医疗器械生物学评价第五部分体外细胞毒性试验
- 格式:doc
- 大小:43.50 KB
- 文档页数:6

标题:医疗器械包装检验参考1. 引言医疗器械包装是医疗器械的重要组成部分,其质量直接关系到医疗器械的安全性和有效性。
因此,医疗器械包装检验在医疗器械生产过程中具有重要意义。
本文将详细介绍医疗器械包装检验的参考标准、检验项目及方法,以期为相关企业和机构提供参考。
2. 医疗器械包装检验参考标准2.1 我国医疗器械包装检验标准我国医疗器械包装检验主要参照以下标准:- GB/T 19633-2005《医用包装、容器和材料环氧乙烷灭菌过程的验证》- YY/T 0681.1-2008《医用包装材料与容器第1部分:通用要求》- YY/T 0681.2-2008《医用包装材料与容器第2部分:可重复使用灭菌容器》- YY/T 0681.3-2008《医用包装材料与容器第3部分:一次性使用无菌容器》- YY/T 0681.4-2008《医用包装材料与容器第4部分:纸制无菌包装》- YY/T 0681.5-2008《医用包装材料与容器第5部分:塑料制无菌包装》2.2 国际医疗器械包装检验标准国际医疗器械包装检验主要参照以下标准:- ISO 11607-1:2006《医疗器械包装第1部分:通用要求》- ISO 11607-2:2006《医疗器械包装第2部分:最终灭菌医疗器械包装的验证》- ASTM D 4169-2004《运输包装件的振动、冲击和堆码压力试验》3. 医疗器械包装检验项目及方法3.1 物理性能检验3.1.1 拉伸强度与断裂伸长率检测方法:参照GB/T 1040.3-2006《塑料拉伸性能的测定第3部分:薄膜和薄板》进行测试。
3.1.2 热封强度检测方法:参照GB/T 8807-1988《塑料薄膜热封强度测定方法》进行测试。
3.1.3 抗摆锤冲击性能检测方法:参照GB/T 8809-1988《塑料薄膜抗摆锤冲击性能试验方法》进行测试。
3.2 阻隔性能检验3.2.1 水蒸气透过率检测方法:参照GB/T 1037-1988《塑料薄膜和薄片水蒸气透过率的测定》进行测试。

医疗器械生物学评价医疗器械生物学评价是指对医疗器械进行生物学方面的测试和评价,以确保其安全性和有效性。
这是一个非常重要的环节,因为医疗器械的使用直接涉及到人体健康和生命安全。
本文将就医疗器械生物学评价的背景、方法和重要性进行探讨。
首先,医疗器械生物学评价的背景。
医疗器械的定义包括各种医疗设备、仪器、用品和材料等,这些器械在接触人体组织和体液时会产生生物学反应。
因此,了解这些器械的生物学特性对确保其安全性和有效性至关重要。
医疗器械生物学评价是基于国际标准和相关规定制定的,旨在对器械进行全面的生物学评价。
医疗器械生物学评价的方法有多种,包括体内和体外实验。
体外实验主要包括细胞毒性测试、细胞增殖和存活测试、基因毒性测试等,这些实验可以分析器械对细胞的影响和毒性反应,以确定器械的生物学安全性。
体内实验主要包括动物实验和人体试验。
动物实验通常用于评估器械对动物生物学系统的影响,例如器械对动物组织的刺激和损伤程度。
人体试验则是最直接和可行的方法,可以更准确地评估器械对人体组织的影响,但在伦理和安全上有一定的限制。
医疗器械生物学评价的重要性不言而喻。
首先,生物学评价可以帮助厂商了解和评估其产品对人体的生物相容性。
这对于医疗器械的设计、研发和改进至关重要,可以确保器械的安全性和有效性。
其次,医疗器械生物学评价也是监管机构对医疗器械的批准和注册的重要依据。
例如,美国FDA要求医疗器械经过严格的生物学评价才能上市销售。
最后,医疗器械生物学评价也是医疗器械使用者选择和使用器械的重要参考,可以帮助他们了解器械的生物相容性和安全性。
因此,医疗器械生物学评价应该是医疗器械研发和生产过程中的重要环节。
厂商应该重视生物学评价的每个步骤,确保评价的全面性和可靠性。
同时,监管机构也应加强对医疗器械生物学评价的监管和指导,确保医疗器械的安全性和有效性。
最后,医疗器械的使用者也应在选择和使用器械时关注其生物学评价报告,以确保人体的安全和健康。

医疗器械生物学评价第5部分:体外细胞毒性试验1 范围GB/T 16886 的本部分阐述了评价医疗器械体外细胞毒性的试验方法。
这些方法规定了下列供试品以直接或通过扩散的方式与培养细胞接触和进行孵育;a)用器械的浸提液,和/或b)与器械接触。
这些方法是用相应的生物参数测定哺乳动物细胞的体外生物学反应。
2 规范性引用文件下列文件中的条款通过GB/T 16886 的本部分的引用而成为本部分的条款。
凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本部分,然而,鼓励根据本部分达成协议的各方研究是否可使用这些文件的最新版本。
凡是不注日期的引用文件,其最新版本适用于本部分。
GB/T 16886. 1 医疗器械生物学评价第1部分:评价与试验(GB/T ,idt ISO 10993- 1:1997)CB/ T 16886. 12-2000 医疗器械生物学评价第12部分:样品制备和参照材料(idt ISO 10993-12 :1996)3 术语与定义GB/ T 16886. 1/ ISO 1993-1中确立的以及下列术语和定义适用于本部分。
阴性对照材料negative control material按照本部分试验时不产生细胞毒性反应的材料。
注:阴性对照的目的是验证背景反应,例如高密度聚乙烯1)已作为合成聚合物的阴性对照材料,氧化陶瓷棒则用作牙科材料的阴性对照物。
阳性对照材料 pos itive control material按照本部分试验时可重现细胞毒性反应的材料。
注:阳性对照的白目的是验证相应试验系统的反应,例如用有机锡作稳定剂的聚氯乙烯2)已用作固体材料和浸提液的阳性对照,酚的稀释液用于浸提液的阳性对照。
_____________________________________1)高密度聚乙烯可从美国药典委员会(Rockvillie, Maryland, USA)和Hatano研究所食品和药品安全中心(Ochiai 729-5 ,)获得。

医疗器械生物学试验审评规范(2010版)医疗器械生物学试验审评规范(2010版)一、目的本规范的目的是为了正确、有效地实施GB/T16886医疗器械生物学评价系列标准。
本规范提出了医疗器械生物学试验中试验资料方面的要求,指导检测机构和北京市生产企业进行医疗器械生物学试验。
二、医疗器械生物学试验的基本要求医疗器械生物学试验的目的是确定与人体直接或间接接触的医疗器械/材料是否引起潜在的毒性。
医疗器械/材料不应直接或通过释放材料中的成分引起:(1)局部或全身不良反应;(2)致畸、致突变、致癌性;(3)生殖和发育毒性。
医疗器械生物学评价是在系统试验数据基础上的评价,是为了确保最终产品的受益超过器械材料产生的潜在风险。
当选择适宜的医疗器械生物学评价试验时,应考虑器械所用材料的化学特性,以及和人体接触的性质、程度、频率和时间。
通常这些试验包括:急性毒性、亚慢性和慢性毒性;对皮肤、眼睛和黏膜的刺激;致敏性;血液相容性;遗传毒性;致癌性;以及发育和生殖毒性等。
然而,根据器械的特性、用途以及与人体接触的特性,这些试验也不能充分表明某些特殊器械的安全性。
对于特殊靶器官毒性的附加试验,例如神经毒性和免疫毒性,对某些器械是必须的。
(例如,直接和脑实质以及脑脊液接触的神经器械,应要求动物植入试验,评价对脑实质的影响、癫痫易感性、以及对脉络丛和蛛网膜绒毛分泌和吸收功能机理的影响。
)对于特殊用途和改变用途的医疗器械/材料,应确定哪些试验是适宜的。
对于不同材料构成的多部件医疗器械,宜对所用的每种材料或部件根据临床使用的情况分别进行生物学试验和评价。
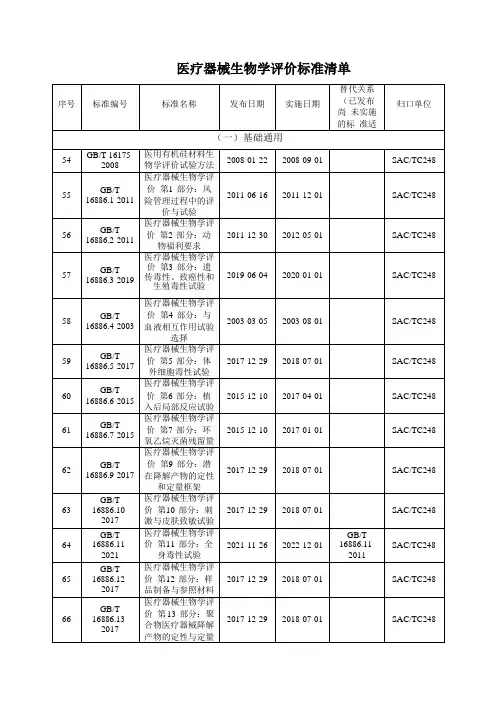
三、医疗器械生物学评价标准目前,医疗器械生物学试验主要参考以下国家标准和行业标准:GB/T 16886.1-2001医疗器械生物学评价——第1部分:生物学评价和试验GB/T 16886.2-2000医疗器械生物学评价——第2部分:动物保护要求GB/T 16886.3-2008医疗器械生物学评价——第3部分:遗传毒性、致癌性和生殖毒性试验GB/T 16886.4-2003医疗器械生物学评价——第4部分:和血液相互作用试验选择(DIS)GB/T 16886.5-2003医疗器械生物学评价——第5部分:体外细胞毒性试验GB/T 16886.6-1997医疗器械生物学评价——第6部分:植入后局部反应试验GB/T 16886.7-2001医疗器械生物学评价——第7部分:环氧乙烷灭菌残留量GB/T 16886.9-2001医疗器械生物学评价——第9部分:潜在降解产物的定性和定量框架GB/T 16886.10-2005医疗器械生物学评价——第10部分:刺激和迟发型超敏试验(DIS)GB/T 16886.11-1997医疗器械生物学评价——第11部分:全身毒性试验GB/T 16886.12-2000医疗器械生物平价第12部分:样品制备与参照样品GB/T 16886.13-2001医疗器械生物学评价第13部分:聚合物医疗器械的降解产物的定性与定量GB/T 16886.14-2003医疗器械生物学评价第14部分:陶瓷降解产物的定性与定量GB/T 16886.15-2003医疗器械生物学评价第15部分:金属与合金降解产物的定性与定量GB/T 16886.16-2003医疗器械生物学评价第16部分:降解产物和可溶出物的毒代动力学研究设计GB/T 16886.17-2005医疗器械生物学评价第17部分:可沥滤物允许限量的建立YY/T 0268-2008牙科学口腔医疗器械生物学评价第1单元:评价与试验YY/T 0127.1-1993口腔材料生物试验方法:溶血试验YY/T 0127.2-1993口腔材料生物试验方法:静脉注射急性全身毒性试验YY/T 0127.3-1998口腔材料生物评价第2单元:口腔材料生物试验方法根管内应用试验YY/T 0127.4-1998口腔材料生物评价第2单元:口腔材料生物试验方法骨埋植试验YY/T 0127.5-1999口腔材料生物评价第2单元:口腔材料生物试验方法吸入毒性试验YY/T 0127.6-1999口腔材料生物评价第2单元:口腔材料生物试验方法显性致死试验YY/T 0127.7-2001口腔材料生物评价第2单元:口腔材料生物试验方法牙髓牙本质应用试验YY/T 0127.8-2001口腔材料生物评价第2单元:口腔材料生物试验方法皮下植入试验YY/T 0127.9-2001口腔材料生物评价第2单元:口腔材料生物试验方法细胞毒性试验:琼脂覆盖法及分子滤过法YY/T 0127.10-2001口腔材料生物评价第2单元:口腔材料生物试验方法鼠伤寒沙门氏杆菌回复突变试验(Ames试验) YY/T 0127.11-2001牙科学用于口腔的医疗器械生物相容性临床前评价2单元:口腔材料生物试验方法盖髓试验医疗器械生物学评价标准GB/T16886.1给出了生物学评价试验及生物学评价流程,但是对于某些新的高风险医疗器械可能还需要补充其它的试验项目。

医疗器械生物相容性测试国家标准前言医疗器械生物相容性测试国家标准是为了确保医疗器械与人体组织之间的相容性,保障患者使用安全而制定的。
本标准旨在提供一套统一的测试方法和评估指标,以评估医疗器械在生物体内的相容性,为医疗器械制造商和监管机构提供参考。
适用范围本标准适用于各类医疗器械的生物相容性测试,包括但不限于植入性医疗器械、体外诊断试剂和辅助诊断试剂等。
测试项目本标准规定了医疗器械生物相容性测试的以下项目:1. 细胞毒性测试:通过评估医疗器械对细胞的毒性,判断其对细胞生长和功能的影响。
2. 局部组织刺激性测试:评估医疗器械与局部组织接触后可能引起的刺激反应,包括炎症反应和组织损伤等。
3. 过敏原性测试:评估医疗器械是否存在过敏原性,判断其是否会引起变态反应。
4. 免疫毒理学测试:评估医疗器械对机体免疫系统的影响,包括免疫功能的影响和免疫毒性的评估。
5. 慢性系统毒性测试:通过长期观察医疗器械在动物体内引起的系统毒性反应,评估其潜在的长期影响。
6. 遗传毒性测试:评估医疗器械对遗传物质的影响,判断其是否存在基因毒性。
评估指标本标准规定了以下评估指标,用于对医疗器械的生物相容性进行评估:1. 毒性评估:根据细胞存活率、细胞形态变化和细胞功能等指标,对医疗器械的细胞毒性进行评估。
2. 刺激性评估:根据炎症反应、组织损伤程度和修复能力等指标,对医疗器械的局部组织刺激性进行评估。
3. 过敏原性评估:通过皮肤接触试验和变态反应指标,对医疗器械的过敏原性进行评估。
4. 免疫毒理学评估:通过免疫功能指标、细胞因子水平和抗体产生等指标,对医疗器械的免疫毒理学进行评估。
5. 慢性系统毒性评估:通过长期观察动物的生理指标、组织病理学和血液指标等,对医疗器械的慢性系统毒性进行评估。
6. 遗传毒性评估:通过基因突变和染色体畸变等指标,对医疗器械的遗传毒性进行评估。
结论医疗器械生物相容性测试国家标准提供了一套全面、科学的测试方法和评估指标,可帮助制造商和监管机构评估医疗器械在生物体内的相容性。

医疗器械生物学评价医疗器械的生物学评价是医疗器械开发和使用的一个重要环节。
它主要是对医疗器械在生物体内的生物相容性、安全性和有效性进行评估,以确保它对人体没有不良的影响,达到设计预期的治疗效果。
医疗器械的生物学评价通常包括以下几个方面:生物相容性、毒理学评估、细胞毒性、致敏反应、细胞增殖和差异化等。
生物相容性是评价医疗器械在生物体内引起的生物学反应的能力,其中包括局部和全身反应。
局部反应可以是炎症、纤维组织形成和血管新生等,全身反应可以是免疫反应和全身毒性等。
这些反应的评价通常通过动物实验和体外实验来进行。
毒理学评估是评价医疗器械对生物体产生的毒性作用的能力。
这包括急性毒性、亚急性毒性、慢性毒性等。
通常通过动物实验和体外实验,如细胞毒性和基因毒性实验来评估医疗器械的毒性。
细胞毒性是评价医疗器械对细胞的直接杀伤作用和影响细胞功能的能力。
一般通过体外实验来评估医疗器械对细胞的毒性。
致敏反应是评价医疗器械引起的变态反应的潜在能力。
通过动物实验和体外实验,如小鼠皮肤致敏实验和人类淋巴细胞转化试验等来评估医疗器械的致敏性。
细胞增殖和差异化是评价医疗器械对细胞的增殖和分化的能力。
这可以通过细胞培养实验来评估。
医疗器械的生物学评价需要根据不同的器械特性和使用方式来确定具体的评价方法。
通常的评估流程包括:确定评估项目和方法、收集相关数据、分析数据、评价结果和风险分析、确定产品的生物安全性和有效性。
生物学评价结果对于医疗器械的研发和使用具有重要的指导意义。
它可以帮助设计人员选择合适的材料和生产工艺,以及优化设备的结构和功能,来提高医疗器械的生物相容性和安全性。
对于临床使用的医疗器械,生物学评价可以评估其治疗效果和副作用,为临床决策提供依据。
总之,医疗器械的生物学评价是保障医疗器械安全有效的重要环节。
只有通过全面、系统的生物学评价,才能确保医疗器械对人体没有不良的影响,保证其在临床应用中能够发挥预期的治疗效果。

《医疗器械生物学评价》
《医疗器械生物学评价》是指对医疗器械进行生物学安全性评价的一项重要工作。
医疗器械是指用于医疗诊断、预防、治疗或缓解疾病的设备、器具、材料、试剂等。
用于人体内或与人体接触的医疗器械必须通过生物学评价来确保其对人体的安全性。
医疗器械生物学评价主要从以下几个方面进行评估:
1. 细胞毒性:评估医疗器械对人体细胞的毒性影响。
常用的方法包括细胞培养实验、细胞生存率测试等。
2. 致敏性:评估医疗器械是否会引发人体过敏反应。
常用的方法包括过敏原性测试、原位皮肤试验等。
3. 细菌毒力:评估医疗器械对细菌的生长和毒力的影响。
常用的方法包括抗菌性测试、细菌毒力测试等。
4. 免疫反应:评估医疗器械是否会引发人体免疫反应。
常用的方法包括淋巴细胞激活测试、抗体产生测试等。
5. 植入性:评估医疗器械在人体内的植入性能。
常用的方法包括组织刺激性测试、植入皮肤刺激性测试等。
医疗器械生物学评价对于确保医疗器械的安全性和有效性具有重要意义。
只有通过严格的生物学评价,才能保证医疗器械不会对人体造成损害,确保患者的健康和安全。
因此,医疗器械
生物学评价是医疗器械注册、上市许可等过程中必不可少的一环。

生物学评价报告生物学评价是对与人体接触的医疗器械、生物材料等产品进行的一系列评估,以确定其对人体的安全性和有效性。
本报告旨在对某一特定产品进行全面的生物学评价,为其临床应用提供科学依据。
一、产品概述本次评价的产品是一款新型的心脏支架,由特殊的合金材料制成,表面经过特殊处理以提高生物相容性。
该支架设计用于治疗冠状动脉狭窄,通过介入手术置入患者体内,以支撑血管并保持血流通畅。
二、评价目的确定该心脏支架在预期使用条件下是否具有生物安全性,是否会引起局部或全身性的不良反应,以及是否能够有效地实现其预期的治疗效果。
三、评价依据1、相关的国家标准和行业标准,如 GB/T 16886 系列标准。
2、国际上公认的生物学评价指南和规范。
3、已有的同类产品的生物学评价数据和临床应用经验。
四、材料和方法1、材料表征对支架的材料成分进行化学分析,确定其主要元素和杂质含量。
对材料的物理性能进行测试,包括硬度、弹性模量等。
2、体外细胞毒性试验使用人源细胞系(如内皮细胞、平滑肌细胞等)进行培养。
将支架材料的提取物与细胞共同培养,观察细胞的形态、生长和增殖情况,以评估细胞毒性。
3、致敏试验采用豚鼠最大剂量法进行致敏试验。
观察动物在接触支架材料后的皮肤反应,判断是否引起过敏反应。
4、刺激试验进行皮肤刺激试验和眼刺激试验。
将支架材料直接接触动物的皮肤或眼睛,观察是否引起刺激症状。
5、血液相容性试验检测支架材料对血液凝固、血小板黏附和聚集等的影响。
评估材料与血液接触后的溶血情况。
6、体内植入试验将支架植入动物(如大鼠或兔)的冠状动脉。
在预定的时间点进行组织学检查,观察支架周围组织的炎症反应、血管内皮化情况等。
五、结果与讨论1、材料表征化学分析结果显示,支架材料的主要成分符合设计要求,杂质含量在可接受范围内。
物理性能测试表明,材料的硬度和弹性模量等参数适合预期的应用。
2、体外细胞毒性试验细胞培养结果显示,在支架材料提取物存在的情况下,细胞形态正常,生长和增殖未受到明显抑制,表明无明显的细胞毒性。

医疗器械的生物学评价医疗器械的生物学评价是保证器械安全和有效性的重要环节。
本文将介绍医疗器械的生物学评价流程和相关标准要求,并探讨生物学评价在医疗器械研发和监管中的重要性。
一、医疗器械的生物学评价流程医疗器械的生物学评价是一项系统、科学的流程,通过不同的实验和测试来评估器械对人体组织和生物体的生物相容性。
一般来说,医疗器械的生物学评价流程可以分为以下几个步骤:1. 制定评估计划:根据相关标准和法规要求,制定评估计划,明确评估的目标、方法和实验方案。
2. 材料评估:对医疗器械使用的材料进行评估,包括原材料和制成品的化学成分分析、物理性能测试等。
3. 生物相容性测试:通过实验和测试评估医疗器械对人体组织和生物体的生物相容性。
常用的测试项目包括细胞毒性测试、皮肤刺激测试、组织刺激测试、植入反应测试等。
4. 数据分析和评估:对测试结果进行数据分析和评估,根据评估结果判断医疗器械的生物学安全性和生物相容性。
5. 编写生物学评价报告:根据评估结果编写生物学评价报告,包括评估流程、测试方法、结果分析和风险评估等内容。
6. 安全性评估:将生物学评价结果与其他评估结果(如临床试验结果)相综合,进行医疗器械的安全性评估。
二、医疗器械生物学评价的标准要求医疗器械的生物学评价需要符合相关的标准和法规要求,以确保评价的科学性和可靠性。
以下是一些常用的标准要求:1. ISO 10993系列标准:国际标准化组织发布的关于医疗器械生物学评价的一系列标准,包括ISO 10993-1(生物学评价的原则和指南)、ISO 10993-3(植入物的生物学评价)、ISO 10993-5(细胞毒性测试)等。
2. FDA指南:美国食品药品监督管理局发布的关于医疗器械生物学评价的指南,包括《Use of International Standard ISO 10993-1, "Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process"》等。

医疗器械技术评估的生物相容性与毒理学评价医疗器械的技术评估对于确保其安全性和有效性至关重要。
其中,生物相容性和毒理学评价是评估医疗器械使用过程中的重要环节。
本文将以技术评估的角度,介绍医疗器械生物相容性与毒理学评价的相关内容。
一、生物相容性评价生物相容性评价是评估医疗器械与人体组织或生物系统之间相互作用的过程。
它能够揭示出医疗器械对人体的生物相容性及可能引发的不良反应,为医疗器械的研发和使用提供重要依据。
1. 生物相容性试验生物相容性试验是通过体外或动物实验的方法,对医疗器械的生物相容性进行评价。
常用的试验包括细胞毒性试验、局部组织刺激试验、皮肤过敏试验等。
通过这些试验可以评估医疗器械与人体组织的相互作用情况,发现潜在的生物相容性问题。
2. 生物相容性评价指标生物相容性评价指标主要包括细胞毒性、致敏性、局部刺激性、体内毒理性等。
细胞毒性是评估医疗器械对人体细胞的毒性作用;致敏性是评估医疗器械对人体引发过敏反应的能力;局部刺激性是评估医疗器械对人体局部组织的刺激作用;体内毒理性是评估医疗器械在体内引发的毒性反应。
3. 生物相容性评价标准生物相容性评价标准是根据国家、国际相关标准制定的,如美国FDA的生物相容性评价标准,欧洲药典和ISO标准等。
它们规范了医疗器械生物相容性评价的方法和指标,确保医疗器械的生物相容性符合国际要求,保障患者的安全。
二、毒理学评价毒理学评价是通过对医疗器械成分或材料进行毒理学实验,评估医疗器械对人体的潜在毒性。
毒理学评价有助于揭示医疗器械可能引发的毒性反应,为医疗器械的设计和选择提供依据。
1. 毒理学试验毒理学试验包括急性毒性试验、亚慢性和慢性毒性试验、基因毒性试验等。
急性毒性试验用于评估医疗器械对人体的一次性毒性效应;亚慢性和慢性毒性试验用于评估医疗器械长期使用所引发的潜在毒性效应;基因毒性试验用于评估医疗器械对遗传物质的损伤程度。
2. 毒理学评价指标毒理学评价指标包括急性毒性指标、亚慢性和慢性毒性指标、基因毒性指标等。
涂层导尿管的体外细胞毒性试验研究周小婷;徐玉茵;田林奇;周静;韩颖【摘要】目的分析国内市场上常用的新型涂层导尿管的细胞毒性,给临床导尿管的选择提供指导.方法参考GB/T 16886.5-2017/ISO10993.5:2009的试验方法,将涂层导尿管浸提液与小鼠成纤维细胞L929培养接触,采用MTT细胞毒性试验计算细胞毒性,计算细胞存活率.结果不同材质和不同涂层的导尿管对L929细胞显示出不同的细胞毒性.结论聚氯乙烯(PVC)基材的涂层导管的体外细胞毒性小,生物相容性高;乳胶基材的涂层导尿管,细胞毒性结果有差异.【期刊名称】《中国医疗设备》【年(卷),期】2019(034)003【总页数】3页(P45-47)【关键词】涂层导尿管;细胞毒性;乳胶;聚氯乙烯【作者】周小婷;徐玉茵;田林奇;周静;韩颖【作者单位】河南省医疗器械检验所河南省医疗器械检验检测工程技术研究中心生物检测室,河南郑州 450000;河南省医疗器械检验所河南省医疗器械检验检测工程技术研究中心生物检测室,河南郑州 450000;河南省医疗器械检验所河南省医疗器械检验检测工程技术研究中心生物检测室,河南郑州 450000;河南省医疗器械检验所河南省医疗器械检验检测工程技术研究中心生物检测室,河南郑州 450000;河南省医疗器械检验所河南省医疗器械检验检测工程技术研究中心生物检测室,河南郑州 450000【正文语种】中文【中图分类】R608引言导尿管是指通过尿路插入膀胱腔以排尿和冲洗膀胱为目的的管状器械。
按《医疗器械分类目录》,根据涂层不同,其管理类别也不同。
润滑涂层导尿管管理类别为Ⅱ类,银盐和抗菌成分等涂层导尿管管理类别为Ⅲ类。
随着临床上导尿管的应用日益广泛,由导尿管引起的导尿管相关尿路感染及损伤日益得到重视,如细菌定植、抗生素耐药性、慢性感染、肾脏和膀胱损伤、肾结石、膀胱假息肉、败血症、尿道损伤等[1]。
由于导尿管的原材料天然乳胶中含有对人体致敏性蛋白、生产工艺控制不严、添加剂或助剂使用比例不正确易导致生物相容性尤其是细胞毒性试验不符合要求。
生物学评价项目类型及产品检测项目1、遗传毒性试验遗传毒性试验的主要作用是采用试验细胞或生物体来研究来研究生殖细胞和体细胞基因改变的风险。
试验主要有:细菌回复突变、体外哺乳动物染色体畸变、小鼠淋巴瘤细胞(TK)基因突变试验微核试验这四类。
2、急性全身毒性急性毒性试验可作为亚急性/亚慢性和其他试验确定剂量接触方式的初试步骤,并且可提供物质预期临床接触途径毒性作用模式方面的信息。
急性全身毒性是指在24小时内一次、多次或持续接触试验样品后在任何时间发生的不良作用。
3、亚慢性全身毒性许多医疗器械更常见的人体接触方式是重复或持续接触形式,重复或持续接触可能会由于化学物在组织内的积聚或其他机制产生反应,长期试验(亚急性、亚慢性、慢性)对于鉴别此类作用是非常重要的。
4、植入后局部反应试验试验样品植入适宜种属的动物和部位以评价材料的生物安全性,这些植入方法预期不用于评价或测定试验样品在机械或功能负荷方面的性能。
GB/T16886的本部分可能也适用于临床上预期用于损伤表面或损伤内表面的医疗器械,以评价局部组织反应。
5、血液相容性血液相容性试验主要评价血液或血液成分与器械间的相互作用所导致的对血液、器官、组织或器械的影响。
根据检测的主要过程或系统将血液相互作用分成五大类:血栓形成、凝血、血小板、血液学、补体系统。
6、异常毒性异常毒性有别于药物本身所具有的毒性特征,是指由生产过程中引人或其他原因所致的毒性。
本法系给予动物一定剂量的供试品溶液,在规定时间内观察动物出现的异常反应或死亡情况,检查供试品中是否污染外源性毒性物质以及是否存在意外的不安全因素。
7、溶血试验该方法是非常有意义的一项帅选试验,在试验操作规范的情况下,血浆血红蛋白量升高指示溶血并反应出在材料与器械接触中红细胞膜的易破裂性。
8、热原试验将一定剂量的供试品,静脉注入家兔体内,在规定时间内,观察家兔体温升高的情况,以判断供试品中所含热原的限度是否符合规定。
9、皮内反应用作植入物的医疗器械采用皮内反应试验。
无菌医疗器械质量检验相关标准(一)医疗器械生物相容性试验GB/T 16886.3 医疗器械生物学评价 第3部分:遗传毒性、致癌性和生殖毒性试验GB/T 16886.4 医疗器械生物学评价 第4部分:与血液相互作用试验选择GB/T 16886.5 医疗器械生物学评价 第5部分:体外细胞毒性试验GB/T 16886.6 医疗器械生物学评价 第6部分:植入后局部反应试验GB/T 16886.7 医疗器械生物学评价 第7部分:环氧乙烷灭菌残留量GB/T 16886.8 医疗器械生物学评价 第8部分:生物学试验参照样品的选择和定性GB/T 16886.10 医疗器械生物学评价 第10部分:刺激与致敏试验GB/T 16886.11 医疗器械生物学评价 第11部分:全身毒性试验GB/T 16886.12医疗器械生物学评价 第12部分:样品制备和参照样品GB/T 16886.13 医疗器械生物学评价 第13部分:聚合物医疗器械降解产物的定性与定量GB/T 16886.14 医疗器械生物学评价 第14部分:陶瓷降解产物的定性与定量分析GB/T 16886.15 医疗器械生物学评价 第15部分:金属与合金降解产物的定性与定量GB/T 16886.16 医疗器械生物学评价 第16部分:降解产物与可溶出物的毒代动力学研究设计) GB/T 16886.17 医疗器械生物学评价 第17部分:根据健康风险评价确定可溶出物允许极限的方法 GB/T 16886.18 医疗器械生物学评价 第18部分:材料化学定性GB/T 16886.19 医疗器械生物学评价 第19部分:材料理化、机械和形态定性GB/T 16886.20 医疗器械生物学评价 第20部分:医疗器械免疫毒理学试验原理和方法GB/T16175医用有机硅材料生物学评价试验方法GB/T 16886.9医疗器械生物学评价 第9部分:潜在降解产物的定性与定量总则YY/T 0473外科植入物 聚交酯共聚物和共混物 体外降解试验YY/T 0474外科植入物用聚L-丙交酯树脂及制品 体外降解试验YY/T XXXX 一次性使用橡胶手套生物学评价要求与试验方法GB /TXXXX 医疗器械 微生物法 第3部分:细菌内毒素试验方法 用于常规监测和跳批检验YY/T 0567.1 医疗产品的无菌加工 第1部分:通用要求YY/T 0567.2 医疗产品的无菌加工 第2部分:过滤GB XXXX.1 医疗器械生产用动物组织及其衍生物 第1部分:风险分析与管理GB XXXX.2 医疗器械生产用动物组织及其衍生物 第2部分:索源、收集和处置GB XXXX.3 医疗器械生产用动物组织及其衍生物 第3部分:病毒和传播媒介的去除与灭活GB XXXX.4 医疗器械生产用动物组织及其衍生物 第4部分:液体灭菌剂灭菌的确认和常规控制 (二)医用输液(血)、注射器具标准GB/T14233.1医用输液、输血、注射器具检验方法 第1部分:化学分析方法GB/T14233.2医用输液、输血、注射器具检验方法 第2部分:生物试验方法GB15593输血(液)器具用软聚氯乙烯塑料YY0114医用输液、输血、注射器用聚乙烯专用料YY0242医用输液、输血、注射器用聚丙烯专用料GB 8368一次性使用输液器GB 18671一次性使用静脉输液针YY 0286.1专用输液器 第1部分:一次性使用精密过滤输液器YY 0286.2专用输液器 第2部分:一次性使用滴定管式输液器YY 02868.3专用输液器 第3部分:一次性使用避光式输液器,重力输液式YY 02868.4专用输液器 第4部分:一次性使用压力输液装置用输液器YY 0286.5专用输液器 第5部分:一次性使用吊瓶式和分液袋式输液器YY 0286.6专用输液器 第6部分: 一次性使用非PVC输液器YY 0286.7 专用输液器 第7部分: 一次性使用流量可设定输液器YY XXXX 一次性使用静脉营养袋YY 0451 一次性使用输注泵YY XXXX一次性使用高压造影输注器YY 0585.1压力输液设备用一次性使用液路及附件 第1部分: 液路YY 0585.2压力输液设备用一次性使用液路及附件 第2部分: 附件YY 0585.3压力输液设备用一次性使用液路及附件 第3部分: 过滤器YY0585.4 压力输液设备用一次性使用液路及附件 第4部分: 防回流阀YY 0586输液用肝素帽YY/T 0582.1输液瓶悬挂装置 第1部分:一次性使用悬挂装置YY/T 0582.2输液瓶悬挂装置 第2部分:多用悬持装置GB8369一次性使用输血器GB 14232.1人体血液及血液成分袋式塑料容器 第1部分:传统型血袋GB 14232.2 人体血液及血液成分袋式塑料容器 第2部分:图形符号GB 14232.3 人体血液及血液成分袋式塑料容器 第3部分:带特殊组件的血袋系统 YY 0327一次性使用紫外线透疗血液容器GB 19335一次性使用血路产品通用技术条件YY0113一次性使用采血器YY 03282一次性使用机用采血器YY 0326.1一次性使用离心式血浆分离器 第1部分:血浆分离杯YY 0326.2一次性使用离心式血浆分离器 第2部分:血浆管路YY 0326.3 一次性使用离心式血浆分离器 第3部分:血浆袋YY 0329-2009一次性使用去白细胞过滤器YY0031硅橡胶输液(血)管YY 0584一次性使用离心杯式血液成分分离器材YY XXXX一次性使用离心袋式血液成分分离器材YY/T 0289一次性使用微量采血吸管YY 0314一次性使用人体静脉血样采集容器YY 0464-2009 一次性使用血液灌流器YY 0465-2009一次性使用空心纤维血浆分离器GB 15810 一次性使用无菌注射器GB 15811 一次性使用无菌注射针YY/T 0243 一次性使用无菌注射器用橡胶活塞YY/T 0282-2009 注射针(三)医用导管、插管标准YY 0285.1一次性使用血管内导管 第1部分:通用要求YY 0285.2一次性使用无菌血管内导管 第2部分:造影导管YY 0285.3一次性使用无菌血管内导管 第3部分:中心静脉导管YY 0285.4一次性使用无菌血管内导管 第4部分:球囊扩张导管YY 0285.5一次性使用无菌血管内导管 第5部分:套针外周导管YY/T 0586医用高分子制品X透射线不透性试验方法YY 0450.1一次性使用无菌血管内导管辅件 第1部分:导引器械YY 0450.2一次性使用无菌血管内导管辅件 第2部分:套针外周导管管塞YY 0450.3一次性使用无菌血管内导管辅件 第3部分:球囊扩张导管扩张泵GB/T15812.1非血管内导管 第1部分: 一般性能试验方法GB/T15812.2非血管内导管 第2部分: 弯曲性能试验方法YY 0030 腹膜透析管YY 0325一次性使用无菌导尿管YY 0489一次性使用无菌引流导管及其辅助器械YY 0488一次性使用无菌直肠导管YY 0483一次性使用肠营养导管、肠给养器及其连接件 设计与试验方法YY 1040.1 麻醉和呼吸设备 圆锥接头 第1部分:锥头与锥套YY 0337.1气管插管 第1部分:常用型插管及接头YY 0337.2气管插管 第2部分:柯尔型插管YY 0338.1气管切开插管第1部分:成人用插管及接头YY 0338.2气管切开插管第2部分:小儿用气管切开插管YY 0339-2009 呼吸道用吸引导管YY 0461 麻醉机和呼吸机用呼吸管路(四)无菌植入物标准YY 0484外科植入物 双组分加成型硫化硅橡胶YY 0334硅橡胶外科植入物通用要求YY 0332植入式给药装置YY 0333软组织扩张器YY 0487一次性使用无菌脑积水分流器及其组件YY0308医用透明质酸钠凝胶YY/TXXXX医用天然高分子降解多糖材料:通用要求及试验方法(五)卫生材料、敷料YY/T 0471.1接触性创面敷料试验方法 第1部分:液体吸收性YY /T 0471.2接触性创面敷料试验方法 第2部分:透气膜敷料水蒸汽透过率YY/T 0471.3接触性创面敷料试验方法 第3部分:阻水性YY/T 0471.4 接触性创面敷料试验方法 第4部分:舒适性YY /T 0471.5接触性创面敷料试验方法 第5部分:阻菌性YY /T 0471.6接触性创面敷料试验方法 第6部分:气味控制YY/T 0471.7 创伤敷料试验方法 第7部分:颗粒脱落YY /T0471.8 创伤敷料试验方法 第8部分:关于粘连(创面和皮肤)YY/T 0472.1医用非织造敷布试验方法 第1部分:敷布生产用非织造布YY/T 0472.2医用非织造敷布试验方法 第2部分:成品敷布YY 0330医用脱脂棉YY 0331脱脂棉纱布和脱指棉与粘胶纱布性能要求和试验方法YY 0594 外科纱布敷料通用要求YY/T 0148医用粘贴胶带通用要求YY/T 0506.2-2009病人、医护人员和器械用手术单、手术衣和洁净服 第2部分:性能要求和性能水平 YY/T 0506.3病人、医护人员和器械用手术单、手术衣和洁净服 第3部分:试验方法YY/T 0506.4病人、医护人员和器械用手术单、手术衣和洁净服 第4部分:干态落絮试验方法YY/T 0506.5-2009病人、医护人员和器械用手术单、手术衣和洁净服 第5部分:干态阻菌试验方法YY/T 0506.6-2009病人、医护人员和器械用手术单、手术衣和洁净服 第6部分:湿态阻菌试验方法YY/T 0506.7病人、医护人员和器械用手术单、手术衣和洁净服 第7部分:阻污染气溶胶穿透试验方法YY/T 0506.8病人、医护人员和器械用手术单、手术衣和洁净服 第8部分:抗激光性试验方法(六)其他标准YY/T 0720-2009 一次性使用产包 自然分娩用YY 0321.1-2009一次性使用麻醉穿刺包YY 0321.2-2009一次性使用麻醉用针YY 0583—2005《一次性使用胸腔引流装置 水封式》YY 0167非吸收性外科缝线YY 1116可吸收外科缝合线GB 7543-1996 橡胶医用手套GB 10213-1995 一次性使用橡胶检查手套GB 7544-2004 天然胶乳橡胶避孕套技术要求和试验方法*(非无菌医疗器械)YY /T0698.1-2009 最终灭菌医疗器械包装材料 第1部分:吹塑包装复合塑料膜 要求和试验方法YY /T0698.2-2009最终灭菌医疗器械包装材料 第2部分:灭菌包裹材料 要求和试验方法YY /T0698.3-2009最终灭菌医疗器械包装材料 第3部分:纸袋、组合袋和卷材生产用纸 要求和试验方法 YY /T0698.4-2009 最终灭菌医疗器械包装材料 第4部分:纸袋 要求和试验方法;YY /T0698.5-2009 最终灭菌医疗器械包装材料 第5部分:纸与塑料膜组合的热封和自封袋和卷材要求和试验方法;YY /T0698.6-2009 最终灭菌医疗器械包装材料 第6部分:用于低温灭菌过程或辐射灭菌的无菌屏障系统生产用纸 要求和试验方法;YY /T0698.7-2009 最终灭菌医疗器械包装材料 第7部分:环氧乙烷或辐射灭菌的医用无菌屏障系统生产用可密封涂胶纸 要求和试验方法;YY /T0698.8-2009 最终灭菌医疗器械包装材料 第8部分:蒸汽灭菌器用重复性使用灭菌容器 要求和试验方法;YY /T0698.9-2009 最终灭菌医疗器械包装材料 第9部分:可密封组合袋、卷材和盖材生产用无涂胶聚烯烃非织造布材料 要求和试验方法;YY/T0698.10-2009 最终灭菌医疗器械包装材料 第10部分:可密封组合袋、卷材和盖材生产用涂胶聚烯烃非织造布材料 要求和试验方法。
医疗器械生物学评价第5部分:体外细胞毒性试验1 范围GB/T 16886 的本部分阐述了评价医疗器械体外细胞毒性的试验方法。
这些方法规定了下列供试品以直接或通过扩散的方式与培养细胞接触和进行孵育;a)用器械的浸提液,和/或b)与器械接触。
这些方法是用相应的生物参数测定哺乳动物细胞的体外生物学反应。
2 规范性引用文件下列文件中的条款通过GB/T 16886 的本部分的引用而成为本部分的条款。
凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本部分,然而,鼓励根据本部分达成协议的各方研究是否可使用这些文件的最新版本。
凡是不注日期的引用文件,其最新版本适用于本部分。
GB/T 16886. 1 医疗器械生物学评价第1部分:评价与试验(GB/T 16886.1-2001,idt ISO 10993- 1:1997)CB/ T 16886. 12-2000 医疗器械生物学评价第12部分:样品制备和参照材料(idt ISO 10993-12 :1996)3 术语与定义GB/ T 16886. 1/ ISO 1993-1中确立的以及下列术语和定义适用于本部分。
3.1阴性对照材料negative control material按照本部分试验时不产生细胞毒性反应的材料。
注:阴性对照的目的是验证背景反应,例如高密度聚乙烯1)已作为合成聚合物的阴性对照材料,氧化陶瓷棒则用作牙科材料的阴性对照物。
3.2阳性对照材料 pos itive control material按照本部分试验时可重现细胞毒性反应的材料。
注:阳性对照的白目的是验证相应试验系统的反应,例如用有机锡作稳定剂的聚氯乙烯2)已用作固体材料和浸提液的阳性对照,酚的稀释液用于浸提液的阳性对照。
_____________________________________1)高密度聚乙烯可从美国药典委员会(Rockvillie, Maryland, USA)和Hatano研究所食品和药品安全中心(Ochiai 729-5 ,Hanagawa.257-Japan)获得。
医疗器械生物学评价医疗器械生物学评价是指通过一系列的实验和测试,对医疗器械与人体生物系统的相互作用进行评估和分析。
它是确保医疗器械安全可靠性和有效性的重要环节。
本文将就医疗器械生物学评价的背景、内容和方法进行探讨。
一、背景随着医疗器械在临床应用中起着越来越重要的作用,对其质量和安全性的要求也越来越高。
医疗器械生物学评价是一种全面、科学、合理的评估方法,它对医疗器械和人体的相互作用进行系统研究,可以评估医疗器械对人体的生物相容性、毒性和免疫反应,从而为医疗器械的应用提供依据。
二、内容医疗器械生物学评价的内容包括生物相容性、毒性和免疫反应等方面。
1. 生物相容性生物相容性是评估医疗器械对人体组织和生物体的相容性能力,其主要考察医疗器械在人体中进入、停留和排除过程中是否能引起有害反应。
一般来说,生物相容性评价包括细胞相容性、组织反应和免疫反应三个方面。
(1)细胞相容性:通过体外实验,评估医疗器械与细胞的相互作用,确定是否有细胞毒性或细胞刺激性等不良反应。
(2)组织反应:通过体内实验,评估医疗器械与组织的相互作用,观察是否引起炎症、纤维化等组织反应。
(3)免疫反应:通过体内和体外实验,评估医疗器械是否会引起免疫反应,包括过敏反应和免疫抑制等。
2. 毒性医疗器械的毒性评价主要考察其对人体有无直接或间接的毒性反应。
毒性评价一般分为急性毒性和亚慢性/慢性毒性两个方面。
(1)急性毒性:评估医疗器械在短时间内对人体的毒性反应。
主要测试项目包括皮肤刺激、眼刺激和吸入刺激等。
(2)亚慢性/慢性毒性:评估医疗器械在长期接触后对人体的毒性反应。
主要测试项目包括皮肤变态反应、致癌性和生殖毒性等。
3. 免疫反应免疫反应评价主要考察医疗器械与人体免疫系统的相互作用。
它包括机体的免疫功能、器官的免疫排斥、特异性免疫应答和免疫毒理学等方面。
三、方法医疗器械生物学评价的方法多种多样,一般包括实验室试验、动物试验和临床试验等。
1. 实验室试验实验室试验是医疗器械生物学评价的基础,通过体外实验评估医疗器械与细胞、组织和免疫系统的相互作用。
医疗器械生物学评价第5部分:体外细胞毒性试验1 范围GB/T 16886 的本部分阐述了评价医疗器械体外细胞毒性的试验方法。
这些方法规定了下列供试品以直接或通过扩散的方式与培养细胞接触和进行孵育;a)用器械的浸提液,和/或b)与器械接触。
这些方法是用相应的生物参数测定哺乳动物细胞的体外生物学反应。
2 规范性引用文件下列文件中的条款通过GB/T 16886 的本部分的引用而成为本部分的条款。
凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本部分,然而,鼓励根据本部分达成协议的各方研究是否可使用这些文件的最新版本。
凡是不注日期的引用文件,其最新版本适用于本部分。
GB/T 16886. 1 医疗器械生物学评价第1部分:评价与试验(GB/T 16886.1-2001,idt ISO 10993- 1:1997)CB/ T 16886. 12-2000 医疗器械生物学评价第12部分:样品制备和参照材料(idt ISO 10993-12 :1996)3 术语与定义GB/ T 16886. 1/ ISO 1993-1中确立的以及下列术语和定义适用于本部分。
3.1阴性对照材料negative control material按照本部分试验时不产生细胞毒性反应的材料。
注:阴性对照的目的是验证背景反应,例如高密度聚乙烯1)已作为合成聚合物的阴性对照材料,氧化陶瓷棒则用作牙科材料的阴性对照物。
3.2阳性对照材料 pos itive control material按照本部分试验时可重现细胞毒性反应的材料。
注:阳性对照的白目的是验证相应试验系统的反应,例如用有机锡作稳定剂的聚氯乙烯2)已用作固体材料和浸提液的阳性对照,酚的稀释液用于浸提液的阳性对照。
_____________________________________1)高密度聚乙烯可从美国药典委员会(Rockvillie, Maryland, USA)和Hatano研究所食品和药品安全中心(Ochiai 729-5 ,Hanagawa.257-Japan)获得。
提供这一信息是为本部分的使用者提供方便,但ISO对使用该产品不提供担保。
2)有机锡聚氯乙烯阳性对照材料可从SIMS Portex Ltd,Hythe, Kent,CT21 6JL,UK(产品号码499-300-000)获得。
ZDEC和ZDBC 聚氨甲酸乙酯可从Hatano 研究所食品和药品安全中心(Ochiai 729-5 , Hanagawa257-Japan) 获得。
提供这一信息是为本部分的使用者提供方便,但ISO 对使用该产品不提供担保。
3)3.3试剂对照 reagent control在不加试验材料的条件下,按浸提条件和试验步骤得到的浸提介质。
注:在本部分中,该定义取代GB/T 16886. 12/ lSO 10993-12中3.1给出的定义。
3.4培养器皿 culture vessels适用于细胞培养的器皿,包插玻璃培养皿、塑料培养瓶或塑料多孔培养板和微量滴定板等器皿。
注,在这些试验方法中,这些器皿只要符合组织培养级别的要求,并适用于哺乳动物细胞培养.可以互换使用。
3.5近汇合 subconfluency在对数生长期末,约80% 的细胞汇合。
4 样品制备4.1 总则供试品可选用:a)材料浸提液;和/或B)材料本身。
样品制备应符合CB/T 16886. 12/ISO 10993-12。
4.2 材料浸提液的制备4.2.1 浸提原则为了测定潜在的毒理学危害,浸提条件应模拟或严于临床使用条件,但不应导致试验材料发生,诸如熔化、溶解或化学结构改变等明显变化。
注:浸提液中任何内源性或外源性物质的浓度及其接触试验细胞的量取决于接触面积、浸提体积、pH、化学溶解度、扩散率、溶质度、搅拌、温度、时间和其他因素。
4.2.2 浸提介质哺乳动物细胞检测中应使用下列一种或几种溶剂。
对浸提介质的选择应进行验证:a) 含血清培养基;b) 无血清培养基;c) 生理盐水溶液;d) 其他适宜的溶剂。
注:溶剂的选择应反映出浸提的目的,并应考虑使用极性和非极性两种溶剂。
适宜的溶剂包括纯水、植物油、二甲基亚砜(DMSO)。
在所选择的测试系统中.如DMSO 浓度大于0.5% (体积分数),则有细胞毒性。
4.2.3 浸提条件4.2.3.1 浸提应使用无菌技术,在无菌、化学惰性的封闭容器中进行,应符合GB/ T 16886. 12/lSO 10993-12 的基本要求。
4.2.3.2 推荐的浸提条件是:a) 37℃±2℃下不少于24 h;b) 50℃±2℃下72 h±2 h;c) 70℃±2℃下24 h±2 h;d) 121℃±2℃下1 h士0. 2 h。
可根据器械特性和具体使用情况选择推荐的条件。
当浸提过程使用含血清培养基时,只能来用4. 2. 3. 2 a)规定的浸提条件。
4.2.3.3 浸提液用于细胞之前,如果进行过滤、离心或其他处置方法,最终报告(见第9章)中应予以说明,对浸提液pH值的调整也应在提告中说明。
对浸提撞的处理,例如对pH值的调整会影响试验结果。
4.3 直接接触试验材料的制备4.3.1 在细胞毒性检测中,多种形状、尺寸或物理状态(即液态或固态)的材料未经修整即可进行测试。
固体样品应至少有一个平面。
其他形状和物理状态的样品应进行调整。
4.3.2 应考虑试验样品的无菌性。
4.3.2.1 无菌器械试验材料的试验全过程应按无菌操作法进行。
4.3.2.2 试验材料如取自通常在使用前灭菌的器械,应按照制造商提供的方法灭菌,并且试验全过程应按无菌操作法进行。
用于试验系统之前,制备试验材料时应考虑灭菌方法或灭菌剂对器械的影响。
4.3.2.3 试撞材料如取自使用中不需要灭菌的器械,则应在供应状态下使用,但在试验全过程中应按无菌操作法进行。
4.3.3 对液体进行试验应:a)直接附着;或b)附着到具有生物惰性和吸收性的基质上。
注:滤膜是适用的基质。
4.3.4 对高吸收性材料,如果可能,试验前应用培养基将其浸透,以防吸收试验器皿中的培养基。
5 细胞系5.1 优先采用已建立的细胞系并应从认可的贮源获取3)5.2 在需要特殊敏感性时,只能使用直接由活体组织获取的原代细胞、细胞系和器官型培养物,但需证明其反应的重现性和准确性。
5.3 细胞系原种培养贮存时,应放在相应培养基内,在-80℃或-80℃以下冻存,培养基内加有细胞保护剂,如二甲基亚砜或甘油等,长期贮存(几月至几年)只能在-130℃或-130℃以下冻存。
5.4 试验应使用无支原体污染细胞,使用前采用可靠方法检测是否存在支原体污染。
6 培养基6.1 培养基应无菌。
6.2 含血清或无血清培养基应符合选定细胞系的生长要求。
注:培养基中允许含有对试验无不利影响的抗生素。
培养基的稳定性与其成分和贮存条件有关。
含谷氨酰胺和血清的培养基在2℃~8℃条件下贮存不超过一周,含谷氨酰胺的无血清培养基在2℃~8℃条件下贮存不超过2周。
6.3 培养基的pH 值应为7. 2~7. 4。
7 细胞原种培养制备7.1 用选定的细胞系和培养基制备试验所需足够的细胞。
使用冻存细胞时,如加有细胞保护剂应除去,使用前至少传代培养一次。
7.2 取出细胞,用适宜的酶分散法和/或机械分散法制备成细胞悬浮液。
———————————————3)推荐的细胞系如CCL1(NCTC clone 929),CCL 163(Balb/3T3 clone A31)、CCL171(MRC-5)、CCL 75(Wl-38)、CCL81(Vero)、CCL10[BHK-21(C-l3) 和V-79 379A。
这一信息只是为本部分的使用者提供方便,但ISO 对使用该产品不提供担保。
也可使用其他能得出相同或更佳结果的细胞系。
8 试验步骤8.1 平行样数至少采用三个平行试验样品数和对照数。
8.2 浸提液试验8.2.1 该试验用于细胞毒性定性和定量评价。
8.2.2 从持续搅拌的细胞悬浮液中吸取等量的悬浮液,注入浸提液接触的每只培养器皿内,轻轻转动培养器皿使细胞均匀地分散在器皿的表面。
8.2.3 根据培养基选择含或不含5%(体帜分数)二氧化碳的空气作为缓冲系统,在37℃士2℃温度下进行培养。
试验应在近汇合单层细胞或新鲜悬浮细胞上进行。
如仅是检测克隆形成,应采用较低的细胞密度。
8.2.4 试验前用显微镜检查培养细胞的近汇合和形态情况。
8.2.5 试验可选用:a ) 浸提原液;和b ) 以培养基作稀释剂的系列浸提稀释液。
试验如果用单层细胞,应弃去培养器皿中的培养基,在每只器皿内加等量浸提液或上述稀释液。
如用悬浮细胞进行试验,细胞悬浮液制备好后立即将浸提液或上述稀稀释液加到每只平行器皿中。
8.2.6 采用水等非生理浸提液时,浸提液用培养基稀稀后应在最高生理相容浓度下试验。
注:建议在稀释浸提液时使用浓缩的(如2倍、5倍)培养基。
8.2.7 加等量的空白试剂和阴性及阳性对照液至其他平行器血中。
注:如需要,还可以用新新鲜培养基做对照试验。
8.2.8 器皿按8. 2.3 中所述同样条件进行培养,培养期间应符合选定方法的要求。
8.2.9 经过至少24 h 的培养后,接8. 5 确定细胞毒性反应。
8.3 直接接触试验8.3.1 该试验用于细胞毒性定性和定量评价。
8.3.2 从持续搅拌的细胞悬液中吸取等量的悬浮液,注入与试验样品直接接触的每只器皿内。
轻轻水平转动器皿,使细胞均匀地分散在每只器皿的表面。
8.3.3 根据培养基选择含或不含5% (体积分数)二氧化碳的空气作为缓冲系统,在37℃±2℃温度下进行培养,直至培养细胞生长至近汇合。
8.3.4 试验前用显微镜检查培养细胞的近汇合和形态情况。
8.3.5 弃去培养器皿中的培养基,加新鲜培养基至各器皿内。
8.3.6 在每只器皿中央部位的细胞层上各轻轻放置一个试验样品,应确保样品覆盖细胞层表面约十分之操作时应注意防止样品不必耍的移动,否则可能会导致细胞的物理性损伤,这可从被移动细胞的碎片来判断。
注:如可能,在细胞加入前将样品放入培养器皿内。
8.3.7 同法制备阴性和阳性对照材料平行器皿。
8.3.8 器皿按8.3.3中所述同样条件进行培养,培养期间(最少24h)应符合选定方法的要求。
8.3.9 去除上层培养基,接8.5确定细胞毒性反应。
8.4 间接接触试验8.4.1 琼脂扩散试验8.4.1.1 该试验用于细胞毒性定性评价,该方法不适用于不能通过琼脂层扩散的可沥滤物或与琼脂反应的物质。
8.4.1.2 从持续搅样的细跑悬浮液中吸取等量的悬浮液,注入每只试验用平行器皿内。
轻轻水平转动器皿.使细胞均匀地分散在每只器皿的表面。