高聚物的结晶过程。
- 格式:ppt
- 大小:279.00 KB
- 文档页数:13

聚合物的结构(计算题:均方末端距与结晶度)1.简述聚合物的层次结构。
答:聚合物的结构包括高分子的链结构和聚合物的凝聚态结构,高分子的链结构包括近程结构(一级结构)和远程结构(二级结构)。
一级结构包括化学组成、结构单元链接方式、构型、支化与交联。
二级结构包括高分子链大小(相对分子质量、均方末端距、均方半径)和分子链形态(构象、柔顺性)。
三级结构属于凝聚态结构,包括晶态结构、非晶态结构、取向态结构、液晶态结构和织态结构。
构型:是指分子中由化学键所固定的原子在空间的几何排列。
(要改变构型,必须经过化学键的断裂和重组。
)高分子链的构型有旋光异构和几何异构两种类型。
旋光异构是由于主链中的不对称碳原子形成的,有全同、间同和无规三种不同的异构体(其中,高聚物中全同立构和间同立构的总的百分数称为等规度。
)。
全同(或等规)立构:取代基全部处于主链平面的一侧或者说高分子全部由一种旋光异构单元键接而成间同立构:取代基相间地分布于主链平面的两侧或者说两种旋光异构单元交替键接无规立构:取代基在平面两侧作不规则分布或者说两种旋光异构单元完全无规键接几何异构是由于主链中存在双键而形成的,有顺式和反式两种异构体。
构象:原子或原子基团围绕单键内旋转而产生的空间分布。
链段:把若干个键组成的一段链作为一个独立运动的单元链节(又称为重复单元):聚合物中组成和结构相同的最小单位高分子可以分为线性、支化和交联三种类型。
其中支化高分子的性质与线性高分子相似,可以溶解,加热可以熔化。
但由于支化破坏了高分子链的规整性,其结晶能力大大降低,因此支化高分子的结晶度、密度、熔点、硬度和拉伸强度等,都较相应的线性高分子的低。
交联高分子是指高分子链之间通过化学键形成的三维空间网络结构,交联高分子不能溶解,只能溶胀,加热也不能熔融。
高分子链的构象就是由单键内旋转而形成的分子在空间的不同形态。
单键的内旋转是导致高分子链呈卷曲构象的根本原因,内旋转越自由,卷曲的趋势就越大。

聚合物等温结晶过程的计算机模拟第一章 绪论1.1计算机模拟实验技术的优势计算机模拟实验在一定程度上可以缩短各领域科学技术实验的周期,它对于实际实验的协助程度主要依赖于对实验过程的了解程度(建模的准确性)和计算复杂度(受限于计算机的计算速度)。
理论上,如果确保了模型的准确性,那么计算机模拟实验可以弥补实际实验的一些不足,这一优势已经引起越来越多的关注。
1.2计算机模拟技术在聚合物结晶过程中的运用某些领域,譬如网络仿真和电路仿真等,由于模型结构建立的非常完善所以得以实现。
本文所讨论的内容是计算机模拟技术在聚合物结晶过程中的运用,自从Hay JN 和Przekop ZJ [1]通过结晶过程的计算机模拟实验对Avrami 方程进行评价以来,计算机模拟技术已经成为评估该类模型的有力工具。
Galeski A [2-3]通过模拟二维和三维的球晶生长,获得了不同成核方式下Avrami 指数与球晶的大小分布和形态。
Billon N [4]等人从Evans 理论导出了一个描述聚合物薄膜等温结晶过程的模型,并开发了模拟结晶过程的计算机程序用于对模型的测试。
Pineda [5]等人检测了成核和生长速率的降低以及晶核分布的非无规性对Avrami 结晶动力学过程的影响。
Piorkowska [6]对纤维增强复合材料的结晶过程进行了模拟,以验证导出的表达式和结晶形态。
正是通过学者专家们的不断研究,聚合物结晶过程模型结构体系得以逐步完善。
时至今日,计算机模拟实验在聚合物结晶动力学理论和模型验证及新发现方面发挥着重要作用.1.3高聚物等温结晶动力学的现状(1)考虑结晶后期球晶的相互挤撞一级增长动力学模型周卫华[7]等人用一级增长动力学模型描述高聚物的结晶动力学过程,即()αα-⋅⋅=1S K dtd (1) 式中,K 是不依赖于温度的常数,与结晶体的线生长速率成正比;S 是结晶体的总表面积。
该模型认为,二次结晶阶段由于结晶体相互挤撞使可供晶体生长的总表面积减少,从而导致Avrami 方程与实验数据发生偏离。

高分子物理考试重点一、名词解释:等效自由连接链:若干个键组成的一链段算作一个独立的单元,称之为“链段”,链段间自由结合,无规取向,这种链的均方末端距与自由连接链的计算方式等效。
高分子θ溶液:Avrami 方程: 用数学方程描述聚合物等温结晶过程。
测定结晶度随时间的变化,这种方法测定的是结晶总速率(包括成核速率和生长速率)。
通常用膨胀计法,由于结晶时有序排列而体积收缩,若比容在时间为0,t 和∞时分别为V 0,V t 和V ∞,则结晶过程可用Avrami 方程描述:(V t -V ∞)/(V 0-V ∞)=()t n k W o W L -=exp 通过双对数作图,从斜率求n ,从截距求k ,n 称Avrami 指数,n=生长的空间维数+时间维数,异相成核的时间为0,均相成核为1,。
k 用来表征结晶速率,k 越大,结晶速率越快。
平衡熔点:熵弹性: 理想高弹性等温形变过程,只引起熵变,对内部保持不变,即只有熵的变化对理想高弹性的弹性有贡献,这种弹性称为熵弹性。
粘弹性:是材料对外界作用力的不同响应情况。
对于聚合物,其力学性质可同时兼有不可回复的永久形变和可回复的弹性形变,介于理想弹性体和理想粘性体之间,形变与时间有关,但不是线性关系。
此性质就是粘弹性。
力学损耗: 聚合物在应力作用下,形变的变化落后于应力的变化,发生滞后现象,每一个循环变化中就要消耗功,这个功就是力学损耗。
滞后现象: 一定温度与循环(交变)应力作用下,试样应变滞后于应力变化的现象。
Boltzmann 叠加原理:对于聚合物材料的蠕变过程,形变是整个负荷历史的函数,每一次阶跃式加负荷对以后应变的贡献是独立的,最终形变等于各个所加负荷所贡献的形变的加和。
时温等效原理:升高温度和延长观察时间对分子运动是等效的,对于聚合物的粘弹性行为也是等效的。
这种等效性即被称为时温等效原理。
构型:是指分子中由化学键所固定的原子在空间的排列。
构象:由于σ单键内旋转而产生的分子在空间的不同形态。


高分子结晶和熔融的过程和小分子有哪些不同一、结晶过程1、透明度(透明的原因就是光在介质中不发生折射、反射。
完全结晶和完全非晶的物质,由于其密度和折射率完全均匀、一致,没有光的折射,所以往往透明。
)小分子是完全晶体,透明(图1);高聚物不可以完全结晶,由于晶区和非晶并存,晶区和非晶区折射率不同,光产生双折射被吸收,所以一般不是很透明(图2)。
完全非晶的则透明,比如有机玻璃(图3)。
其次小分子结晶直径小于高分子,光线通过更有利。
2、高聚物是否都能结晶?聚合物按其能否结晶可以分为两大类:结晶性聚合物和非结晶性聚合物(图4)。
后者是在任何条件下都不能结晶的聚合物,而前者是在一定条件下能结晶的聚合物,即结晶性聚合物可处于晶态,也可以处于非晶态。
聚合物结晶能力和结晶速度的差别的根本原因是不同的高分子具有不同的结构特征,而这些结构特征中能不能和容易不容易规整排列形成高度有序的晶格是关键。
主链含有不对称碳原子的分子链,如果具有空间构型的规整性,则仍然可以结晶,否则就不能结晶。
3、结晶速率的不同,由于高分子的分子量大,分子链长,分子链间的相互作用大,导致高分子链的运动比小分子困难,尤其是对刚性分子链或带庞大侧基的、空间位阻大的分子链,所以,高分子的结晶速度一般比小分子慢。
复习:影响高分子结晶速率的因素:(1)分子链结构(2)相对分子质量(3)温度(4)小分子液体(5)外力(6)结晶成核剂4、结晶形态晶系(图5):立方、四方。
斜方、单斜、三斜、六方、三方高聚物结晶中没有立方晶系。
30%是斜方,30%是单斜高分子的结晶形态(图6):(1)单晶(小分子或是稀溶液、刚性聚合物缓慢结晶)(2)球晶(3)树枝晶(4)串晶和纤维状晶(5)伸直链片晶5、构成晶体的基本质点小分子:原子、分子、离子。
它们晶胞中的排列是相互分离的高分子:大分子长链沿c轴排列,在c轴方向上是连续的。
在c轴方向上分子链构象重复出现的“分子链段”为基本单元6、二次结晶小分子:①成核、②增长高分子:①成核(慢)、②主期结晶(快)、③二次结晶(慢)(图7)二、熔融过程相同点:都是热力学一级相变的过程(图8 P29 第四PPT的两图)小分子:熔融过程中体系的热力学函数随温度变化围很窄,只有0.2℃左右聚合物:呈现一个较宽的熔融温度围,即存在一个“熔限”结晶聚合物熔融时出现熔限的原因:聚合物结晶形态和完善程度不同(1)在结晶过程中,随温度的降低,溶体黏度迅速增加,分子链的活动性减小,在砌入晶格时来不及做充分的位置调整,使形成的晶体停留在不同的阶段(2)在熔融过程中,尺寸较小,比较不完善的晶体将在较低的温度下熔融,尺寸较大的、较完善的晶体需要在较高的温度下熔融,因而出现一个“熔限”。
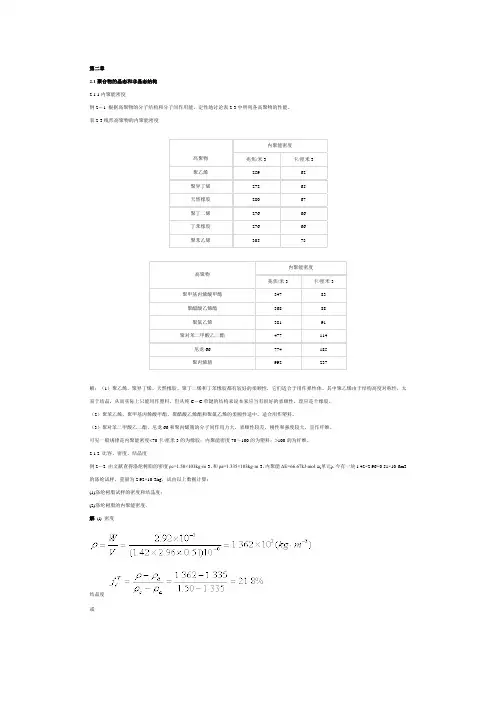
第二章2.1聚合物的晶态和非晶态结构2.1.1内聚能密度例2-1 根据高聚物的分子结构和分子间作用能,定性地讨论表2-3中所列各高聚物的性能。
表2-3线形高聚物的内聚能密度高聚物内聚能密度兆焦/米3 卡/厘米3聚乙烯259 62聚异丁烯272 65天然橡胶280 67聚丁二烯276 66丁苯橡胶276 66聚苯乙烯305 73高聚物内聚能密度兆焦/米3 卡/厘米3聚甲基丙烯酸甲酯347 83聚醋酸乙烯酯368 88聚氯乙烯381 91聚对苯二甲酸乙二酯477 114尼龙66 774 185聚丙烯腈992 237解:(1)聚乙烯、聚异丁烯、天然橡胶、聚丁二烯和丁苯橡胶都有较好的柔顺性,它们适合于用作弹性体。
其中聚乙烯由于结构高度对称性,太易于结晶,从而实际上只能用作塑料,但从纯C-C单键的结构来说本来应当有很好的柔顺性,理应是个橡胶。
(2)聚苯乙烯、聚甲基丙烯酸甲酯、聚醋酸乙烯酯和聚氯乙烯的柔顺性适中,适合用作塑料。
(3)聚对苯二甲酸乙二酯、尼龙66和聚丙烯腈的分子间作用力大,柔顺性较差,刚性和强度较大,宜作纤维。
可见一般规律是内聚能密度<70卡/厘米3的为橡胶;内聚能密度70~100的为塑料;>100的为纤维。
2.1.2 比容、密度、结晶度例2-2 由文献查得涤纶树脂的密度ρc=1.50×103kg·m-3,和ρa=1.335×103kg·m-3,内聚能ΔΕ=66.67kJ·mol-1(单元).今有一块1.42×2.96×0.51×10-6m3的涤纶试样,重量为2.92×10-3kg,试由以上数据计算:(1)涤纶树脂试样的密度和结晶度;(2)涤纶树脂的内聚能密度.解(l) 密度结晶度或(2) 内聚能密度文献值CED=476(J·cm-3)例2-3 试从等规聚丙烯结晶(α型)的晶胞参数出发,计算完全结晶聚丙烯的比容和密度。



聚合物的结晶度的名词解释聚合物是由许多重复单元组成的大分子化合物,它们通常具有高度的分子量和复杂的结构。
结晶度是聚合物中具有规律有序排列的部分的比例,是衡量聚合物结晶程度的重要参数。
1. 聚合物的结晶过程聚合物的结晶过程可以分为两个阶段:核化和生长。
在核化阶段,小分子或聚合物链段聚集形成核心结构,这些核心结构在接下来的生长阶段发展成为晶粒。
结晶的速度受到溶剂、温度和浓度等因素的影响。
2. 结晶的类型根据聚合物分子排列的有序性程度,结晶可以分为完全结晶、部分结晶和无结晶三类。
完全结晶聚合物具有高度有序分子排列,形成紧密堆积的晶体结构。
例如,高密度聚乙烯(HDPE)和聚苯乙烯(PS)。
部分结晶聚合物中,只有一部分聚合物链可以形成结晶区域,其余部分仍然为无序状态。
这种结构常见于低密度聚乙烯(LDPE)和聚丙烯(PP)。
无结晶聚合物则完全没有有序的结晶结构,整个聚合物呈无规则状态。
例如,天然橡胶和软质聚氯乙烯(PVC)。
3. 结晶度的影响因素结晶度受到多种因素的影响。
首先,分子链的长度对结晶度起着关键作用。
较长的分子链使得形成有序结构的机会更多,因此结晶度更高。
其次,溶剂的选择也会影响结晶度。
合适的溶剂能够促进分子链的有序排列,从而增加结晶度。
此外,温度和降温速率也会对结晶度产生影响。
较高的温度和较慢的降温速率有利于结晶的形成。
4. 结晶度的测量方法常用的结晶度测量方法有热分析法、X射线衍射法和差示扫描量热法。
热分析法通过测量聚合物熔点和熔融热来计算结晶度。
X射线衍射法利用X射线通过晶体结构产生的衍射图案来测量结晶度。
差示扫描量热法则通过测量聚合物在加热和冷却过程中的热量差来确定结晶度。
5. 结晶度的意义和应用结晶度对聚合物的性能和应用具有重要影响。
结晶度高的聚合物通常具有较高的力学性能和热稳定性,适用于制造强度要求高的产品,如塑料零件、纤维和薄膜等。
例如,高结晶度的聚乙烯被广泛用于制作各种塑料袋。
相反,结晶度低的聚合物具有较好的柔韧性和可加工性,适用于制备拉伸性和弯曲性要求较高的产品。


第二章高分子的链结构1.聚合物的层次结构聚合物的结构包括高分子的链结构和聚合物的凝聚态结构,高分子的链结构包括近程结构一级结构和远程结构二级结构;一级结构包括化学组成,结构单元连接方式,构型,支化于交联;二级结构包括高分子链大小相对分子质量,均方末端距,均方半径和分子链形态构象,柔顺性;三级结构属于凝聚态结构,包括晶态结构,非晶态结构,取向态结构,液晶态结构和织态结构; 2.结构单元的键接方式,许多实验证明自由基或离子型聚合产物中大多数是头—尾键接的,链接方式对聚合物的性能有比较明显的影响;例1:纤维要求分子链中单体单元排列规整,结晶性能好,强度高,便于抽丝和拉伸例2:维尼纶纤维缩水性较大的根本原因:聚乙烯醇PVA做维尼纶只有头—尾键接才能使之与甲醛缩合生成聚乙烯醇缩甲醛;如果是头—头键接额,羟基就不易缩醛化,是产物中保留一部分羟基,羟基的数量太多会使纤维的强度下降;3.聚合物的空间构型概念:结构单元为—CH2—CHR—型的高分子,在每一个结构单元中都有一个手性碳原子,这样,每一个链节就有两种旋光异构体,高分子全部由一种旋光异构体键接而成称为全同立构,由两种旋光异构单元交替键接,称为间同立构,两种旋光异构单元完全无规键接时,则称为无规立构全同立构和间同立构的高聚物有时统称为等规高聚物高聚物中含有全同立构和间同立构的总的百分数是指等规度由于内双键的基团在双键两侧排列的方向不同而有顺式构型与反式构型之分,他们称为几何异构体例:几何构型对聚合物的影响聚丁二烯1,2-加成的全同立构或间同立构的聚丁二烯PB,由于结构规整,容易结晶,弹性很差,只能作为塑料使用;顺式1,4-聚丁二烯链的结构也比较规整,容易结晶,在室温下是一种弹性很好的橡胶,反式1,4-聚丁二烯分子链的结构也比较规整,容易结晶,在室温下是弹性很差的塑料;4. 高分子共聚物共聚物的序列结构常用参数平均序列长度L和嵌段数R;当R=100时表明是交替共聚,R=0时表明是嵌段共聚物例1:聚甲基丙烯酸甲酯PMMA分子带有极性酯基是分子间作用力比聚苯乙稀PS大,因此在高温的流动性差,不宜采取注塑成型法加工;需将PMMA和少量PS共聚可以改善树脂的高温流动性,适用于注塑成型ps. 和少量的丙烯晴AN共聚后,其冲击强度,耐热性,耐化学腐蚀性都有所提高,可供制造耐油的机械零件例2:ABS树脂在结构组成制备工艺上可提高产品的力学性能的方法ABS树脂是丙烯晴,丁二烯和苯乙烯的三元共聚物;其中丙烯晴有CN基,能使聚合物耐化学腐蚀,提高制品的抗张强度和硬度;丁二烯使聚合物呈现橡胶状韧性,这是材料抗冲击强度增高的主要因素;苯乙烯的高分流动性能好,便与加工成型,而且可以改善制品表面光洁度.,ps. ABS是一类性能优良的热塑性塑料例3:SBS在结构和组成上的特点及加工方法概述用阴离子聚合法制得的苯乙烯与丁二烯的嵌段共聚物SBS树脂;丁二烯常温是一种橡胶,而聚苯乙烯是硬性塑料,两者不相容,因此SBS具有两项结构;聚丁二烯段形成连续的橡胶相,聚苯乙烯是热塑性的,聚苯乙烯起交联作用高温下可以破坏也可以重组,所以SBS是一种可以注塑方法进行加工而不需要硫化的橡胶;聚氨酯与其相似,统称热塑性弹性体;5.高分子链的支化例:为什么高压聚乙烯的冲击强度好于低压聚乙烯的冲击强度支化对物理性能的影响有时相当显著,高压聚乙烯低密度聚乙烯LDPE由于支化破坏了分子的规整性,使其结晶度大大降低,低压聚乙烯高密度聚乙烯HDPE是线型分子,易于结晶,故在密度,熔点,结晶度和硬度方面都高于强者;分子链支化程度增加,分子间的距离增加,分子间的作用力减小,因而使拉伸强度降低,但冲击强度会提高;6.高分子链的交联支化高分子能够溶解,交联高分子不熔不熔,只有交联度不大的时候能在溶剂中溶胀;热固性塑料和硫化橡胶都是交联高分子例:硫化橡胶未经硫化交联的橡胶分子之间容易滑动,受力后会产生永久变形,不能回复原状,经硫化的橡胶分子间不能滑移,才有大的可逆弹性变形,所以橡胶一定要经过硫化变成交联结构后才能使用;交联度小的橡胶含硫5%一下弹性较好,交联度大的橡胶含硫20%~30%弹性就差,交联度再增加,机械强度和硬度都将增加,最终失去弹性而变脆;7.高分子链的构象概念:构象:单间内旋转而产生的分子在空间的不同排列形态,由于热运动分子的构象在时刻改变,因此高分子的键的构象是统计性的,由此可知,这种构象是不固定的;构型:大分子链中由化学键所固定的原子在空间的几何排列,这种排列是稳定的要改变构型必经过化学键的断裂和重组;构型包括单体单元的键合顺序,空间构型的规整性,支化度,交联度以及共聚物的组成及序列结构;无规线团:单键内旋转是导致分子链呈蜷曲构象的原因,内旋转愈自由,蜷曲的趋势越大,我们称这种不规则的蜷曲高分子链的构象为无规线团;理想链理想柔性链,自由链接链:高分子键的一种理想化的简单模型,假定高分子的主链由足够多的不占体积的化学键自有链接而成,这些键的取向不受键角以及相邻旋转交的限制,没有位垒的障碍,在空间上的取向几率都相等;自由旋转链:每个链都能在键角限制范围内自由旋转,不考虑空间位阻影响,有足够多的不占体积的化学键自有链接而成,这些键的取向受键角及相邻旋转交的限制,没有位垒障碍;受阻旋转链:同自由旋转链,除不能自由旋转;末端距:对于线性高分子,分子链的一端至另一端的直线距离即为末端距;均方末端距:末端距的平方的平均值,通常用来表征高分子链的尺寸;高斯链:把真实的高分子末端距模型化的一种由n个长度为l的统计单元组成,他的末端距大小分布符合高斯统计函数,这种假想链叫做高斯链Ps.末端距的计算见附录例1. 自由连接链和高斯链的区别1.高斯链的统计单元为链段,自由链接链的链接单元为化学键2.高斯链可以产生链段的回转和取向,自有链接连不能产生化学链的旋转和取向3.高斯链是实际存在的,自有链接连是不存在的4.高斯链研究高分子链的共性,自有链接链是理想化的;例2.聚丙烯是否可以通过单键的内旋转由全同立构变成间同立构,为什么答:不可以;因为全同立构和间同立构是属于构型的范畴,构型是指分子中有化学键所固定的原子在空间的排列;单键的内旋转只会改变构象,而改变构型必须经过化学键的断裂才能实现;例3.为什么只有柔性高分子链才适合做橡胶答:橡胶具有高弹性,弹性模量很小,形变量很大的特点;只有处于蜷曲状态的长链分子才能在外力的作用下产生大形变,才能作为橡胶;蜷曲程度与柔性是相对应的,蜷曲程度越高,柔性越好,所以适合做香蕉的高分子必须具备相当程度的柔性;例4.试述近程相互作用和远程相互作用的含义以及它们对高分子链构象以及柔性的影响答:所谓“近程”和“远程”是根据沿大分子链的走向来区分的,并非为三维空间上的远和近;事实上,即使是沿高分子长链很远的枝节也会由于主链单间内旋转而在三维空间上相互靠的很近;近程相互排斥作用的存在使得实际高分子的内旋转受阻,是指在空间可能有的构象数远远小于自由内旋转的情况,受阻程度越大构象数就越少,高分子链的柔性就越小;远程相互作用可为斥力,也可称为引力;当大分子链中相距较远的原子或原子团由于单键的内旋转,可是其间的距离小于范德瓦尔斯半径而表现为斥力,大于范德瓦尔斯半径为引力,五轮哪种力都使单间内旋转受阻构象数减小,柔性下降,末端距变大;例5. 分子链柔顺性大小顺序聚乙烯PE,聚丙烯PP,聚丙烯晴PAN,聚氯乙烯PVC取代基极性越大,取代基之间的相互作用就越强,高分子链内旋转越困难,柔性越小;取代基的极性顺序为—CN>—CL—CH3—H,所以PE>PP>PVC>PAN例6.请排出分子间作用力的大小聚苯乙烯,聚对苯二甲酸乙二酯和尼龙66,聚乙烯尼龙66>据对苯甲酸乙二酯>聚苯乙烯>聚乙烯尼龙66分子间能形成氢键,因此分子间作用力最大;聚对苯二甲酸乙二酯含有强极性基团,分子间作用力比较大,聚苯乙烯含有侧基,连段运动较困难,分子间作用力较小,聚乙烯是非极性分子,又不含侧基,分子间作用力最小;例7. 请排出结晶难易程度的排序1聚对苯二甲酸乙二酯和聚间苯二甲酸乙二酯,聚乙二酸乙二酯2尼龙66,尼龙1010聚己二酸乙二酯>聚对苯二甲酸乙二酯>聚间苯二甲酸乙二酯,这是由于聚己二酸乙二酯的柔顺性好,聚间苯二甲酸乙二酯对称性不高,尼龙66>尼龙1010尼龙66中氢键密度大于尼龙1010第三章高分子溶液1.聚合物溶解过程和溶剂选择概念:内聚能密度:内聚能是将一摩尔液体或固体分子汽化时所需要的能量,单位体积的内聚能即为内聚能密度;δ溶度参数:溶度参数是内聚能密度的平方根;溶质与溶剂的溶度参数越接近越可能相互溶解;冻胶:是由范德瓦尓斯力交联而成的,加热可以拆散范德瓦尓斯力的交联,使冻胶溶解;凝胶:是高分子链之间以化学键形成的交联结构的溶胀体;例1.聚合物的溶解过程答:聚合物的溶解过程分为两个阶段,先是溶剂分子深入聚合物内部,是聚合物体积膨胀,称为溶胀,然后才是高分子均匀分散在溶剂中形成完全溶解的分子分散的均相体系,对于交联聚合物,在与溶剂接触时也会发生溶胀,但因有交联的化学键束缚,不能再进一步使交联分子拆散,只能停留在溶胀阶段,不会溶解;例2.聚合物的溶解度与分子量的关系:溶解度与聚合物的分子量有关,分子量大的溶解度小,分子量小的溶解度大,对交联聚合物来说,交联度大的溶胀度小,交联度小的溶胀度大;例3.非晶聚合物和结晶聚合物对溶解的影响非晶聚合物的分子堆砌比较松散,分子间的相互作用较弱,因此溶剂分子比较容易渗入聚合物内部使之溶胀和溶解;静态聚合物由于分析排列规整,堆砌紧密,分子间相互作用力很强,以致溶剂分子深入聚合物内部非常困难,因此晶态聚合物的溶解比非晶态聚合物困难得多;溶液的热力学性质溶解过程的自发需要满足△Fm=△Hm-T△Sm<0对于极性聚合物在极性溶剂中,由于高分子溶剂强烈相互作用,分子排列趋于混乱所以△Sm增加溶解时放热△Hm<0且使体系△Fm降低所以溶解过程能自发进行非极性聚合物,溶解过程一般是吸热的△Hm>0,故只有在升高温度T或者减小混合热△Hm才能使体系自发溶解;非极性溶液的混合热△Hm的大小取决于溶度参数,如果两种液体溶度参数接近,则混合热越小,两种液体越能互相溶解;Ps.聚丙烯腈不能溶解在溶度参数与他相接近的乙醇,甲醇,苯酚;乙二醇等溶剂中,这是因为这些溶剂的极性太弱了,只有二甲基甲酰胺,二甲基乙酰胺,乙腈,二甲基亚砜,丙二腈才能使其溶解;丙酮不能溶解聚苯乙烯是丙酮极性太强而聚苯乙烯是弱极性的;可以得出结论,极性聚合物,不但要求它与溶剂的溶度参数中的非极性部分接近,还要求极性部分也接近才能溶解;注:如果溶质与溶剂间能形成结晶性非极性聚合物的溶剂选择最困难,它的溶解包括两个过程:其一是结晶部分的熔融;其二是高分子与溶剂的混合,两者都是吸热的过程,所以要提高温度;除非生成氢键,因为氢键的生成是放热反应;例1.溶剂的选择原则:1)极性相近,要求溶剂的极性和高聚物极性相近,极性高聚物选择极性相当的溶剂;2)溶度参数相近原则,参数越接近,溶解可能性越大,非晶态—非极性比较合适,对于晶态的非极性高聚物需加外界条件,对晶态极性不适用;3)溶剂化原则基团的相互作用溶剂分子与高分子链之间相互吸引作用是高分子链与链之间相互分离导致高分子溶解于溶剂形成溶液;理想溶液概念:理想溶液:是指溶液中溶质分子间溶剂分子间和溶质分子间的相互作用能都相等,溶解过程没有体积变化也没有焓的变化;Huggins参数:是表示高分子溶液混合时相互作用能的变化θ温度:是高分子溶液的一个参数,当T=θ时高分子溶液中的过量化学位为零,与理想溶液中溶剂的化学位没有偏差θ条件:通过选择溶剂和温度使高分子溶液中溶剂的过量化学位为零的条件,这种条件称为θ条件或θ状态;无扰状态:高分子在稀溶液中,一个高分子很难进入另一个高分子所占的区域,即每个高分子都有一个排斥体积;如果高分子链段和溶剂分子相互作用能大于高分子链段与高分子链段的相互作用能,则高分子被溶剂化而扩张,使高分子不能彼此接近,高分子的排斥体积就很大;如果高分子链段与溶剂分子相互作用能等于高分子链段与高分子链段的相互作用能;高分子与高分子可以与溶剂分子一样彼此接近,互相贯穿,这样排斥体积为零,相当于高分子处于无扰状态;这种状态的尺寸就称为无扰尺寸;扩张因子:高分子在良溶剂中,由于溶剂化的作用,是卷曲的高分子链伸展,高分子的均方末端距和均方旋转半径扩大;扩张因子α是指高分子链的均方末端距或均方旋转半径与高分子链在θ状态下的均方末端距或均方旋转半径之比,它表示高分子链的扩张程度;溶胀比:交联高聚物在溶胀平衡时的体积与溶胀前的体积之比例1. 根据高分子的混合自由能,推导出其中溶剂的化学位变化,并讨论在什么条件下高分子溶液中溶剂的化学位变化,等与理想溶液中溶剂的化学位变化答:见附录例2. 高分子溶液在什么情况下与理想溶液的一些热力学性质相近当T=θ时;高分子溶液中溶剂的过量化学位为零;χ1=1/2,高分子处于θ状态,此时高分子溶液与理想溶液的一些热力学性质相近;例3. 什么是θ温度当高于,低于或等于θ温度时,大分子的自然构象有何不同为什么θ温度是高分子溶液的一个参数;当T=θ时,高分子溶液中溶剂△μ=0与理想溶液中的溶剂化学位没有偏差;当T>θ时,溶剂为高分子良溶剂,在良溶剂中,高分子链由于溶剂化而扩张,高分子线团伸展,当T<θ时,溶剂为高分子的不良溶剂,在不良溶剂中,高分子链由于溶剂化作用很弱,高分子链紧缩;当T=θ时,溶剂为高分子的θ溶剂,在θ溶剂中,高分子链段与高分子链段的相互作用能等于高分子链段与溶剂的相互作用能,高分子与高分子可以与溶剂分子一样彼此接近,互相贯穿,这样高分子链的排斥体积为零,相当与高分子链处于无干扰的无规线团;例4.试举出可判定聚合物溶解性好坏的三种热力学参数,并讨论当它们分别为何值时,溶剂是良溶剂,θ溶剂,劣溶剂:过量化学位△μ₁,Huggins参数χ₁,第二维利系数A₂可以判定聚合物溶解性的好坏的三种热力学参数,△μ₁<0,χ₁<1/2,A₂>0时为良溶剂;△μ₁=0,χ₁=1/2,,A₂=0时为θ溶剂;μ₁>0,χ₁>1/2,A₂<0时为劣溶剂;Ps.θ状态与真正的理想溶液还是有区别的,真正的理想溶液没有热效应,任何温度下都呈现理想行为,而在θ温度时的高分子稀溶液只是过量化学位等于0而已;偏摩尔混合热和偏摩尔混合熵都不是理想值,只是两者的非理想效应近似相互抵消;例5.临界共溶温度:是聚合物溶解曲线极大处的温度就是Tc;溶质的分子量越大,溶液的临界共溶温度越高;当温度降至Tc一下某一定值时,就会分离成稀相和浓相,当体系分成两相最终达到相平衡时,每种组分在两相间扩散达到动态平衡,这就要求每种组分在两相间的化学未达到相等;相分离的起始点就是临界点,在临界点,两个相浓度相等;简述荣章法测定聚合物的δ的原理和方法溶胀法可以测定交联聚合物的平衡溶胀比,及交联聚合物达到溶胀平衡时的体积与溶胀前的体积之比;若交联聚合物与溶剂的溶度参数越接近,高分子与溶剂的相互作用愈大,及高分子溶剂化程度愈大,交联网链愈能充分伸展,是交联聚合物的平衡溶胀比增大,若用若干种不同溶度参数的溶剂溶胀聚合物,用溶胀法分别测定聚合物在这些溶剂中的平衡溶胀比,以平衡溶胀比对溶剂的溶度参数作图,找出平衡溶胀比极大值所对应的溶度参数,此溶度参数可作为交联聚合物的溶度参数;Ps.增塑剂为了改善聚合物材料的成型加工性能和使用性能,通常在聚合物树脂中加入高沸点,低挥发性的小分子液体或低沸点固体,以降低玻璃化转变温度和粘流温度,改善树脂流动性,降低粘度石制品有较好的柔韧性,和耐寒性;第四章高分子的多组分体系高分子的相容性概念高温临界共溶温度UCST:高温互容低温分相;低温临界共溶温度LCST:低温互容高温分相;曲线分析见附录临界胶束浓度:将嵌段共聚物溶解在小分子溶剂中,如果溶剂溶解共聚物前段时没有很强的选择性,那么嵌段共聚物的溶液性质与一般均聚物的溶液性质没有和大的差别;但如果溶剂对其中的某一嵌段具有很强的相互吸引作用,在固定温度改变浓度或固定浓度改变温度两种条件下,嵌段共聚物类似于小分子的表面活性剂,与溶剂作用强的嵌段倾向于与溶剂混合,而另一嵌段就倾向于与其它链的相似嵌段聚集在一起,形成胶束,形成胶束的临界条件被称为临界胶束浓度,和临界胶束温度;进一步增加浓度,这些胶束逐渐发生交叠,形成物理凝胶几乎不能流动,形成凝胶的临界浓度称为临界胶束浓度静态光散射通过测定溶液中形成结构的平均分子量来估算是否形成了胶束Ps.UCST,LCST曲线见附录第五章聚合物的非晶态非晶态聚合物的结构模型概念无规线团模型:在非晶态聚合物本体中,分子链的构象与在溶液中的一样,成无规线团状,线团的尺寸在θ状态下高分子的尺寸相当,线团分子之间是任意相互贯穿和无规缠结的,前端的堆砌不存在任何有序的结构,因而非晶态聚合物在凝聚态结构上是均相的;玻璃化转变:玻璃态和高弹态之间的转变称为玻璃化转变,对应的转变温度即玻璃化转变温度;玻璃态:当非晶聚合物在较低的温度下受外力时,有与链段运动被冻结,只能使主链的键长和键角有微小的改变,因此从宏观上来说,聚合物形变是很小的,形变与受力的大小成正比,当外力除去后,形变能立刻回复;这种力学性质称虎克型弹性体,又称普弹体,非晶态聚合物处于具有普弹性的状态,称为玻璃态;玻璃化温度:高聚物分子链开始运动或冻结的温度;它是非晶态高聚物作为塑料使用的最高温度,橡胶使用的最低温度;高弹态:在聚合物受到外力时,分子链可以通过单键的内旋转和链段的改变构象以适应外力的作用,由于这种变形是外力作用促使聚合物主链发生内旋转的过程,它需要的外力显然比聚合物在玻璃态时变形所需外力要小得多,而变形量却大得多,这种性质叫做高弹性,它是非晶态聚合物处在高弹态下特有的力学特征;粘流态:整个分子链运动,松弛时间缩短,在外力作用下发生粘性流动,它是整个分子链互相滑动的宏观表现;形变不可逆外力除去后,形变不能再自发回复自由体积理论:Fox和Flory提出,认为液体或固体物质,其体积由两部分组成:一部分是被分子占据的体积;另一部分是未被占据的自由体积;后者以“孔穴”的形式分散于整个物质之中,正是由于自由体积的存在,分子链才可能发生运动;自由体积理论认为,当聚合物冷却时,起先自由体积逐渐减少,到某一温度时,自由体积达到一最低值,这是聚合物进入玻璃态;在玻璃态下,有与链段运动被冻结,自由体积也被冻结,并保持一恒定值,自由体积“孔穴”的大小及分布也将基本上维持固定;因此对任何聚合物,玻璃化温度就是自由体积达到某一临界值的温度,在这临界值一下,已经没有足够的空间进行分子链构象的调整了;因而聚合物的玻璃态可视为等自由体积状态;不管什么聚合物,发生玻璃化转变时,自由体积分数都等于2.5%;Ps. WLF方程见附录例1::无规线团模型的实验证据1.橡胶的弹性理论完全是建立在无规线团模型基础上的,而且实验证明,橡胶的弹性模量和应力-温度系数关系并不随稀释剂的加入而有反常的改变,说明在非晶态下,分子链是完全无序的,并不存在可被进一步溶解或拆散的局部有序结构2.在非晶聚合物的本体和溶液中,分别用高能辐射是高分子发生交联,实验结果并未发现本体体系中发生分子内教练的倾向比溶液中更大,说明本体中并不存在诸如紧缩的线团或折叠连那些局部有序的结构;3用X光小角散射的实验结果,提别有力的支持了无规线团;.对于分子量相同的聚甲基丙烯酸甲酯试样,用不同的方法光散射,X光散射和中子散射,不同条件下本体或溶液中,测得分子的回转半径相近;并且本体的数据与θ溶剂氯代正丁烷的数据以及所得指向的斜率更为一致,证明非晶态本体中,分子的形态与它在θ溶剂中一样,它们的尺寸都是无扰尺寸例2.两相球粒模型1模型包含了一个无序的粒间相,从而能为橡胶弹性变形的回缩力提供必要的构象熵,因而可以解释橡胶的弹性回缩力;2实验测得许多聚合物的非晶和结晶密度比按分子链成无规线团形态的完全无序的模型计算的密度高,说明有序的粒子相与无序的粒间相并存,两相中由于嵌段的堆砌情况有差别,导致了密度的差别;3模型例子中嵌段的有序堆砌,为洁净的迅速发展准备了条件,这就不难解释许多聚合物结晶速度很快的事实;4某些非晶态聚合物缓慢冷却或热处理后密度增加,电镜下还观察到球粒的增大,这可以用粒子相有序程度的增加和粒子相的扩大来解释;例3.非晶态聚合物形变-温度曲线如果取一块非晶聚合物试样,对它施加一恒定的力,观察试样发生的形变与温度的关系,我们将所得到的曲线称为形变-温度曲线或热机械曲线;当温度较低时,试样呈刚性固体状,在外力作用下只发生非常小的形变;温度升到某一范围后,式样的形变明显的增加,并随后,并在随后的温度区间达到一相对稳定的形变,在这一个区域中,试样变成柔软的弹性体,温度继续升高,形变基本上保持不变;温度再进一步升高,则形变量又逐渐加大,试样最后完全变成粘性流体; Ps.形变温度曲线见附录例4.试用分子运动的观点说明非晶聚合物的三种力学状态和两种转变在玻璃态下,由于温度较低,分子运动的能量很低,不足以克服主链内旋转的位垒,因此不足以激发起链段的运动,链段处于被冻结的状态,只有那些较小的运动单元,如侧基,支链和小链节能运动,当收到外力时,由于链段处于冻结状态,只能使主链的键长和键角有微小的改变,形变很小,当外力除去后形变能立刻回复;随着温度的升高,分子热运动的能量增加,当达到某一温度Tg时,链段运动被激发,聚合物进入高弹态,在高弹态下,链段可以通过单键的内旋转和链段的运动不断地改变构象,但整个分子仍然不能运动;当受到外力时,分子链可以从蜷曲状态变为伸直状态,因而可发生较大形变;温度继续升高,整个分子链也开始运动,聚合物进入粘流态,这时高聚物在外力作用下便发生粘性流动,它是整个分子链互相滑动的宏观表现,外力去除后,形变不能自发回复;玻璃化转变就是链段有运动到冻结的转变,流动转变使整个分子链由冻结到运动的转变;例5.为什么聚合物通常有一份相对确定的玻璃化温度,却没有一个确定的粘流温度随着相对分子量的增加,玻璃化温度会升高,特别是在较低的相对分子质量范围内,这种影响较为明显,但是当相对分子质量增加到一定程度后,玻璃化温度随着相对分子质量的变化很小;而聚合物的粘流温度是整个分子链开始运动的温度,相对分子质量对粘流温度的影响比较明。
高聚物结晶的原理高聚物结晶是指高分子化合物在适当的条件下从溶液或熔融态转变为固态结晶态的过程。
结晶是高聚物在空间有序的方式排列成晶体,使高聚物有了规则的晶格结构和有序的长程排列,从而具有更好的物理和化学性质。
高聚物结晶的原理可以分为两个方面,即核心形成与晶体生长。
首先,核心形成是高聚物结晶的起始过程。
高聚物在溶液中或熔融态时,分子经常发生热运动,分子之间的相互作用力将分子分散在空间中。
当有适当的外界因素作用于高聚物分散体系时,例如降温、溶剂挥发、添加结晶助剂等,分子将逐渐更趋于有序,形成结晶核心。
核心形成的过程可以分为三个阶段,即原核形成、生长期和稳定期。
原核形成是核心形成的起始阶段,该阶段中核心数量很少,大小较小。
生长期是在原核的基础上,进一步成长,核心的大小逐渐增大。
稳定期是达到一定生长程度后,核心形成的速率与消失的速率趋于相等,此时晶核数量和大小趋于稳定。
核心形成的主要机制有三种,即自由基、链端末和核心网络。
自由基机制是指高聚物链的末端包围住某个小分子,在随后的降温或挥发过程中形成核心。
链端末机制是指高聚物链的末端具有较高的结晶活性,使其在最初性核心周围聚集形成核心。
核心网络机制是指高聚物链在三维空间中相互交织形成核心。
其次,晶体生长是高聚物结晶过程中的另一重要环节。
晶体生长是指核心周围的高聚物分子进一步转移到核心上,使得核心逐渐增大和发展成固态结晶态。
晶体生长的过程可以分为三个阶段,即形态发生、方向生长和外观生长。
形态发生是指核心周围的高聚物分子沿着一定的方向有序排列,形成晶体的初始形态。
方向生长是指晶体沿着其主要生长方向不断增长,使得晶体的长度逐渐增加。
外观生长是指晶体的横向生长,使得晶体的面积逐渐增大。
晶体生长的速率取决于多种因素,包括温度、浓度、溶剂性质、晶体结构等。
晶体生长的速率与温度成正相关,即温度越高,生长速率越快。
溶液或熔融态中高聚物的浓度也会影响晶体的生长速率,例如浓度越高,生长速率越快。
结晶高聚物结晶度与密度关系公式的简易推导以结晶高聚物结晶度与密度关系公式的简易推导为标题结晶高聚物是一类重要的聚合物材料,其结晶度与密度之间存在着一定的关系。
本文将对这一关系进行简易推导,以加深对结晶高聚物性质的理解。
我们需要明确结晶度和密度的定义。
结晶度是指聚合物中结晶区域的比例,而密度是指单位体积内的质量。
结晶高聚物的结晶度和密度是由分子之间的排列方式决定的。
在推导结晶度与密度关系之前,我们先了解一下结晶高聚物的结晶过程。
当聚合物处于高温状态下时,分子呈现无序排列,形成无定形结构;当温度降低到临界结晶温度以下时,聚合物分子开始有序排列,形成结晶区域,同时无定形结构逐渐减少。
当温度进一步降低时,结晶区域的大小将达到平衡状态。
根据上述结晶过程,我们可以推导出结晶度与密度之间的关系。
结晶度越高意味着结晶区域的比例越大,而无定形结构的比例越小。
由于结晶区域中分子的排列更加紧密有序,因此结晶度越高,密度也就越大。
进一步推导结晶度与密度之间的具体关系,我们需要考虑结晶高聚物的分子量以及分子的形态。
事实上,结晶高聚物的分子量越大,分子之间的相互作用力越强,结晶度也就越高。
而结晶度的增加会导致分子之间的排列更加紧密,因此密度也会增加。
结晶高聚物的分子形态也会对结晶度与密度之间的关系产生影响。
分子形态的不同会导致结晶区域的大小和形状不同,进而影响结晶度和密度的数值。
例如,线性结构的聚合物分子更容易形成有序排列的结晶区域,因此线性结构的聚合物通常具有较高的结晶度和密度。
相反,支化结构的聚合物分子由于分子链的扭曲和交叉,结晶度和密度较低。
总结以上推导,我们可以得出结晶高聚物结晶度与密度之间的关系:结晶度越高,密度也就越大。
而结晶度的增加与分子量的增加和分子形态的特征有关。
这一关系对于材料工程师和研究人员在设计和控制结晶高聚物材料的性能和应用方面具有重要的指导意义。
结晶度与密度关系的推导不仅为我们理解结晶高聚物的性质提供了一种简单的方法,还为材料设计和开发提供了理论依据。