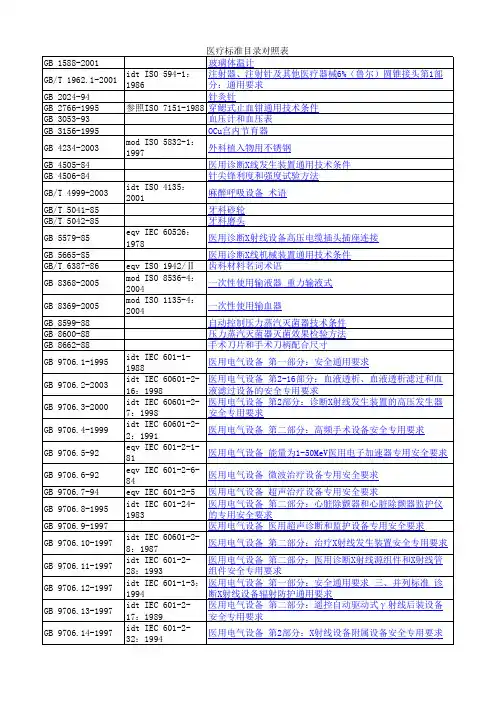
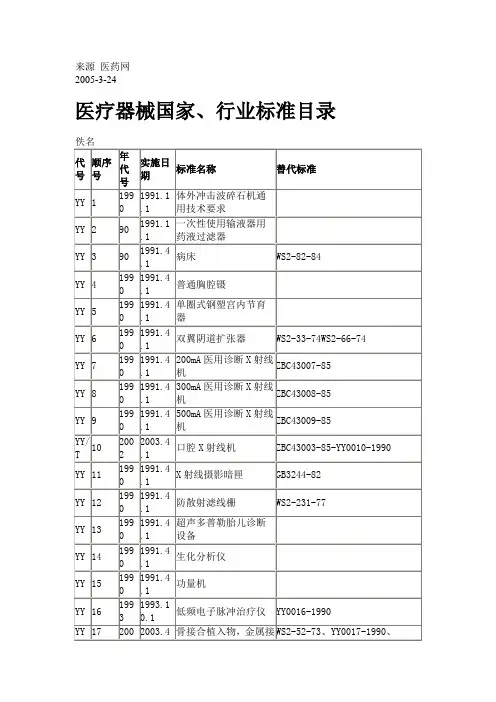
各国医疗器械标准目录(中英文对照)
- 格式:xls
- 大小:661.50 KB
- 文档页数:16

美国FDA 医疗器械体系法规QSR820中文版之巴公井开创作Part 820——质量体系法规——目录Subpart A- 总则820.1 范围820.3 定义 820.5 质量体系Subpart B –质量体系要求820.20 管理职责820.22 质量审核 820.25 人员Subpart C- 设计控制 820.30 设计控制Subpart D- 文件控制 820.40 文件控制Subpart E- 推销控制 820.50 推销控制Subpart F- 标识与可追溯性820.60 标识 820.65 可追溯性Subpart G - 生产和过程控制820.70 生产和过程控制820.72 检验、丈量和试验设备 820.75 过程确认Subpart H - 验收活动: 820.80 进货、过程和成品器械检验 820.86 检验状态Subpart I –分歧格品 820.90 分歧格品Subpart J - 纠正和预防措施 820.100 纠正和预防措施Subpart K –标识和包装控制820.120 设备标签 820.130 设备包装Subpart L –搬运/储存/分销和装置820.140 搬运820.150 贮存820.160 分销 820.170 装置Subpart L –记录820.180 记录的通用要求820.181 设备主要记录820.184 设备历史记录820.186 质量体系记录 820.198 投诉文件Subpart M –服务 820.200 服务Subpart N –统计技术820.250 统计技术Subpart A——总则Subpart A--General Provisions Sec.820.1 范围Sec. 820.1 Scope. (a)适用性Applicability。
(1)实质量体系法规说明了当前良好制造法规Current good manufacturing practice(CGMP)的要求。

美国FDA 医疗器械体系法规QSR820中文版Part 820——质量体系法规——目录Subpart A- 总则820.1 范围820.3 定义820.5 质量体系Subpart B –质量体系要求820.20 管理职责820.22 质量审核820.25 人员Subpart C- 设计控制820.30 设计控制Subpart D- 文件控制820.40 文件控制Subpart E- 采购控制820.50 采购控制Subpart F- 标识与可追溯性820.60 标识820.65 可追溯性Subpart G - 生产和过程控制820.70 生产和过程控制820.72 检验、测量和试验设备820.75 过程确认Subpart H - 验收活动:820.80 进货、过程和成品器械检验820.86 检验状态Subpart I –不合格品820.90 不合格品Subpart J - 纠正和预防措施820.100 纠正和预防措施Subpart K –标识和包装控制820.120 设备标签820.130 设备包装Subpart L –搬运/储存/分销和安装820.140 搬运820.150 贮存820.160 分销820.170 安装Subpart L –记录820.180 记录的通用要求820.181 设备主要记录820.184 设备历史记录820.186 质量体系记录820.198 投诉文件Subpart M –服务820.200 服务Subpart N –统计技术820.250 统计技术Subpart A——总则Subpart A--General ProvisionsSec.820.1 范围Sec. 820.1 Scope.(a)适用性Applicability。
(1)本质量体系法规阐明了当前良好制造法规Current good manufacturing practice (CGMP)的要求。
本标准适用于所有预期用于人类的成品器械的设计、制造、包装、标识、储存、安装和服务中所使用的管理方法、设施和控制。



美国FDA 医疗器械体系法规QSR820中文版Part 820——质量体系法规——目录Subpart A- 总则820.1 范围820.3 定义 820.5 质量体系Subpart B –质量体系要求820.20 管理职责820.22 质量审核 820.25 人员Subpart C- 设计控制 820.30 设计控制Subpart D- 文件控制 820.40 文件控制Subpart E- 采购控制 820.50 采购控制Subpart F- 标识与可追溯性820.60 标识 820.65 可追溯性Subpart G - 生产和过程控制820.70 生产和过程控制820.72 检验、测量和试验设备 820.75 过程确认Subpart H - 验收活动: 820.80 进货、过程和成品器械检验 820.86 检验状态Subpart I –不合格品 820.90 不合格品Subpart J - 纠正和预防措施 820.100 纠正和预防措施Subpart K –标识和包装控制820.120 设备标签 820.130 设备包装Subpart L –搬运/储存/分销和安装820.140 搬运820.150 贮存820.160 分销 820.170 安装Subpart L –记录820.180 记录的通用要求820.181 设备主要记录820.184 设备历史记录820.186 质量体系记录 820.198 投诉文件Subpart M –服务 820.200 服务Subpart N –统计技术820.250 统计技术Subpart A——总则Subpart A--General Provisions Sec.820.1 范围Sec. 820.1 Scope. (a)适用性Applicability。
(1)本质量体系法规阐明了当前良好制造法规Current good manufacturing practice(CGMP)的要求。

Information suppliedby the manufacturer ofmedical devices医疗器械厂商提供的信息I C S 01.110; 11.040.01; 11.120.01B R I T I S H S T A N D A R D B S E N1041:2008N O COPYING W I T H O U T BSI PERMISSION EXCEPT AS PERMITTED BY COPYRIGHT L A WB S E N 1041:2008National foreword国家前言This British Standard is the U K implementation of E N 1041:2008. Itsupersedes BS E N1041:1998, which will be withdrawn on 3 August 2011.此英国标准是英国执行的EN1041:2008,取代了英国标准EN 1041:1998,将于2011年8月3日被孤立。
The U K participation in its preparation was entrusted to TechnicalCommittee CH/210/3, General terminology and symbols.A list of organizations represented on this committee can be obtained on requestto its secretary.This publication does not purport to include all the necessary provisions ofa contract. Users are responsible for its correct application.Compliance with a British Standard cannot confer immunity fromlegal obligations.英国参与此制定是委托了技术委员会CH/210/3,常规术语和符号。
Information suppliedby the manufacturer ofmedical devices医疗器械厂商提供的信息I C S 01.110; 11.040.01; 11.120.01B R I T I S H S T A N D A R D B S E N1041:2008N O COPYING W I T H O U T BSI PERMISSION EXCEPT AS PERMITTED BY COPYRIGHT L A WB S E N 1041:2008National foreword国家前言This British Standard is the U K implementation of E N 1041:2008. Itsupersedes BS E N1041:1998, which will be withdrawn on 3 August 2011.此英国标准是英国执行的EN1041:2008,取代了英国标准EN 1041:1998,将于2011年8月3日被孤立。
The U K participation in its preparation was entrusted to TechnicalCommittee CH/210/3, General terminology and symbols.A list of organizations represented on this committee can be obtained on requestto its secretary.This publication does not purport to include all the necessary provisions ofa contract. Users are responsible for its correct application.Compliance with a British Standard cannot confer immunity fromlegal obligations.英国参与此制定是委托了技术委员会CH/210/3,常规术语和符号。

欧盟mdcg医疗器械法规中英文对照全文共3篇示例,供读者参考篇1The European Union Medical Device Regulation (MDR) provides a comprehensive regulatory framework to ensure the safety and effectiveness of medical devices marketed in the EU. The MDR replaces the existing Medical Device Directive (MDD) and introduces several new requirements to enhance the oversight of medical devices throughout their lifecycle.In this article, we will provide a side-by-side comparison of key provisions in the MDR in both English and Chinese to help stakeholders understand the changes and implications of the new regulation.Title/标题欧盟医疗器械法规/ European Medical Device RegulationScope/范围The MDR applies to medical devices and in vitro diagnostic medical devices intended for use by humans for the purpose of diagnosis, prevention, monitoring, treatment or alleviation ofdisease./ 欧盟医疗器械法规适用于用于诊断、预防、监测、治疗或减轻疾病目的的医疗器械和体外诊断医疗器械。

医疗器械中英对照节号中译文原英文总则(General Provisions)866.1 范围 Scope866.3 有关上市前批准生效日期的要求 Effective dates of requirement for premarket approval866.9 联邦食品、药品和化妆品法(Federal Food, Drug, and Cosmetic Act,FDCA)第510(k)节的豁免限制 Limitations of exemptions from section 510(k) of the Federal Food, Drug, and Cosmetic Act (the act).诊断器械(Diagnostic Devices)866.1620 抗菌敏感性测试盘 Antimicrobial susceptibility test disc866.1640 抗菌敏感性测试粉 Antimicrobial susceptibility test powder866.1645 全自动短期孵化周期抗菌敏感性系统 Fully automated short-term incubation cycle antimicrobial susceptibility system866.1700 抗菌敏感性测试用培养基 Culture medium for antimicrobial susceptibility tests微生物学器械(Microbiology Devices)866.2050 葡萄糖球菌类抗生素 Staphylococcal typing bacteriophage866.2120 厌氧室 Anaerobic chamber866.2160 凝固酶血浆 Coagulase plasma866.2170 自动菌落计数器 Automated colony counter866.2180 手动菌落计数器 Manual colony counter866.2300 多用途培养基 Multipurpose culture medium866.2320 鉴别培养基 Differential culture medium866.2330 增菌(富集)培养基 Enriched culture medium866.2350 微生物鉴定培养基 Microbiological assay culture medium866.2360 选择性培养基 Selective culture medium866.2390 转运培养基 Transport culture medium866.2410 致病奈瑟球菌培养基 Culture medium for pathogenic Neisseria spp 866.2420 淋病氧化酶筛选检测 Oxidase screening test for gonorrhea866.2440 自动介质分发和堆积器械 Automated medium dispensing and stacking device866.2450 培养基补充装置 Supplement for culture media866.2480 培养基质量控制试剂盒 Quality control kit for culture media866.2500 微量滴定稀释和分配装置 Microtiter diluting and dispensing device 866.2540 微生物培养器 M生物染色(Biological Stains)864.1850 染料和化学溶液染色剂 Dye and chemical solution stains864.1860 免疫组织化学试剂和试剂盒 Immunohistochemistry reagents and kits细胞和组织培养产品(Cell and Tissue Culture Products)864.2220 人造细胞和组织培养基及成分 Synthetic cell and tissue culture media and components864.2240 细胞和组织培养供应品及设备 Cell and tissue culture supplies and equipment864.2260 染色体培养试剂盒 Chromosome culture kit864.2280 培养的动物和人体的细胞 Cultured animal and human cells864.2360 支原体检测介质和成分 Mycoplasma detection media and components 864.2800 动物和人的血清 Animal and human sera864.2875 平衡盐溶液或配方 Balanced salt solutions or formulations病理学检测设备和附件(Pathology Instrumentation and Accessories)864.3010 组织处理装置 Tissue processing equipment864.3250 标本运送和储存容器 Specimen transport and storage container864.3260 用于药物滥用检测的OTC检测样品收集系统 OTC test sample collection systems for drugs of abuse testing864.3300 细胞离心机 Cytocentrifuge864.3400 显微切片封接器械 Device for sealing microsections864.3600 显微镜及其附件 Microscopes and accessories864.3800 自动载玻片着色器 Automated slide stainer864.3875 自动组织处理器 Automated tissue processor标本制备试剂(Specimen Preparation Reagents)864.4010 通用试剂 General purpose reagent864.4020 被分析物特效试剂 Analyte specific reagents。
医疗设备中英文对照表AABSS 自动(磁带)空白部分扫描ABL(automatic bright limiting)自动亮度限制ABL ON OFF 自动黑电平开/关ABL SW ON 自动黑电平开关接通ABO 自动电子束最佳化ABO ADJ 自动电子束最佳化调整ABO VIDEO 自动电子束最佳化视频ABO VIDEO ADJ 自动电子束最佳化视频调整ABO VIDEO IN 自动电子束最佳化视频输入AC (alternating current)交流电AC IN 交流输入AC MOTOR 交流电机AC MOTOR SWAC 交流电机开关AC 自动色(饱和度)控制AC mains input 交流电输入ACC AMP ACC放大ACC AMP (REC) ACC放大录制ACC/APC BURST FLAG 自动色度控制/自动相位控制旗脉冲ACC (automatic chrominance control) 自动色度控制ACC AMP (automatic chrominance control amplifier) 自动色度控制ACC BF PHASE 自动控制旗脉冲相位ACC LEVEL 自动色度控制电平ACC LEVEL SW 自动色度控制电平开关ACC BURST GATE ACC色同步选通门ACC DC AMP ACC直流放大ACC DET 自动消色放大ACTION 作用ADAPTOR 适配器ADC(automatic degaussing circuit)自动消磁电路ADD CIRUIT 相加电路ADD RESSING 寻址ADJ (ADJUSTMENT) 调整ADV (一桢一桢)步进AERIAL 天线AFC (automatic frequency control) 自动频率控制AFC BALANCN 自动频率控制平衡调节AFC CENTER AFC中心AFC DC 自动频率控制(AFC)直流AFC DC BIAS AFC 直流偏置AFC (DC) OUT 自动频率控制(DC)输出AFC DRIVE 自动频率控制推动AFC ERROR 自动频率控制误差信号AFC ERROR BUFFER AFC误差缓冲AFC FH TUNING AFC行频调谐AFC FH TUNING AMP AFC行频调谐放大AFC GAIN AFC增益AFC GATE 自动频率控制门AFC IN 自动频率控制输入AFC OUT AFC输出AFC PULSE AMP 自动频率控制脉冲放大AFC SET 自动频率控制设定AFC VCO AFC压控振荡器AFC VCO FREQ AFC压控振荡器频率AFPC (automatic frequency phase control) 自动频率相位控制AFS(automatic frequency stabilization)自动频率稳定AFTER CLOCK 时钟后AFTER CLOCK PULSE 时钟脉冲之后AGC 自动增益控制AGC AMP AGC放大AGC DETECTOR 自动增益控制检测AGC ERROR BUFFER 自动增益控制误差缓冲器AGC PROT AGC保护AH(AUDIO/CTLHEAD) AH(音频控制磁头)ALARM TONE BURST 告警音频缓冲ALT 行交替ALT PULSE 行交替脉冲ALTERNATEDSC 交替的副载波ALU 运算器AMP(amplifier)放大器AMPLIFIER DETECTOR 放大器/检波器AMPLITUED LIMIER 限幅器ANALOG SWITCH 模拟开关ANODE 阳极ANC 自动消噪电路ANTENNA 天线APC(automatic phase control)自动相位控制APC BF INV APC 旗脉冲倒相APC 自动相位控制(检波)ARC(automatic resolution control)自动清晰度控制AT(Ampere turns)安(培)匝数ATT (ATTENUATOR) 衰减器AUTOMATIC 地自动BB(blue)蓝色B(brightness)亮度BA(buffer amplifier)缓冲放大器BALANCE 平衡BALUN 平衡-不平衡转换器BRIGHT 亮度BRIGHTNESS 亮度调节BLLE OUT OFF 蓝枪截止调节BLUE OUT 蓝色输出BURST 色同步信号BURST GATE 色同步选通电路BURST PHASE 色同步信号相位CCURRENT LIMITTER 电流限制器CEN 中心CHROMA 色度CHROMA AMP 色度放大器CHROMA BURST AMP 色度、色同步信号放大器CHROMA BOARD 色通道板CHROMA FILTER 色度滤波器CHROMINANCE 色度通道CLAMPER 钳位器CMOS (complementary metal -oxide-semiconductor) 互补型金属-氧化物半导体COLOUR CONT(color controller) 彩色控制器COLOUR DIFFERENCE 色差COLOUR SYNC 彩色同步调节COLORKILLER 消色器COLORTONE 色调CONT 对比度、控制CONTRAST 对比度CONTROL 控制CONSOLE-控制柜CPT (color picture tube) 彩色显像管CPT BOARD 彩色显像管座板CRT (cathode - ray tube) 阴极射线管(显像管)CRT DRIVE BOARD 显像管激励电路板DDC (direct current)直流电DAMPER 阻尼器DGC (degaussing coil) 消磁线圈DL (delay line ) 延时线DRIVE 激励、推动DRIVE TRANSF 推动变压器DY (deflection yoke)偏转线圈EEHT (extra -high tension) 极高压EMERGENCY-急停装置ERROR AMP (error amplifier) 误差电压放大器E-W CORRECTION(east - west correction) 东西向校正FFBT (fly back transformer) 逆程变压器FILTER 滤波器FLIP FLOP 双稳态触发器FIYEACK BLANKING 回扫消隐FOCUS 焦点FOCUS VR (focus variable rheostat) 聚焦电位器f.(fuse) (fuse) 保险丝GGANTRY-机架G (green) 绿色的GND (ground) 接地GREEN CUT OFF 绿枪截止调节GREEN OUT 绿色输出GREY 灰度G - Y MATRIX (G - Y )矩阵HH. BLK (horizontal blanking) 行消隐H.DY (horizontal deflection yoke) 行偏转线圈HFC (high frequency choke) 高频扼流圈H.HOLD (horizontal hold) 行同步调节H (L).DRIVE (horizontal driver) 行推动放大器HLIN (horizontal linearity) 行线性H(L)OUT BOARD 行输出板H .M(module)厚膜电路HOR AFC (horizontal automatic frequency control) 行自动频率控制HOR DRIVE TRANS 行激励变压器HORIZONTAL 行(水平)扫描部分HORIZ O/P (horizontal out put ) 行脉冲输出H.OSC(horizontal oscillator) 行振荡器H.PHASE (horizontal phase) 行(同步)相位调节H.SIZE 水平幅度调节器HV (high voltage)高压IIC (integrated circuit)集成电路INPUT 输入KKC (kilohertz) 千周KHz(kilohertz) 千赫KILLER AMP (killer amplifier)消色放大器LLEVER 电平LINE-FILTER-滤波器LOW 低的LPF(low-pass filter)低通滤波器MMAIN BOARD 主电路板MC (megacycles per second)兆赫MF (ceramic filter) 陶瓷滤波器MFD;mfd(microfarad) 微法ms (millisecond) 毫秒mV (mill volt) 毫伏OON/OFF 开/关operating point 工作点OSC (oscillator) 振荡器OUT 输出端OVERLOAD -FUSE 过载保险丝OVERLOAD -PROTECTION 过载保护OVER-VOLTGE PROTECTION 过压保护PPOSITIVE THERMISTOR 正温度系数热敏电阻POWER BOARD 电源板POWER CORD 电源线POWER DRIVE 功率激励POWER RECT(power rectifier) 电源整流器POWER REG(power regulation) 功率调整POWER REG RM (power regulation reluctance) 功率调整管散热片POWER SUPPLY 电源POWER TRANS (power transformer) 电源变压器PROTECTOR-保护装置PEDESTAL CLAMP 消隐脉冲钳位PEDESTAL CLAMPER 消隐脉冲钳位电路PF (Pico farad) 微微法PHASE CONT (phase controller)相位控制器RR (red) 红色的R.GBL( red background) 红色背景(暗平衡)调节RECT (rectifier) 整流器R.DRIVE (red drive) 红色驱动(白平衡)调节RED CUT OFF 红色截止调节RED OUT 红色输出REFERENCE VOLTAGE 基准电压REGULATOR 稳压器SSHIELD-防护SIDE-PINAMP 左校正放大器STACK-硅堆SUB BRIGHT 副亮度调节SYNC (synchonization) 同步(信号)SAW (surface acoustic wave) 声表面滤波器SCREEN 帘栅极(加速器)电压调节SYNC (synchronous separator)同步信号分离器TTF (temperature fuse) 温度保险丝THERMO-SENSOR 温度探头TO CPT BOARD 接到显像管印刷板VOUT SWITCH 垂直泵浦开关V.BLK (vertical blanking) 场消隐V.DY (vertical deflection yoke) 场消隐线圈VERT CENT (vertical center) 场中心调节VERT DRIVE AMP 场推动放大器VERT OSC (vertical oscillator) 场振荡器VERT SIZE (vertical size) 垂直幅度调节VERT TRIGG (vertical trigger) 场触发V HOLD 场同步调节VIDEO 视频放大VIDEO& CHROMA BOARD 视频与色度印制板VOLTAGE DIVIDER 分压器XX (crystal) 石英晶体谐振器YY AMP 亮度放大器yoke 偏转线圈。
Sterilization of health-care products ̶ Ethyleneoxide ̶Requirements for the development, validation and routine control of a sterilizationprocess for medical devices医疗保健产品灭菌——环氧乙烷——医疗器械灭菌过程开发、确认和常规控制要求1 Scope范围1.1 Inclusions 包含内容This International Standard specifies requirements for the development, validation and routine control of an ethylene oxide sterilization process for medical devices in both the industrial and health care facility settings, and it acknowledges the similarities and differences between the two applications. 本标准规定了医疗器械产品在工业与医疗保健机构的环氧乙烷灭菌过程的开发、验证和常规控制的要求,并承认这两个领域之间灭菌过程开发、确认的常规控制的异同。
NOTE 1. Among the similarities are the common need for quality systems, staff training, and proper safety measures. The major differences relate to the unique physical and organizational conditions in health care facilities, and to the initial condition of reusable medical devices being presented for sterilization.注1.其中, 相同之处在于质量体系、人员培训及适当的安全措施的通用要求。
美国FDA 医疗器械体系法规QSR820中文版Part 820——质量体系法规——目录Subpart A- 总则820.1 范围820.3 定义 820.5 质量体系Subpart B –质量体系要求820.20 管理职责820.22 质量审核 820.25 人员Subpart C- 设计控制 820.30 设计控制Subpart D- 文件控制 820.40 文件控制Subpart E- 采购控制 820.50 采购控制Subpart F- 标识与可追溯性820.60 标识 820.65 可追溯性Subpart G - 生产和过程控制820.70 生产和过程控制820.72 检验、测量和试验设备 820.75 过程确认Subpart H - 验收活动: 820.80 进货、过程和成品器械检验 820.86 检验状态Subpart I –不合格品 820.90 不合格品Subpart J - 纠正和预防措施 820.100 纠正和预防措施Subpart K –标识和包装控制820.120 设备标签 820.130 设备包装Subpart L –搬运/储存/分销和安装820.140 搬运820.150 贮存820.160 分销 820.170 安装Subpart L –记录820.180 记录的通用要求820.181 设备主要记录820.184 设备历史记录820.186 质量体系记录 820.198 投诉文件Subpart M –服务 820.200 服务Subpart N –统计技术820.250 统计技术Subpart A——总则Subpart A--General Provisions Sec.820.1 范围Sec. 820.1 Scope. (a)适用性Applicability。
(1)本质量体系法规阐明了当前良好制造法规Current good manufacturing practice(CGMP)的要求。
美国FDA 医疗器械体系法规QSR820中文版欧阳引擎(2021.01.01)Part 820——质量体系法规——目录Subpart A- 总则820.1 范围820.3 定义 820.5 质量体系Subpart B –质量体系要求820.20 管理职责820.22 质量审核 820.25 人员Subpart C- 设计控制 820.30 设计控制Subpart D- 文件控制 820.40 文件控制Subpart E- 采购控制 820.50 采购控制Subpart F- 标识与可追溯性820.60 标识 820.65 可追溯性Subpart G - 生产和过程控制820.70 生产和过程控制820.72 检验、测量和试验设备 820.75 过程确认Subpart H - 验收活动: 820.80 进货、过程和成品器械检验 820.86 检验状态Subpart I –不合格品 820.90 不合格品Subpart J - 纠正和预防措施 820.100 纠正和预防措施Subpart K –标识和包装控制820.120 设备标签 820.130 设备包装Subpart L –搬运/储存/分销和安装820.140 搬运820.150 贮存820.160 分销 820.170 安装Subpart L –记录820.180 记录的通用要求820.181 设备主要记录820.184 设备历史记录820.186 质量体系记录 820.198 投诉文件Subpart M –服务 820.200 服务Subpart N –统计技术820.250 统计技术Subpart A——总则Subpart A--General Provisions Sec.820.1 范围Sec. 820.1 Scope. (a)适用性Applicability。
(1)本质量体系法规阐明了当前良好制造法规Current good manufacturing practice(CGMP)的要求。
A Absolute Temperature 绝对温度A Acceleration 加速度A Ammeter 安培计,电流表A Ampere 安培,电流,SI基本单位A Amplifier 放大器,扩大器A Amplitude 振幅,幅度A Angstrom 埃,10-10mA Anode 阳极A Antenna 天线A Atom 原子A atto 阿(托)10-18A-D-T Admission-discharge-transfer System 入院-出院-转院系统A-H Atrio-His Bundle 房室束A-P Ausculation and Percussion 听诊与叩诊A-VO2 Arteriovenous Oxygen Difference 动静脉氧分压差A/A Analysis of Accounts 计数分析A/B Acid-base Ratio 酸碱比A/DV Arteriao/Deep Venous 动脉/深静脉A/G Albumin /Globulion (Ratio) 白蛋白/球蛋白(比)A/SV Arterio/Superficial Venous 动脉/浅静脉A/T Action Time 作用时间A/W Actual Weight 实际重量AA Acrylic Amide 丙烯酰胺AA Activation Analysis 自动分析AA Adaptive Amplifier 自适应放大器AA Amino-acid Analyzer 氨基酸分析仪AA Atomic Absorption 原子吸收AA Audiometric Assistant 电测听辅助装置AA Auto-alarm 自动报警AA Automatic Absorption 自动吸收aaa Amalgam 汞合金AAA Atomic Absorption Analysis 原子吸收分析AAAS American Association for the Advancement of Science 美国科学发展协会AAC Acoustical Absorption Coefficient 吸音系数AAC Acoustical Attenuation Constant 声衰减常数AAC Amplitude Absorption Coefficient 振幅吸收系数AAC Atomic Absorption Coefficient 原子吸收系数AAC Automatic Absorption Coefficient 自动吸收指数AAC Automatic Amplitude Control 自动幅度控制AAFS Atomic Absorption Flame Spectrometer 原子吸收火焰光度计AAL Acoustical Absorption Loss 声吸收损耗AAMI Association for the Advancement of Medical Instrumentation (美国)医疗器械促进协AAPD Alveolar Arterial Pressure Difference 肺泡动脉压差AAS Atomic Absorption Spectrometer 原子吸收光谱仪AAS Atomic Absorption Spectrometry 原子吸收光谱测定AAS Automatic Addressing System 自动寻址系统AAT Accelerated Aging Test 加速老化试验AAU Automatic Addressing Unit 自动寻址装置AB Air Break 空气断路器AB Audio Bandwidth 声频带宽ABA Acid-base Analyzer 血液酸碱分析仪ABB Air-blood Barrier 气血屏障ABBI Advanced Breast Biopsy Instrumentation 先进乳腺活检器械ABC Acid Base Calculator 血气酸碱计算器ABC Actual Bicarbonate Radical 实际碳酸氢根ABC Automatic Bandwidth Control 自动带宽控制ABC Automatic Beam Control 自动波束控制,自动射束控制ABC Automatic B-gain Contrl 自动B增益控制ABC Automatic Brake Control 自动制动控制ABC Automatic Brightness Control 自动亮度控制ABCC Automatic Brightness Contrast Control 自动亮度反差调整ABCE American Board of Clinical Engineering 美国临床工程委员会ABD Automated Border Detection 内心膜自动边缘检测ABD Average Body Dose 平均全身剂量ABDC Automatic Baseline Drift Correction 自动基线漂移校正ABE Actual Base Excess 实际碱过剩ABER Auditory Nerve and Brainstem Electric Response 听神经脑干电反应ABEST Asymmetric Blipped Echoplanar Single Pulse TechniqueABF Aortic Blood Flow 主动脉血流量ABF Audio Band Pass Filter 音频带通滤波器ABG Air Bone Gap 气骨导间距,气骨隙ABG Arterial Blood Gas 动脉血气ABI Aorta Balloon Inflate 主动脉气囊充气ABL Automatic Brightness Limit 自动亮度限制电路ABM Alarm Box Module 报警信号传送器(盒)ABM Anesthesia and Brain Monitor 麻醉与脑功能监测仪ABMD Area Bone Mineral Density 面骨密度ABP Absolute Boiling Point 绝对沸点ABP Arterial Blood Pressure 动脉血压ABP Artificial Brainstem Perfusion 人工脑干灌注ABPM Ambulatory Blood Pressure Monitoring 动态血压监测ABR Acrylonitrile-butadiene Rubber 丁腈橡胶ABR Auditory Brain Response 听力脑干响应Abs Absorbent 吸收剂,吸光度Abs Absorption 吸收(作用)ABS Acrylonitrile-butadiene-styrene Copolymer 丙烯腈-丁二烯-苯乙烯共聚物ABS Automatic Brightness Stabilization 自动辉度稳定ABT Automatic Bur-translator 自动电源转换器AC Accelerator 加速剂,加速器Ac Accumulator 累加器,蓄电池,贮存器Ac Actinium 锕AC Activated Charcoal 活性炭AC Adaptive Control 自适应控制AC Air Condenser 空气冷凝器,空气电容器AC Air Conduction 空气传导AC Alternating Current 交流电AC Analogue Computer 模拟计算机AC Attenuation Correction 衰减校正AC Automatic Computer 自动计算机ACA Automatic Chemical Analyzer 自动化学分析器ACA Automatic Clinical Analyzer 自动临床分析仪ACAC Albumin Collodion Activated Charcoal 白蛋白-火棉胶活性炭ACAC Automatic Circuit Analyzer and Verifier 自动电路分析器和检测器Acc Accommodation 视觉调焦Accel Accelerator 加速器Access Accessory 零件ACCESS Automatic Computer Controlled Electronic Scanning SystemACD Annihilation Coincidence Detection 湮没符合探测ACD Anode-cathode Distance 极距,阴阳极距离ACD Anterior Chest Diameter 前胸胸径计ACE Antenatal Cell Extractor 胎儿细胞提ACE Automatic Calling Equipmnt 自动呼叫装置ACE Automatic Checkout Equipment 自动检测设备ACE Automatic Computing Equipment 自动计算装置ACES Automatic Checkout and Evaluation System 自动检测和鉴定系统ACF Auto-correlation Function 自相关函数ACFG Automatic Continuous Function Generation 自动连续函数生成ACG Angiocardiograph 心血管造影装置ACG Angiocardiography 心血管造影术ACG Apexcardiogram 心尖搏动图ACG Autocorrelogram 自相关图Ach Achromatic Objective 消色差透镜ACH Autocorrelation Histogram 自相关直方图ACI Allowable Concentration Index 容许浓度指数ACI Atrial Contraction Index 心房收缩指数Acld Air Cooled 气冷式ACLS Advanced Cardiac Life Support 高级心脏生命支持ACM Automatic Cardioflow Measurement 自动心脏血流测量Acn Acrylonitrile 丙烯腈ACOE Automatic Check-out Equipment 自动检测设备ACOM Automatic Coding Machine 自动编码机ACP Anodal Closing Picture 阳极通电图像ACP Automatic Catholic Protector 自动阴极防护器ACPD Ambulant Continuous Peritoneal Dialysis 非卧床持续性腹膜透析ACQSIM Acro-quality Similator 高品质模拟定位器ACR American College of Radiology 美国放射协会ACR Automatic Controller 自动控制器ACS Audio Communication System 声频通信系统ACS Automatic Control System 自动控制系统ACSF Artificial Cerebrospinal Fluid 人工脑脊液ACT Activated Clotting Time 激活凝血时间ACT Adaptive Current Tomography 自适应电流激励体层成像ACT Anticoagulation Therapy 抗凝结治疗ACT Atrial Contraction Time 心房收缩时间ACT Automatic Calibration Technique 自动校正技术ACTAS Automatic Computerized Transverse Axial Scanner 计算机横断层轴向自动扫描器ACTER Active Filter 有源滤波器ACTS Acoustic Control and Telemetry System 声控和遥测系统ACU Arithmetic and Control Unit 运算和控制装置ACU Automatic Calling Unit 自动呼叫装置ACV Automatic Control Valve 自动控制阀Acw Anticlockwise 逆时针AD Absorbed Dose 吸收剂量AD Accidental Death 事故死亡,意外死亡AD Acquisition Densitometry 声学密度定量技术Ad Adapter 接头,拾音器AD Admitting Diagnosis 入院诊断AD Advanced Development 试制样机,试制样品Ad Afterdischarge 后放电AD Alternate Display 备用显示器Ad Analog-digital 模拟-数字AD Assistive Device 辅助装置AD Automatic Detection 自动检测,自动探测AD Average Deviation 平均偏差ADA Action Data Automation 自动数据处理系统ADAC Analog-digital-analog Converter 模拟-数字-模拟转换器ADAPS Automatic Display and Plotting System 自动显示和标图系统ADAS Automatic Data Acquisition System 自动数据获取系统ADAS Auxiliary Data Annotation Sets 辅助数据解读装置ADAT Automatic Data Accumulator and Transfer 自动数据储存和传输装置ADC Analogue-to-digital Converter 模数变换器ADC Apparent Diffusion Coefficient 表观弥散系数ADC Automatic Drift Control 自动漂移控制ADD Analog Data Digitizer 模拟数据数字读出器ADES Automatic Digital Encoding System 自动(数字)编码系统Adh Adhesive 粘合剂ADHS Automatic Data Handling System 自动数据处理系统ADI Acceptable Daily Intake 日容许摄入量ADIOS Automatic Digital Input-output System 自动数字输入输出系统ADIS Automatic Data Interchange System 自动数据互换系统ADIT Analog Digital Integrating Translator 模拟数字综合转换器Adj Adjustment 调整,调节,校准,对准ADL Amplitude Difference Limen 幅度辨别阈ADL Analog Delay Line 模拟延迟线ADM Air Detector Module (透析液内)空气检测单元ADMIS Automated Data Management Information System 自动数据管理信息系统ADP Advanced Data Processing 先进的数据处理ADP Air Drive Pump 气动泵ADP Automatic Data Processing 自动数据处理ADPE Automatic Data Processing Equipment 自动数据处理装置ADPP Automatic Data Perfection Process 自动数据典型处理ADPS Automatic Data Processing System 自动数据处理系统Adr Adder 加法器ADR Automatic Dose Rate 自动计量率ADRA Automatic Dynamic Response Analyzer 自动动态响应分析器ADRS Analog-to-digital Recording System 模拟数字记录系统ADTU Automatic Data Test Unit 自动数据测试系统ADX Automatic Data Exchanger 自动数据交换系统AE Absolute Error 绝对误差AE Accuracy Error 精确误差AE Acoustic Emission 声发射AE Atrial Electrogram 心房电图AEA Averaged Electroencephalic Audiometry 平均脑电测听法AEBSR Auditory Evoked Brain Stem Response 听诱发脑干反应AEC Automatic Exposure Control 自动曝光控制AECD Automatic External Cadioverter Defibrillator 体外自动复律除颤器AECG Abdominal Electrocardiograph 腹部心电图机AECG Active Electrocardiography 动态心电图AED Acceptable Emergency Dose 应急容许剂量AED Auditory Evoked Magnetic Field 听觉诱发磁场AED Automated External Defibrillator 体外自动除颤器AEEG Active EEG 动态脑电图AEF Auditory Evoked Field 听觉诱发野AEG Air Encephalography 气脑造影术AEI Atrial Emptying Index 心房排空指数AELS Advanced Emergency Life Support 高级急救生命支持AEM Analytical Electron Microscope 分析电子显微镜AEM Anodic Electrophoretic Mobility 阴极电泳泳动度AEMB Alliance for Engineering in Medicine and Biology 医学与生物工程联合会AEP Acoustially Evoked Potential 声诱发电位AEP Auditory Evoked Potential 听觉诱发电位AEP Average Evoked Potential 平均诱发电位AER Acoustic Evoked Response 声诱发反应AER Anion Exchange Resin 阴离子交换树脂AER Auditory Evoked Response 听觉诱发反应AER Average Electroencephalic Response 平均脑电反应AER Average Evoked Response 平均诱发反应AERP Atrial Effective Refractory Period 心房有效不应期AES Auger Electron Spectroscopy 俄歇电子光谱AET Absorption Equivalent Thickness 吸收当量厚度AET Approximate Exposure Time 近似曝光时间AF Aortic Flow 主动脉血流(量)AF Arithmetic formula 运算公式,算术公式AF Atomic Fluorescence 原子荧光AF Atrial Fibrillation 心房纤维颤动AF Audio Frequency 声频,音频AF Auto Fluorescence 自发荧光,自体荧光AF Automatic Focus 自动聚焦AF Automatic Following 自动跟踪AF Axis Field 轴射野AFA Audio Frequency Amplifier 声频放大器AFAQ Association Francaise pour l'Assurance de la qualite=French Association for Quality Assurance 法国质量保证协会AFC Audio Frequency Choke 音频扼流圈AFC Automatic Frequency Control 自动频率控制AFD Alkali Flame Detector 碱性火焰检测器AFD Amplitude Frequency Distortion 幅频失真AFDR Automatic Frequency Drift Rejector 自动频率漂移抑制器aFECG Abdominal Fetal Electrocardiogram 腹部胎儿心电图AFG Analog Function Generator 模拟函数发生器AFHT Antepartum Fetal Heart Rate Testing 产前胎儿心率检测AFI Atrial Filling Index 心房充盈指数AFM Atomic Force Microscope 原子力显微镜AFP Adiabatic Fast Passage 绝热快速通道AFP Alpha Fetoprotein 甲胎蛋白AFPC Automatic Frequency and Phase Control 自动频率和相位控制AFR Acceptable Failure Rate 容许故障率AFT Audio Frequency Transformer 音频变压器AFT Automatic Fine Tuning 自动微调AFT Automatic Frequency Tuner 自动频率调谐器AFTAAS Advanced Fast Time Acoustic Analysis System 高级快速声学分析系统AG Air Gap 空气隙AG Arteriography 动脉造影术AG Available Gain 可用增益Ag Silver 银AGC Automatic Gain Control 自动增益控制AGC Automatic Gauge Controller 自动测量调整装置AGE Angle of Greatest Extension 最大牵伸角度AGF Angle of Greatest Flexion 最大屈曲角度AGS Alternating Gradient Synchrotron 交变磁场梯度同步加速器AGS American Gastroscopic Society 美国胃镜学会AGSS Anesthesia Gas Scavenging System 麻醉气体清除系统AH Amplitude Histogram 振幅直方图,振幅度数分布AH Arterial Hypertention 动脉高血压AHD After Hemodialysis 血液透析后AHD Atherosclerotic Heart Disease 动脉粥样硬化性心脏病AHE Adaptive Histogram Equalization 适应直方图均衡AHI Apnea Hyponea Index 呼吸紊乱指数AHIS Automated Hospital Information System 自动化医院信息系统AHP Artificial Heart Pump 人工心脏泵AHVC Automatic High-voltage Control 自动高压控制AI Aortic Insufficiency 主动脉瓣闭锁不全AI Artificial Intelligence 人工智能AI Automatic Input 自动输入AI Automatic Inspection 自动检查,自动检验AICD Automatic Implantable Cardioverter Defibrillator 植入式自动心脏复律除颤器AID Argon Ionization Device 氩离子检查器AID Automatic Implantable Defibrillator 植入式自动除颤器AIEC Advanced Ion Exchange Cellulose 高级离子交换纤维素AII Average Image Intensity 图像平均强度AIM Artificial Intelligence in Medicine 医学人工智能,医学智能模拟AIM Automated Information Management 自动信息管理AIMD Active Implantable Medical Devices 有源植入医疗器械AIMDD Active Implantable Medical Device Directive 有源植入医疗器械指令(欧共体) AIOP Analog Input/Output Package 模拟输入/输出组件AIP Average Intravascular Pressure 平均血管内压AIRS Automatic Image Retrieval System 自动影像检索系统AIS Abbreviated Injury Scale 简略创伤分度法,简略创伤定级标准AIT Artificial Intelligence Technique 人工智能技术AIUM American Institute of Ultrasound in Medicine 美国医用超声学会AJ Antijamming 抗干扰AJD Antijamming Display 抗干扰显示器AL Adaptation Level 适应电平,适应响度级Al Aluminium 铝ALC Adaptive Logic Circuit 自适应逻辑电路ALC Automatic Light Compensation 自动光量补偿ALC Automatic Load Control 自动负荷控制ALC Automatic Light Control 自动亮度调整ALC Analytical Liquid Chromatography 分析用液相色谱仪ALCr Cr Aluminium Crown 铝冠(齿科) ALE Average Life Expectency 平均期望工作寿命,平均寿命预期值AlEq Aluminium Equivalent 铝过滤当量AlgoL Algorithmic Language 算法语言ALK Alkyd-resin 聚酯树脂,醇酸树脂ALK Automated Lamellar Keratoplasty 角膜层屈光性手术,自动角膜板层磨镶术Alm Alarm 报警器ALP Argon Laser Photocoagulation 氩激光凝固,氩激光光灼止血ALR Automatic Load Regulator 自动负载调节器ALS Advanced Life Support 高级生命支持ALVAD Abdominal Left Ventricular Assist Device 腹部左心室辅助装置ALVR Artificial Lung Ventilation Regime 人工肺通气方案AM Aeromedical Monitor 航空医学监测器AM Agile Manufacturing 敏捷制造术Am Americium 镅Am Amperemeter 安培计AM Amplitude Modulation 调幅AM-LCD Active Matrix Liquid Crystal Display 有源矩阵液晶显示器Amalg Amalgam 汞合金Amb Ambulance 救护车,救护船,救护飞机AMD Analog Memory Device 模拟存储器AME Average Magnitude of Error 误差平均值AMH Automated Medical History 自动问诊AMHS Automatic Message Handling System 自动信息处理系统AMHTS Automated Multiphasic Health Testing System 自动多项健康检查系统AML Automated Multitest Laboratory 自动多项检查实验室AMP Ampere 安培AMP Amplifier 放大器,扩大器AMP Amplitude 振幅,幅度AMPA Adaptive Multibeam Phased Array 自适应多波束相控阵AMRS Automated Medical Record System 自动化医学档案系统AMS Acoustic Measurement System 声学测量系统,音响测量系统AMS Automicrobic System 微生物自动诊检仪AMT Advanced Manufacturing Technology 先进制造技术AMV Assisted Mechanical Ventilation 机械辅助通气AN Anacrotic Notch 颈动脉搏动图(升支切迹)An Normal Atemosphere 常压,标准大气压ANACOM Analog Computer 模拟计算机Anesth Anesthetic 麻醉剂Ang Angiogram 血管造影照片Ang Angiography 血管造影术Ang Angle 角,角度ANL Automatic Noise Limiter 自动噪声限制器ANN Artificial Neural Network 人工神经网络ANS Automatic Noise Suppressor 自动噪声抑制器ANSI American National Standards Institute 美国国家标准协会AO Acridine Orange 丫啶橙Ao Aortogram 主动脉造影照片,主动脉照片AOC Automatic Output Control 自动输出功率控制AOC Automatic Overload Control 自动过载控制AOD Acridine-orange Diagnosis 丫啶橙诊断AoD Aortic Diastolic Pressure 主动脉舒张压AoD Aortic Dimension 主动脉内径AOG Auditory Electrooculomotogram 听觉性眼球运动电流图AoI Area-of-interest 感兴趣区域AOM Add One to Memory 加1存储器AOP Anode Opening Picture 阳极断电图AoP Aortic Pressure 主动脉压AOS Acquisition of Signal 信号探测,信号获取AoS Aortic Systolic Pressure 主动脉收缩压AOTF Acousto-optic Turnable Filter 声光可调滤光器AOV Automatically Operated Valve 自动阀AP Action Potential 动作电位AP Aortic Pressure 主动脉压AP Arithmetic Progression 算术级数AP Artificial Pacemaker 心脏起搏器AP Artificial Pancreas 人工胰腺AP Automatic Programming 自动程序设计AP Atmospheric Pressure 大气压AP Attached Processor 附加处理器APA Action Potential Amplitude 动作电位振幅APA Annular Phased Array 对称环形相控阵技术APADAS Automatic Phase and Amplitude Data Acquisition System 自动相位和振幅数据获取系统APADS Automatic Programmer and Data System 自动程序设计器和数据系统APATS Automatic Programming and Test System 自动程序设计和试验系统APC Automatic Phase Control 自动相位控制APC Automatic Power Control 自动功率控制APD Action Potential Duration 动作电位时程APD Avalanche Photo Diode 雪崩光敏二极管APD Avalanche Photodetector 雪崩光电探测器APF All Pass Filter 全通滤波器APG Air Pressure Gauge 气压计APG Auricular Plethysmogram 耳垂容积图API Ambulatory Pump Infusion 携带式泵输注API Atmospheric Pressure Ionization 大气压电离作用APL Adjustable Pressure Limiting 可调限压APL Average Picture Level 平均图像电平APN Average Peak Noise 平均峰值噪声APO Apochromatic Objective 复消色差物镜APP Alternating Pressure Pad 变压垫App Appliance 矫正器App Approve 批准,同意APp Artificial Pneumoperitoneum 人工气腹APRV Airway Pressure Release Ventilation 气道压变化通气APS Accessory Power Supply 辅助电源APS Aperiodic Photic Stimulation 不定期光刺激APS Automatic Phase Synchronization 自动相位同步APT Appendant Potential Tomography 外加电位断层图像法APt Artificial Pneumothorax 人工气胸APT Automatic Picture Transmission 自动图像传输APTS Automatic Picture Transmission System 图像自动传输系统APTT Activated Partial Thromboplastin Time 激活部分血栓形成时间APU Auxiliary Power Unit 辅助电源APV Adaptive Pressure Ventilation 自适应压力调节呼吸AQ Acoustic Quantification 声定量化AQ Any Quantity 任意量AQL Acceptable Quality Level 容许质量标准,合格质量水平AR Acrylic Rubber 丙烯酸酯橡胶AR Analytical Reagent 分析试剂Ar Argon 氩AR Artificial Respiration 人工呼吸AR Autorefractor 自动屈光计ARC Automatic Remote Control 自动遥控ARCET Automatic Recording Crystal Electric Tonometer 自动记录晶体电眼压计ARD Average Relative Deviation 平均相对偏差ARG Aorticrheogram 主动脉血流图ARG Autoradiograph 放射自显影ARG Autoradiography 放射自显影术ARI Acute Respiratory Infection 急性呼吸道感染ARI Radio Isotopic Arteriograph 放射性同位素动脉造影术ARL Average Remaining Lifetime 平均剩余寿命ARP Absolute Refractory Period 绝对不应期Arrhy Arrhythmia 心律失常,心律不齐ART Acoustic Response Technique 声学响应技术ART Algebraic Reconstruction Technique 代数重建技术AS Active Sleep 主动睡眠As Arsenic 砷AS Automatic Sprinkler 自动洒水器AS Automatic Synchronizer 自动同步器ASA American Standards Association 美国标准协会ASA Australian Standards Association 澳大利亚标准协会ASAIO American Society for Artificial Internal Organs 美国人工内脏学会ASC Automatic Selectivity Control 自动选择性控制ASC Automatic Sensibility Control 自动灵敏度控制ASCU Automatic Scanning Control Unit 自动扫描控制装置ASD Atrial Septal Defect 心房间隔缺损ASD Automatic Synchronizing Device 自动同步装置ASE Automatic Stabilization Equipment ASN Average Sample Number 平均采样数AST Automatic Starter 自动启动器ASTM American Society for Testing Materials 美国材料试验学会ASTM American Standard of Testing Materials 美国试验材料标准ASTM American Standard Testing Manual 美国标准试验手册ASV Acceleration Switching Valve 快速开关阀ASV Adaptive Support Ventilation 自适应辅助通气ASV Automatic Shuttle Valve 自动关闭阀AT Applanation Tonometry 压平眼压测量法At Astatine 砹AT Atrial Tachycardia 房性心动过速AT Axillary Temperature 腋下温度at.vol Atomic Volume 原子体积at.wt Atomic Weight 原子量AtA Atmosphere Absolute 绝对大气压ATC Amplitude to Time Converter 幅度-时间变换器ATC Analogue to Time Converter 模拟-时间变换器ATC Automatic Temperature Controller 自动温度控制器ATC Automatic Timing Corrector 自动记时校正器ATD Average Temperature Difference 平均温差ATE Automatic Test Equipment 自动测试设备ATK Annular Thermokeratoplasty 环状热角膜成形术ATL Automatic Telling 自动报警ATLS Advanced Trauma Life Support 先进创伤生命支持ATM Asynchronous Transfer Mode 异步转移模式ATR Attenuate Total Reflection 衰减全反射Atr Fib Atrial Fibrillation 心房纤颤性颤动ATRC Automatic Temperature Recorder Controller 自动温度计录控制器ATS Automatic Test System 自动测试系统ATS Automatic Tuning System 自动调谐系统AU Absorbance Unit 吸光度单位AU Alarm Unit 报警装置Au Gold 金AUC Area Under the Curve 曲线下面积AUDRI Automated Drug Identification System 自动化药物鉴定系统AULL Augmented Unipolar Limb Lead 加压单极肢体导联AUS Auxiliary Switch 辅助开关AV Actual Velocity 实际速度AV Arteriovenous 动静脉的AV Artificial Ventilation 人工通气AV Atrioventricular 房室的AV Average value 平均值AVB Auriculo-ventricular Block 房室传导阻滞AVC Automatic Volume Control 自动强度控制,自动音量控制AVD Arteriovenous Difference in Oxygen 动静脉氧分压差AVDO2 Arteriovenous Oxygen Difference 动静脉氧分压差ave Average 平均,平均值AVF Azimmthally Varying Field 方位变频场AVI Aortic Valve Index 主动脉瓣指数AVI Atrioventricular Insufficiency 房室瓣闭锁不全AVID Antiarrhythmics Versus Implantable Difibrillation 抗心律失常药物与埋藏式除颤器avl Average Length 平均长度AVM Arteriovenous Malformation 动静脉畸形AVP Ambulatory Venous Pressure 非卧床静脉压AVPD Atrioventricular Plane Displacement 房室平面位移AVR Aortic Valve Replacement 主动脉瓣置换AVR Automatic Voltage Regulation 自动电压调整AVR Automatic Voltage Regulator 自动稳压器AVSV Aortic Valve Stroke Volume 主动脉瓣排血量AVU Assisted Ventilation Unit 辅助通气装置avw Average Width 平均宽度AW Atomic Weight 原子量AZ Automatic Zero 自动零位调整,自动调零AZM Azotometer 氮素计B Base of Prism 棱镜底B Bicuspid 二尖瓣B Blood 血B Boron 硼B Brightness 亮度B.P Boiling Point 沸点B.wt Body Weight 体重B/S Balance Sheet 平衡表B/S Bits Per Second 比特/秒,位/秒B/U Backup 备用设备,备件,备用的,辅助的B/W Black/White 黑白(信号,图像,照片,影片,电视等)Ba Barium 钡BA Best Amplitude 最佳振幅BA Bioassay 生物测定BA Breathing Apparatus 呼吸器BA Buffer Amplifier 缓冲放大器BAC Bacteria 细菌BAC Biomedical Application of Computers 计算机的生物医学应用BAEP Brainstem Auditory Evoked Potential 脑干听觉诱发电位BAER Brainstem Auditory Evoked Response 脑干听觉诱发反应BAG Bronchial Arteriography 支气管动脉造影术BAIT Bacterial Automatic Identification Technique 细菌自动鉴定技术BAL Balance 平衡,对称,天平BAL Basic Assembler Language 基本汇编语言BAM Basic Access Method 基本存取法BAP Brachial Artery Pressure 肱动脉压BAPG Biauricle Plethysmography 双侧心房体积描记法BAR Barometer 气压计Bar Barometer 气压计BAS Balloon Atrial Septostomy 球囊房间隔造孔术BASIC Beginners All Purpose Symbolic Instruction Code 初学者的通用符号指令码(计算机的一种会话语言)BAV Bicuspid Aortic Valve 双叶式主动脉瓣BB Baseband 基本频带BB Black Body 黑体BB Blood Buffer 血液缓冲剂BB Body Burden 体内(放射性)积存量BB Buffer Base 缓冲碱BB-meter Babies Bilirubin Meter 新生儿胆红素测定仪BBB Blood-brain Barrier 血脑屏障BBBB Bilateral Bundle Branch Block 双侧束支传导阻滞BBR Black Body Radiator 黑体辐射器BBT Basal Body Temperature 基础体温BC Biocompatibility 生物相容性BC Board of Control 控制板,控制盘BC Bone Conduction 骨传导BC Bradycardia 心搏徐缓BC Breathing Capacity 呼吸量BCF Bioconcentration Factor 生物浓度指数BCG Ballistocardiogram 心冲击图BCLS Basic Cardiac Life Support 基本心脏生命支持BCN Bone Conduction Noise 骨导噪声BCR Biological Cleaning Room 生物洁净室BD Beam Divider 分束器BD Brains Death 脑死亡BD Bronchodilator 支气管扩张器BDI Behavioral Disturbance Index 行为障碍指数BDI Below Detectable Limit 低于检出限BDN Blood Deficit Nomogram 血量不足列线图BDP Beginning Diastole Pressure 初始舒张压BDR Biological Decay Rate 生物衰亡率BDU Basic Display Unit 主显示器BDV Blood Dilution value 血液稀释值BDV Breakdown Voltage 击穿电压,破坏电压BE Base Excess 碱过量Be Beryllium 铍BE Biological Engineering 生物工程学BE Bottom Echo 底部回声,底反响BEA Background Equivalent Activity 本底当量放射性,背景等效放射性BEAM Brain Electrical Activity Mapping 脑电地形图BEAR Biological Effects of Atomic Radiation 原子辐射生物效应BEB British Electrotechnical Bureau 英国家用电器局颁发安全质量认证标志BEC Background Equivalent Concentration 本底等效浓度BEC Bio-electrochemistry 生物电化学BEEE Basal Energy Expenditure Equation 基础能量消耗方程式BEF Best Excitatory Frequency 最佳兴奋频率BEI Backscattered Electron Imaging 背散射电子显像BEIR Biological Effect of Ionizing Radiation 电离辐射生物效应BEMF Back Electromotive Force 反电动势BEP Bioelectric Polyurethane 生物电聚氨基甲酸酯BEP Brainstem Evoked Potential 脑干诱发电位BER Basic Electrical Rhythm 基础电节律BER Biologic Effect Ratio 生物效应比BER Brainstem Electric Response 脑干电反应BER Brainstem Evoked Response 脑干诱发反应BERA Brainstem Electric Response Audiometry 脑干电反应测听法BERA Brainstem Evoked Response Audiometry 脑干诱发反应测听法BES Bioluminescence Emission Spectrum 生物发光发射谱BEST Blipped Echoplanar Single Pulse Technique (MRI)回波平面成像序列之一BEV Beam?s Eye View 射野平面BeV Billion Electron Volts 十亿电子伏特BF Band Filter 带通滤波器BF Best Frequency 最佳频率BF Biofeedback 生物反馈BF Blood Filterability 血液滤过性,血液滤过力BF Blood Filtrate 血滤液BF Blood Flow 血流,血流量BF-ERG Bright-flash Electroretinogram 闪光视网膜电(流)图BfArM Bundesinstitut fur Arzneimittel und Medizinprodukte=Federal Institute for Drugs and Medical Devices (Germany) 联邦药品和医疗器械研究所(德国)BFAST Blood Flow Artifact Suppression Technique 血流伪像抑制技术BFHR Baseline Fetal Heart Rate 胎心率基线BFM Blood Flowmeter 血流量计BFM Body Fat Mass 机体脂肪量BFO Beat Frequency Oscillator 拍频振荡器BFP Biologically False Positivity 生物学假阳性BFR Blood Flow Rate 血流速度BFR Bone formation Rate 成骨速度BFS Bronchofiborscope 纤维支气管镜BFT Biofeedback Training 生物反馈训练BG Background 本底,背景BG Blood Glucose 血糖BGA Bundesgesundheitsamt=Federal Health Office (Germany)BGC Bowel Gas Correction 体内气体校正BGO Bi4Ge3O12??Crystal 锗酸铋,闪烁晶体BHC Beam Hardening Correction 声束硬化修正BHDF Bedside Hemodialysis Filtration 床旁血液透析滤过BHDU Bedside Hemodialysis Ultra-filtration 床旁血液透析超滤BHF Bedside Hemo-filtration 床旁血液滤过BHP Bedside Hemoperfusion 床旁血流灌注BHP Blood Hydrostatic Pressure 血液流体静压BHU Bedside Hemo-ultrafiltration 床旁血液超滤Bi Bismuth 铋BIBS Build-in Breathing System 内装式呼吸系统BIL Basic Impulse Level 基本脉冲电平BIMA Bistable Magnetic Core 双稳态磁心BIONICS Biological Electronics 生物电子学,生物电子仪BIOSEM Biological Scanning Electron Microscope 生物扫描电镜BiPAP Bi-level Positive Airway Pressure 双水平气道正压BIPAP Biphasic Positive Airway Pressure 双相气道正压通气BK Below-knee Prothesis 膝下假肢Bk Berkelium 锫BL Base Line 基准线,基线BL Bio-luminescence 生物发光BLN Balloon 气球,气囊,球囊BloDi Block Diagram 方框图BLR Base Line RestorerBLS Basic Life Support 基本生命支持BLT Bio-luminescence Test 生物发光试验BLT Blood-clot Lysis Time 血块溶解时间BLV Base Line value 基准值BM Balance Master System 平衡检测系统BM Basal Metabolism 基础代谢BM Biomarker 生物标记BM Body Mass 体重BM Brightness Modulation 辉度调制BMC Biomedical Computer 生物医学计算机BMD Bone Mineral Density 骨密度,骨无机密度BME Band of Multiple Echoes 多回波带BME Biomedical Engineering 生物医学工程BME Blood Micro Equipment 血液微量(分析)设备BMES Biomedical Engineering Society 生物医学工程学会BMHR Basal Metabolism Heart Rate 基础代谢心率BMR Basal Metabolic Rate 基础代谢率BMS Biomedical Mass Spectrometry 生物医学质谱测定法BMS Blood Micro System 血液微量(分析)系统BMUE Biomedical Ultrasonic Engineering 生物医学超声工程BNCT Boron Neutron Capture Therapy 硼中子俘获治疗BND Band 频带,波段,光谱带,传送带BNR Beam Non-uniformity Ratio 射束非均匀率BO Bubble Oxygenator 气泡氧合器BOA Behavioral Observation Audiometry 行为测听法BOA Blood Oxygen Affinity 血氧亲和力BOD Biochemical Oxygen Demand 生化需氧量BOD Biological Oxygen Demand 生物需氧量BOLD Blood Oxygen Level Dependent 血氧水平依赖性,血氧合度依赖性BOM Biological Oxygen Monitor 生物耗氧监测器BOS Basic Operation System 主操作系统BP Back Pressure 回压,反压BP Bandpass 带通,传送带BP Barrier Pressure 屏障压BP Base Point 基点BP Bichromatic Photometry 双色光度测定法bp Biological Parameter 生物学参数BP Bioprothesis 生物假体BP Biotic Potential 生物电位BP Bipolar 两极的,双相的BP Blood Pool 血池Bp Blood Pressure 血压BP Blueprin 蓝图BP Body Plethysmography 身体体积描记法bp Boiling Point 沸点BP British Patent 英国专利BP Bypass 旁路,支路BP Mean Systematic Blood Pressure 平均全身血压BPC Blood Platelets Count 血小板计数BPD Biparietal Diameter 顶骨间径,双顶径BPEC Bipolar Electrocoagulation 双极电灼止血BPF Band Pass Filter 带通滤波器BPH Benign Prostatic Hypertrophy 良性前列腺肥大BpH Blood pH 血液pH值BPI Bits Per Inch 比特/英寸,位/英寸BPL Band-pass Limiter 带通限幅器BPM Beats Per Minute 每分钟搏动次数(心脏)BPM Blood Pump Module 血泵单元BPM Breaths Per Minute 每分钟呼吸次数BPM Blood Pressure Measuring System 血压测量系统BPO Buffered Physiological Solution 缓冲生理溶液BPS Bits Per Second 比特/秒bpt Base Point 基点BPT Bronchial Provocation Test 支气管激发试验Bq Becquerel 贝克(勒尔),放射性活度,具有专门名称的SI导出单位BR Background Radiation 背景辐射BR Basic Requirements 基本要求BR Bed Rest 卧床休息BR Biological Reagent 生物学试剂BR Biological Response 生物反应BR Birth Rate 出生率Br Breath 呼吸BR Breathing Reserve 换气储备Br Bridge 桥(齿科用语)Br Bromine 溴BR Buffer Register 缓冲寄存器BRCT Brain Resuscitation Clinical Trial 脑复苏临床试验BRD Bilateral Retinal Detachment 双侧视网膜脱离BRIL Brilliance 亮度BRM Barometer 气压计BRM Biological Response Modifier 生物反应调节剂BRO Bronchoscopy 支气管镜检查BRP Brain Retraction Pressure 脑回缩压BRR Baroreceptor Reflex Response 压力感受器反射反应BRS Body Reference System 物体参考系统,物体基准系统Brth Breath 呼吸BRU Bone Reconstructive Unit 骨重建单位BrW Brain Weight 脑重量BS Beam Splitter 射束分裂器BS Blood Sugar 血糖BS Bone Scan 骨扫描BS Bone Scintigraphy 骨闪烁照相术BS Brain Scan 脑扫描BS Brain Stem 脑干BS Breaking Strength 断裂强度BS Breath Sounds 呼吸音BS British Standard 英国标准BS Buffered Saline 缓冲盐水BS Button Switch 按钮开关BSA Body Surface Area 体表面积BSA Burned Surface Area 烧伤体表面积BSAEP Brainstem Audiotory Evoked Potential 脑干听觉诱发电位BSB Baseband 基带,基本频带BSC Back Scatter Coefficient 背散射系数,背散离系数BSC Bone Scintigraphy 骨闪烁照相术。