请分类:选择适用本申请的申请事项,可以根据需要选择多项。
未列入的申请事项,应在“其他”项中简要填写申请事项。
2.本品种属于:按本品种的属性选择相应的选项。
3.药品分类:按本品种的属性选择相应的选项,属于其他分类的应填写其分类名称。
4.药品通用名称:填写原药品批准证明文件载明的相应内容。
5.英文名/拉丁名:填写原药品批准证明文件及其所附药品标准载明的相应内容。
6.其他名称:系指曾经作为药品通用名称使用,但已被正式颁布的国家药品标准或者《中国药品通用名称》及其增补本收载的药品通用名称取代者。
7.商品名称:填写原药品批准证明文件及其所附药品标准、药品说明书、标签内载明的相应内容。
如果申请使用新的药品商品名,应填写新的商品名称。
进口药品如有英文商品名则填写在英文项内。
8.非制剂:填写原药品批准证明文件载明的相应内容。
9.剂型:填写原药品批准证明文件载明的相应内容。
10.规格:填写原药品批准证明文件及其所附药品标准、药品说明书、标签内载明的相应内容。
如果此次申请增加药品规格,应填写新的药品规格。
11.包装规格:填写原药品批准证明文件及其所附药品标准、药品说明书内载明的相应内容。
如果申请变更包装规格,应填写新的规格。
12.药品有效期:填写原药品批准证明文件及其所附药品标准、药品说明书内载明的相应内容,以月为单位。
如果申请变更药品有效期,应填写新的药品有效期。
13.新药证书编号、批准日期:填写原新药证书载明的相应内容。
14.药品批准文号/进口药品注册证书号:填写原药品批准证明文件载明的相应内容,一份申请表只能填写一个药品批准文号或者进口药品注册证书号。
批准日期、有效期截止日期:填写原药品批准证明文件载明的相应内容15.处方内辅料(含处方量):辅料没有变化的,按原批准药品注册时申报的辅料处方填写。
如果申请变更药品处方中已有药用要求的辅料,应填写新的辅料处方。

【原创】2019版最新《进口药材管理办法》知识测试培训试题及答案2019.06姓名:成绩:一、单选题(每题4分,共20分)1、《进口药材管理办法》的施行日期:。
(B)A、2019年1月11日B、2020年1月1日C、2018年12月1日D、2019年12月1日2、进口药材批件编号格式为:。
(A)A、(省、自治区、直辖市简称)药材进字+4位年号+4位顺序号B、(省、自治区、直辖市简称)药字+4位年号+4位顺序号C、(省、自治区、直辖市简称)药材进字+4位年号+2位月号D、(省、自治区、直辖市简称)药材进字+4位年号+2位月号+4位顺序号3、进口单位提供虚假证明、文件资料或者采取其他欺骗手段办理备案的,给予警告,并处1万元以上3万元以下罚款:。
(C)A、1万元以上2万元以下B、2万元以上3万元以下C、1万元以上3万元以下D、1万元以上5万元以下4、口岸药品监督管理部门收到进口药材不予抽样通知书后,对有证据证明可能危害人体健康且已办结海关验放手续的全部药材采取查封、扣押的行政强制措施,并在日内作出处理决定。
(B)A、10B、7C、25D、305、进口单位对检验结果有异议的,可以依照药品管理法的规定申请复验。
药品检验机构应当在复验申请受理后日内作出复验结论,并报告口岸药品监督管理部门,通知进口单位。
(A)A、20B、50C、55D、60二、多选题(每题5分,共20分)A、加强进口药材监督管理B、保证进口药材质量C、保障进口药材在企业的储存量D、监督进口药材在企业的使用过程2、进口药材的包装必须适合进口药材的质量要求,方便储存、运输以及进口检验。
在每件包装上,必须注明唛头号、出口商名称、到货口岸、重量以及加工包装日期等:。
(ABCD)A、药材中文名称B、批件编号(非首次进口药材除外)C、产地D、进口单位名称3、《进口药材管理办法》适用于进口药材:。
(ABCD)A、申请B、审批C、备案D、口岸检验以及监督管理4、变更进口药材批件批准事项的,申请人应当通过信息系统填写进口药材补充申请表,向原发出批件的省级药品监督管理部门提出补充申请。
Application form for Imported Drug Supplementary Registretion 进口药品补充申请表格State Food and Drug Administration Drug Supplementary Registration Application–for Foreign ApplicantsEntry Number:Acceptance No:StatementWe guarantee:①T his application complies with laws and regulations such as Drug Administration Law of The People’s Republic of China, Implementing Regulation of the Drug Administration Law of The People’s Republic of China, and Drug Registration Regulation;②The content of application form, thesubmitted information and the samples are true and legal, without infringing any other’s rights. Any methods and data is results of research and the drug tests conducted on the drugs;②T he accompanied electronic version is in perfect accordance with the printed version.We will take all the legal consequences of any false statements.Other Statement Items in Particular That:Application Items1The Application for: Import registration2 Drug category:3 whether OTC or not :4 Status of the initial registration:5 Registration category:〇Supplemental applications to be approved by SFDA: □Application for Drug Approval Number of a new drug by the New Drug Certificate holder of the drug.□Use of the name of the Trade Name of drugs. □Additional indications or functions of TCM or natural drug, or theindication approved in China for chemical drugor biological products. □Change in the usage or dosage of the drugs, or the group of patient to use the drug, but without change in route of administration.□Change of strength of drugs □Change of the supplementive in the formula of the drugs, where there is a medial requirement for it. □A change in the drug manufacture technology and process affecting drug quality. □Amendment of drug registration standards.□Substitute or removal of the drug material listed in formula of National Drug Standards as toxic or endangered.□Change of the immediate packing material or container of the import drugs, domestic injection, ophthalmologic, spray, powder Aerosol, Inhaler and Spray. Use of new immediate packing material or container.□Application for combined packing of drug.□The transfer of new drug technology.□Addition or amendment of items in insert sheet of TCM or natural drug, such as pharmacology and toxicology, clinical trial and Pharmacokinetic.□A change in items within the import drug registration certificate, such as name of the drug, drug enterprise name, registered location, packing specification.□Change of the location where the import drug is manufactured.□Change of the location where the import drug is packed overseas.□Repacking of import drugs in China.□Change of the location where the raw material for import preparation is manufactured.〇Supplemental applications to be approved by PDA and be filed for record at SFDA, or directly be filed for record at SFDA: □Change of the name of a domestic drug manufacturer.□Internal change of the manufacture workshop of a domestic drug manufacturer.□Change of immediate packing material or container (except for the item 10 as above)□Change of valid period of domestic drugs□Change of manufacture location of import drugs□Change of appearance of the drug withoutchange of drug standards.□Amendment of insert sheet of the drugs according to national drug standards or required by SFDA.□Supplementing and perfecting of the drug safety part of the insert sheet.□Modification of design of packing and label of the drugs according to the regulation.□Change of the agent for import drug registration.□Others〇Supplemental applications to be filed for record at PDA: □Amendment of insert sheet of the domestic drugs according to national drug standards or required by SFDA.□Supplementing and perfecting of the domestic drug safety part of the insert sheet.□Modification of design of packing and label of the domestic drugs according to the regulation.□Change of the packing specification of domestic drugs.□Change of manufacture location of domestic drugs□Change of appearance of the domestic drug without change of drug standards.□othersDrugs Information6 Generic Name:7 Generic Name Source:8 English / Latin name:9 Chinese Phonetic Alphabet:10 Chemical Name:10 Trade Names:11 Product category:12 strength:13 Other accepted or submitted preparation and Strength at the same time:14 Packaging: immediate packing material:Packaging size:15 Date of Expiration: 36 months16 Prescriptions (Including Prescription Volume):API/materials in TCM(TraditionalChinese medicine):Accessories:17 Materials /Accessories SourceSerial NO.Materials/AccessoriesNameApproval No/RegistrationNo/Accepted NoManufacturerImplementationStandardsVariationor notVariationapproved statusand approvedinstitution1218 Chinese Medicinal Materials Standard:Serial Numbe r Materials/Accessories NameWhetherlegal ornotStandardreferenceImplementationStandardsVariation ornotVariationapprovedstatus andapprovedinstitution1219 Indications or Attending Functions: Indications category:Supplementary contents:20 Supplementary contents:21 Rational to propose this supplementary:22 Initial approved registration contents and relevant information:Initial acceptance No:Clinical Trial Approval No:Initial IDL No:Drug specification No.:Relevant Conditions23 Patents:□Have Chinese patent: □chemical compound patent; □formulation patent; □process patent; □other patent;Patent No.: __________________Patentee: __________________Patent licensing/Publication date __________________□Have foreign patentPatent No.: __________________Patentee: __________________Patent licensing/Publication date __________________Patent Ownership Statement: __________________We state that: the application does not cause patent infringement.24 Variety Protection of Chinese Medicine: Variety Protection of Chinese Medicine expirydate:25 Monitoring Time With Same Variety of New Drugs:Expiry date: __________________26 Times for Applications:〇First time application 〇multi-times application the times application □Withdrew before, date__________________ reason: ______□not approved, date__________________ reason: ______The Applicant and Commissioned Research Institutions27. Institutions 1 (Foreign Pharmaceutical Companies):Chinese Name:English Name:Legal Representative: Position: Registered Address:Country or Region:Head of An Application for Registration: Positions:Tel: Fax:E-mail:Legal Representative (Signatures): (Department Official Seal)Month Day, Year28. Institutions 2 (Imported Drugs Production Plant):Chinese Name:English Name:Legal Representative: Position: Registered Address:Country or Region:Head of An Application for Registration: Positions:Tel: Fax:E-mail:Legal Representative (Signatures): (Department Official Seal)Month Day, Year29 Institutions 3 (Imported Drugs Foreign Packaging Factory):Chinese Name:English Name:Legal Representative: Position: Registered Address:Country or Region:Head of An Application for Registration: Positions:Tel: Fax:E-mail:Legal Representative (Signatures): (Department Official Seal)Month Day, Year30 Institutions 4 (Imported Drugs sub- Packaging Factory):Chinese Name:English Name:Organization code:Pharmaceutical production license No.: Legal Representative: Position: Registered Address: zip code: Postal address: zip code: Head of An Application for Registration: Positions:Tel: Fax:E-mail:Mobile phone:Legal Representative (Signatures): (Department Official Seal)Month Day, Year31 Institutions 5 (Registration Agency of Imported Drugs):This agency is responsible for paymentChinese Name:English Name:Organization Code:Legal Representative: Position: Registered Address:Zip Code:Contact Address:Zip Code:Head of an Application for Registration:Position:Contact: Position: Phone : Fax :E-mail: phone:Legal Representative (Signatures):(Department Official Seal)Month Day, Year32 Commissioned Research Institutions:No program forResearch Name oftheinstitutionResponsiblepersonTel.uthoritiesAfter reviewed, the table is in line with the formwith the request.Authorities: Reviewer (Signatures) Date:。
一、引言随着我国医药市场的不断发展和国际化进程的加快,越来越多的国外优质药品进入我国市场。
为保障人民群众用药安全,促进我国医药产业的健康发展,根据《中华人民共和国药品管理法》及其实施条例的相关规定,现就某国外某药品的进口申请报告如下:二、药品基本信息1. 药品名称:XX(国际非专利名称)2. 药品规格:XX(mg/粒、mg/片等)3. 药品剂型:XX(片剂、胶囊剂、注射剂等)4. 药品成分:XX(主要活性成分及含量)5. 药品适应症:XX(疾病名称及用途)6. 药品生产厂家:XX(国外制药企业名称)7. 注册证号:XX(国外药品注册证号)8. 生产企业地址:XX(国外制药企业地址)三、药品质量及安全性评价1. 质量评价:根据我国药品管理法及其实施条例的相关规定,国外药品进口前需进行质量评价。
经查阅相关资料,该药品在国外已通过严格的质量控制体系,符合国际质量标准。
2. 安全性评价:根据国外药品注册资料及临床研究数据,该药品在国内外上市后,经大量临床应用,未发现严重不良反应。
我国药品审评中心对该药品的安全性进行了综合评估,认为该药品在我国上市使用是安全的。
四、药品市场及临床需求1. 市场需求:随着我国人民生活水平的提高和医疗保健意识的增强,对药品的需求日益增长。
该药品针对我国市场存在的某种疾病具有显著疗效,市场需求较大。
2. 临床需求:经临床实践证明,该药品对某种疾病具有较好的治疗效果,能够满足我国临床用药需求。
五、药品价格及供应情况1. 价格:根据我国药品管理法及其实施条例的相关规定,国外药品进口价格需合理。
经与国外制药企业协商,该药品进口价格符合我国市场及消费者承受能力。
2. 供应情况:国外制药企业承诺,将按照我国市场需求,保证药品的稳定供应。
六、进口申请事项1. 申请人:XX(我国药品生产企业名称)2. 申请事项:申请进口某国外某药品,用于治疗某种疾病。
3. 申请人承诺:申请人承诺,在进口该药品过程中,严格遵守我国药品管理法律法规,确保药品质量及安全性,切实保障人民群众用药安全。
1. 申请和审批1.1首次进口药材:1.2非首次进口药材:3. 口岸检验和监督管理4. 所需提交资料4.1 首次进口药材上报SFDA4.1.1 物料部申请人需报送下列资料一式两份,分别提交国家食品药品监督管理局和中国药品生物制品检定所。
(一)《进口药材申请表》。
(二)本公司《药品经营许可证》或《药品生产许可证》、《营业执照》(复印件)。
(三)供货方合法登记证明文件(如《营业执照》等)(复印件)。
(四)购货合同(复印件)。
(五)药材质量标准及其来源。
(六)药材基源研究证明资料(研究证明资料应由中国境内具有动、植物基源鉴定资质的机构提供)。
4.1.2 如进口药材的质量标准来源于省、自治区、直辖市药材标准,物料部申请人除报送上述资料外,还应根据具体情况,对该标准作相应的提高工作,并报送有关研究资料;如进口药材无法定标准,物料部申请人除报送上述资料外,还应报送下述资料:(一)药材生态环境、生长特征、形态描述、栽培或者培植(培育)技术、产地加工等。
(二)药材质量标准起草说明。
(三)药理毒理研究资料综述。
(四)主要药效学试验资料及文献资料。
(五)一般药理研究的试验资料及文献资料。
(六)急性毒性试验资料及文献资料。
(七)我国批准的中成药处方中含有该药材的证明资料。
4.2 非首次进口药材上报SFDA物料部申请人需报送下述资料一式一份:(一)《进口药材申请表》。
(二)本公司《药品经营许可证》或者《药品生产许可证》、《营业执照》(复印件)。
(三)供货方合法登记证明文件(如《营业执照》等)(复印件)。
(四)购货合同(复印件)。
(五)药材质量标准及其来源。
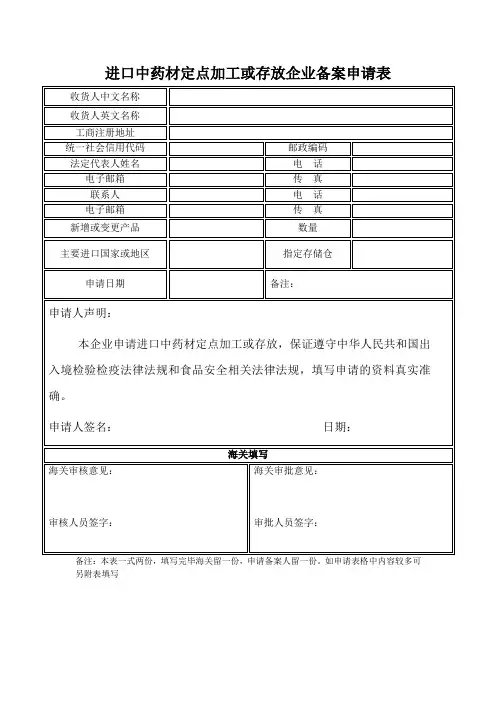
4.3 登记备案上报口岸药监局物料部申请人需报送下列资料一式两份:(一)《进口药材批件》复印件(和《进口药材补充申请批件》复印件)。
(二)本公司《药品经营许可证》或者《药品生产许可证》复印件。
(三)原产地证明复印件。
(四)购货合同复印件。
(五)装箱单、提运单和货运发票复印件。