E.Z.N.A Gel Extraction Kit 琼脂糖凝胶回收试剂盒 中文 说明书
- 格式:doc
- 大小:27.50 KB
- 文档页数:1

琼脂糖凝胶DNA回收试剂盒(离心柱型)QC标准操作流程1. 实验试剂和耗材1.2 实验试剂1.3 耗材2. 实验操作流程胶回收试剂盒的检测须进行两个实验验证,第一个实验为GelRed染色法,第二个实验为EB染色法。
2.1 GelRed染色法2.1.1 1.5%琼脂糖凝胶的制备(1)准确称取1.5g琼脂糖置于250ml三角锥形瓶中,添加125ml 1×TAE溶液。
(2)轻微混匀后将其置于微波炉中,高火加热6min至溶液沸腾,取出锥形瓶放置室温2min。
(3)添加1×TAE溶液至110ml,再次于微波炉中高火加热3min至溶液沸腾。
(4)室温放置10min后将溶液倒入插有梳子的胶槽内,待胶块凝固后使用。
2.1.2 胶回收实验(1)取6个2ml离心管,依次编号为1-6号,使用分析天平准确称取空离心管的重量。
(2)用干净锋利的手术刀片切下不含核酸染料的1.5%琼脂糖凝胶胶块,放入已称重的2ml离心管中,称取胶块与离心管总重,计算凝胶块的重量。
要求胶块重量不超过200mg 为宜。
(3)添加溶胶液Binding Solution B,要求1、2号管中每100mg琼脂糖凝胶加入200ul Binding Solution B,3、4号管中每100mg琼脂糖凝胶加入300ul Binding Solution B,5、6号管中每100mg琼脂糖凝胶加入400ul Binding Solution B.(4)每管中添加50ul DL2502,混匀后置于60℃水浴5min,期间间断混合,直至凝胶块完全融化。
【注意事项】观察各管内溶胶液溶解胶块的快慢程度。
(5)将上述混合液转移至套放于2ml收集管的GenClean柱中,室温放置2min,10000rpm 室温离心1min,取出GenClean 柱,并倒掉收集管中废液。
(6)将GenClean 柱重新放回收集管中,加入500ul Wash Solution,于12000rpm,室温离心1min,倒掉收集管中废液。


琼脂糖凝胶回收试剂盒E.Z.N.A Gel Extraction Kit适合于D2501-**,D2500-**&D2502-**准备工作1. 浓缩的SPW Buffer需用乙醇按如下稀释:D2500-00&D2501-00&D2502-00 加入20ml 100%的乙醇;D2500-01/02&D2501-01/02&D2502-01/02 每瓶加入100ml 100%的乙醇;注意:稀释后的DNA Wash Buffer需室温保存;所有步骤必须在室温下进行.操作方案配制琼脂糖EB凝胶,电泳以分离DNA片段.任何类型或等级的琼脂糖都可以使用.我们强烈的推荐使用新鲜的TAE/TBE电泳缓冲液.不要重复使用电泳缓冲液,旧的电泳缓冲液PH会增加而降低DNA的回收产量;2. 电泳足够时间后,在紫外灯下小心地把所需的DNA片段切下来.并尽量去除多余的凝胶.注意:DNA在紫外灯下的曝光的时间不要超过30秒,同时在紫外灯下操作的时候一定要戴保护眼镜.3.称取空离心管的重量,切下带目的片段的凝胶装在1.5ml离心管中并称其重量,求出凝胶块的重量,近似地确定其体积.一般情况下,凝胶的密度为1g/ml,于是凝胶的体积与重量的关系可按下面换算:凝胶薄片的重量为0.2g 则其体积为0.2ml;加入等倍凝胶体积的Binding Buffer,把混合物置于55℃~65℃水浴中温浴7min至凝胶完全融化,其间每隔2-3分钟混匀一次;重要提醒:在凝胶完全溶解之后,注意凝胶-Binding Buffer混合物的pH值.如果其pH值大于8的话,DNA的产量将大大减少.观察混合物的颜色,如果是橙色或红色,则要加入5μl 浓度为5 M,pH为5.2的醋酸钠,以调低其pH值.经过这一调节,该混合物的颜色将恢复为正常的浅黄色.一般情况下,使用新鲜的电泳缓冲液,凝胶-Binding buffer混合物的PH值的不会升高;4.转移700μl的DNA-琼脂糖溶液到一个HiBindTM DNA柱子,并把柱子装在一个干净的2ml收集管内,室温下,10,000×g离心1min,弃去液体.一个HiBind DNA柱子最多可容纳700μl的溶液,如果DNA-琼脂糖混合物的体积大于700μl,可先转移700μl溶液至柱子,离心完后,将余下的溶液继续加上柱子上.但是每一个HiBindTM柱子最多可以结合25~30μg DNA.如果预期产量较大,则把样品分别加到合适数目的柱中.5. 将柱子重新套回收集管中,加300μl Binding Buffer至HiBind DNA 柱子中;室温下,10,000×g离心1分钟,去弃滤出液;这一步相当关键,不要忽略此步.6.将柱子重新套回收集管中,加入700μl SPW Wash buffer至HiBind DNA柱子中,室温下,10,000×g离心1分钟,去弃滤出液;注:SPW Wash buffer在使用前必须按瓶子标鉴要求用无水乙醇进行稀释.7. 将柱子重新套回收集管中,重复加入700μl SPW Wash buffer至HiBind DNA柱子中,室温下,10,000×g离心1分钟, 弃去滤出液;,将空柱子重新套回收集管中,10,000×g离心1min以甩干柱基质残余的液体.这步可以去除柱子基质上残余的乙醇,不要省略此步―――对得到好的DNA产量是十分重要的.8. 把柱子装在一个干净的1.5ml离心管上,加入30~50μl洗脱液或灭菌水上柱子膜上,10,000×g离心1分钟,离心管中的溶液就是纯化的DNA产物,保存于-20度.如果再洗脱一次的话可以把残余的DNA洗脱出来,不过那样的浓度就会较低.DNA的产量及质量:把纯化产物的样品稀释一定的倍数后,分别在260nm和280nm下测其光吸收值,回收得到的DNA的浓度可按以下公式来计算: DNA浓度=光吸收260×50×稀释倍数μg/ml长度大于500bp的片段通常能纯化得到80%的产量. 而50bp~500bp的带则可达到55%~80%的回收率.(光吸收260/光吸收280)的比率是核酸纯度的一个标记.如果此值1.8,则意味着核酸的纯度>90%.另一方面,如果纯化产物的产量较低时,可以用琼脂糖EB电泳估算产物的浓度.载体。

实验一RT-PCR法钓取小鼠肝脏GAPDH基因一、TRlzol试剂提取小鼠肝脏总RNATRIzol RNA提取试剂盒是由GIBCOBRL公司推出的产品,其操作方法简捷、方便,lh之内即可完成,所制备的RNA可用于cDNA合成及Northern blot等。
【试剂】TRlZOL试剂氯仿异丙醇75%乙醇(DEPC水配制)无RNase水【操作方法】(1)收获细胞1~5×106;或50-100mg组织,加TRlzol试剂lml匀浆。
(2),移入1.5ml Ep管中,室温静置5min。
(3)每1ml TRIZOL中加氯仿0.2m1,摇振15s,置室温2~3min。
(4)4℃离心,12,000g×15min。
(5)仔细吸取上层水相,移至另一Ep管中。
(6)加0.5ml异丙醇,混匀,置室温10min。
(7)4℃离心,12,000g×10min。
(8)弃上清,加75%乙醇1m1,轻轻摇振,充分洗涤沉淀,4℃离心,7500g×5min。
(9)弃上清,真空干燥后,沉淀重悬于50μl无RNase水中。
一70℃保存备用。
二、核酸的定量(1)分光光度法测定核酸浓度。
组成核酸分子的碱基,均具有一定的吸收紫外线特性,最大吸收波长为250~270nm之间。
例如腺嘌呤的最大紫外线吸收值在260.5nm,胞嘧啶:267nm 鸟嘌呤:276nM胸腺嘧啶:264.5 nm尿嘧啶259nm。
这些碱基与戊糖、磷酸形成核苷酸后,其最大吸收峰不会改变,但核苷酸最大吸收波长是260nm,吸收低谷在230nm,这个物理特性为紫外分光光度法测定核酸溶液浓度提供了基础。
在波长260nm紫外线下,1OD值的光密度相当于双链DNA浓度为50ug/ ml;单链DNA或RNA为40ug/ml;单链寡核苷酸为20ug/ml。
另外,还可以通过测定260nm和280nm的紫外线吸收值,然后根据其比值来估计核酸的纯度。
DNA样品的比值为1.8,RNA样品的比值为2.0。
琼脂糖凝胶回收试剂盒E.Z.N.A Gel Extraction Kit适合于D2501-**,D2500-**&D2502-**准备工作1. 浓缩的SPW Buffer需用乙醇按如下稀释:D2500-00&D2501-00&D2502-00 加入20ml 100%的乙醇;D2500-01/02&D2501-01/02&D2502-01/02 每瓶加入100ml 100%的乙醇;注意:稀释后的DNA Wash Buffer需室温保存;所有步骤必须在室温下进行.操作方案配制琼脂糖EB凝胶,电泳以分离DNA片段.任何类型或等级的琼脂糖都可以使用.我们强烈的推荐使用新鲜的TAE/TBE电泳缓冲液.不要重复使用电泳缓冲液,旧的电泳缓冲液PH会增加而降低DNA的回收产量;2. 电泳足够时间后,在紫外灯下小心地把所需的DNA片段切下来.并尽量去除多余的凝胶.注意:DNA在紫外灯下的曝光的时间不要超过30秒,同时在紫外灯下操作的时候一定要戴保护眼镜.3.称取空离心管的重量,切下带目的片段的凝胶装在1.5ml离心管中并称其重量,求出凝胶块的重量,近似地确定其体积.一般情况下,凝胶的密度为1g/ml,于是凝胶的体积与重量的关系可按下面换算:凝胶薄片的重量为0.2g 则其体积为0.2ml;加入等倍凝胶体积的Binding Buffer,把混合物置于55℃~65℃水浴中温浴7min至凝胶完全融化,其间每隔2-3分钟混匀一次;重要提醒:在凝胶完全溶解之后,注意凝胶-Binding Buffer混合物的pH值.如果其pH值大于8的话,DNA的产量将大大减少.观察混合物的颜色,如果是橙色或红色,则要加入5μl 浓度为5 M,pH为5.2的醋酸钠,以调低其pH值.经过这一调节,该混合物的颜色将恢复为正常的浅黄色.一般情况下,使用新鲜的电泳缓冲液,凝胶-Binding buffer混合物的PH值的不会升高;4.转移700μl的DNA-琼脂糖溶液到一个HiBindTM DNA柱子,并把柱子装在一个干净的2ml收集管内,室温下,10,000×g离心1min,弃去液体.一个HiBind DNA柱子最多可容纳700μl的溶液,如果DNA-琼脂糖混合物的体积大于700μl,可先转移700μl溶液至柱子,离心完后,将余下的溶液继续加上柱子上.但是每一个HiBindTM柱子最多可以结合25~30μg DNA.如果预期产量较大,则把样品分别加到合适数目的柱中.5. 将柱子重新套回收集管中,加300μl Binding Buffer至HiBind DNA 柱子中;室温下,10,000×g离心1分钟,去弃滤出液;这一步相当关键,不要忽略此步.6.将柱子重新套回收集管中,加入700μl SPW Wash buffer至HiBind DNA柱子中,室温下,10,000×g离心1分钟,去弃滤出液;注:SPW Wash buffer在使用前必须按瓶子标鉴要求用无水乙醇进行稀释.7. 将柱子重新套回收集管中,重复加入700μl SPW Wash buffer至HiBind DNA柱子中,室温下,10,000×g离心1分钟, 弃去滤出液;,将空柱子重新套回收集管中,10,000×g离心1min以甩干柱基质残余的液体.这步可以去除柱子基质上残余的乙醇,不要省略此步―――对得到好的DNA产量是十分重要的.8. 把柱子装在一个干净的1.5ml离心管上,加入30~50μl洗脱液或灭菌水上柱子膜上,10,000×g离心1分钟,离心管中的溶液就是纯化的DNA产物,保存于-20度.如果再洗脱一次的话可以把残余的DNA洗脱出来,不过那样的浓度就会较低.DNA的产量及质量:把纯化产物的样品稀释一定的倍数后,分别在260nm和280nm下测其光吸收值,回收得到的DNA的浓度可按以下公式来计算: DNA浓度=光吸收260×50×稀释倍数μg/ml长度大于500bp的片段通常能纯化得到80%的产量. 而50bp~500bp的带则可达到55%~80%的回收率.(光吸收260/光吸收280)的比率是核酸纯度的一个标记.如果此值1.8,则意味着核酸的纯度>90%.另一方面,如果纯化产物的产量较低时,可以用琼脂糖EB电泳估算产物的浓度.载体。




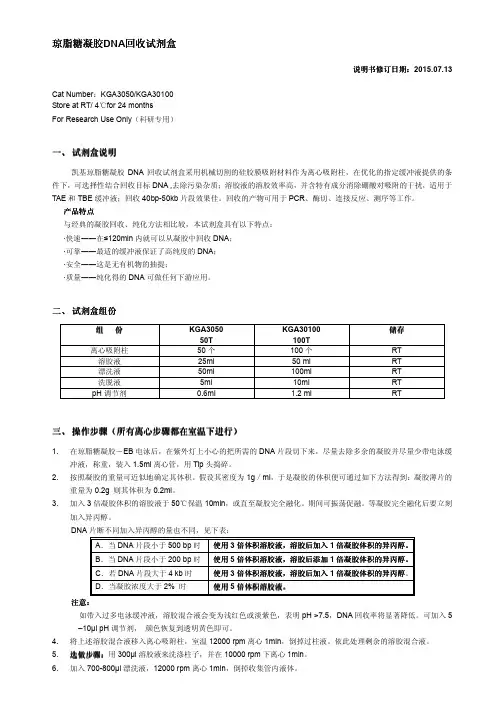
琼脂糖凝胶DNA 回收试剂盒说明书修订日期说明书修订日期::2015.07.13Cat Number :KGA3050/KGA30100 Store at RT/ 4for 24 months ℃For Research Use Only (科研专用)一、 试剂盒说明凯基琼脂糖凝胶DNA 回收试剂盒采用机械切割的硅胶膜吸附材料作为离心吸附柱,在优化的指定缓冲液提供的条件下,可选择性结合回收目标DNA ,去除污染杂质;溶胶液的溶胶效率高,并含特有成分消除硼酸对吸附的干扰,适用于TAE 和TBE 缓冲液;回收40bp-50kb 片段效果佳。
回收的产物可用于PCR 、酶切、连接反应、测序等工作。
产品特点与经典的凝胶回收、纯化方法相比较,本试剂盒具有以下特点:·快速――在≤120min 内就可以从凝胶中回收DNA ; ·可靠――最适的缓冲液保证了高纯度的DNA ; ·安全――这是无有机物的抽提;·质量――纯化得的DNA 可做任何下游应用。
二、 试剂盒组份组 份 KGA3050 50T KGA30100 100T 储存 离心吸附柱50个 100个 RT 溶胶液 25ml 50 ml RT 漂洗液 50ml 100ml RT 洗脱液 5ml 10ml RT pH 调节剂0.6ml1.2 mlRT三、 操作步骤操作步骤((所有离心步骤都在室温下进行所有离心步骤都在室温下进行))1. 在琼脂糖凝胶-EB 电泳后,在紫外灯上小心的把所需的DNA 片段切下来,尽量去除多余的凝胶并尽量少带电泳缓冲液,称重,装入1.5ml 离心管,用Tip 头捣碎。
2. 按照凝胶的重量可近似地确定其体积。
假设其密度为1g /ml ,于是凝胶的体积便可通过如下方法得到:凝胶薄片的重量为0.2g 则其体积为0.2ml 。
3.加入3倍凝胶体积的溶胶液于50℃保温10min ,或直至凝胶完全融化。
期间可振荡促融。
杭州昊鑫生物科技股份有限公司 htpp://AidQuick Gel Extraction Kit琼脂糖凝胶纯化回收试剂盒目录号:DR01❖适用范围:适用于琼脂糖凝胶DNA回收❖试剂盒组成、储存、稳定性:试剂盒组成保存50次(DR0101)100次(DR0102)200次(DR0103)平衡液室温5ml 10ml 20ml 溶胶液DD 室温50 ml 50ml×2 200 ml漂洗液WB 室温15 ml 25 ml 50 ml 第一次使用前按说明加指定量乙醇洗脱缓冲液EB 室温10 ml 15 ml 15 ml吸附柱EC 室温50个100个200个收集管(2ml)室温50个100个200个本试剂盒在室温储存12个月不影响使用效果。
储存事项:1.所有的溶液应该是澄清的,如果环境温度低时溶液可能形成沉淀,此时不应该直接使用,可在37℃水浴加热几分钟,即可恢复澄清。
使用前应该恢复到室温。
2.储存于低温(4℃或者-20℃)会造成溶液沉淀,影响使用效果,因此运输和储存均在室温下(15℃-25℃)进行。
3.避免试剂长时间暴露于空气中产生挥发、氧化、pH值变化,各溶液使用后应及时盖紧盖子。
❖产品介绍:在高离序盐存在的情况下,DNA片断选择性的吸附于离心柱内的硅基质膜上,再通过一系列快速的漂洗-离心的步骤,漂洗液将引物、核苷酸、蛋白、酶等杂质去除,最后低盐、高pH值的洗脱缓冲液将纯净DNA从硅基质膜上洗脱。
❖产品特点:1.离心吸附柱内硅基质膜全部采用进口世界著名公司特制吸附膜,柱与柱之间吸附量差异极小,可重复性好。
克服了国产试剂盒膜质量不稳定的弊端。
2.使用了优质溶胶液,不含传统溶胶液的碘化钠和高氯酸盐,不抑制回收后酶切、连接克隆等下游反应。
3.溶胶液加酚红调制成为了黄颜色,便于观察溶胶效果和监测pH值变化从而达到最佳结合效果,大大提高回收效率。
4.改进的溶胶液配方,大大提高了缓冲能力和稳定性,即使样品变化很大也能将PH缓冲在最佳结合范围内。
DNA凝胶回收QIAquick Gel Extraction Kit① (提前记录1.5ml离心管重量)在紫外灯光下切下含有目的DNA的琼脂糖凝胶,用纸巾吸干凝胶表面液体并切碎(方便融化),计算凝胶重量(重量作为凝胶体积,100mg≈100μl体积)②入3个凝胶体积的Buffer QG ,每个吸附柱的最大量为400mg。
对于>2%凝胶浓度,加入6个凝胶体积的Buffer QG。
③混个均匀后于50℃加热10min(或者直至凝胶全部溶解),每2-3min间断混合以助溶解,凝胶完全融化后,确认混合液的颜色为黄色(类似于Buffer QG没加入溶解的凝胶之前的颜色)。
如果混合液的颜色是橙色的或者紫色的,加入10μL 3M sodium acetate(醋酸钠), pH 5.0,混合。
混合液转变为黄色。
④加入一个体积的异丙醇于样品中,混合。
⑤将QIAquick吸附柱置于2ml 收集管中,为了吸附DNA,将样品置于吸附柱中, 17900×g(13000rpm) 离心1min。
弃去液体,将QIAquick吸附柱置于相同的收集管上。
⑥如果DNA用于后续测序,体外转录,显微注射,加入500μL Buffer QG于吸附柱上,离心1min,弃去液体,将吸附柱置于新的收集管上。
⑦洗涤,加入750μL Buffer PE于吸附柱中,离心1min,弃去液体,将吸附柱置于新的收集管上。
注意:如果将DNA用于盐敏感型应用(例如,测序,平末端连接反应中),加入Buffer PE后静置2-5min。
为了去除残留液体,继续离心1min。
⑧将收集柱置于新的1.5mL离心管中。
⑨为了洗提DNA,加入50μL Buffer EB(10mM Tris·Cl, pH 8.5),加入膜中央,离心1min。
为了增加DNA浓度,加入30μL Buffer EB于膜中央,静置1min,离心1min。
孵育50℃最多4min,可提高DNA提取效率。
凝胶DNA回收试剂盒使用说明一、试剂盒组成1.碱性ELUTION缓冲液:用于回收DNA片段。
2.DNA绑定胶柱:用于吸附DNA片段。
3.洗涤缓冲液:用于去除琼脂糖凝胶残渣和其他杂质。
4.电子式试剂盒设备:用于快速洗脱DNA片段。
二、凝胶DNA回收试剂盒使用步骤1.准备工作:a.将碱性ELUTION缓冲液加热至65℃,并保存在试剂盒中。
b.手套和面具是使用过程中必需的,以确保实验环境的干净。
c.严格遵守实验室的安全操作规程。
2.凝胶切割:a. 切割需回收的DNA片段:将琼脂糖凝胶切割为片段大小约为50-5000 bp的目标DNA。
b.将切割好的琼脂糖凝胶片段放入微量离心管中,注意不要弄混不同大小的DNA片段。
c.记录每个DNA片段的位置和大小。
3.DNA回收:a.将切割好的琼脂糖凝胶片段放入DNA绑定胶柱中。
b.将绑定胶柱放入收集管中,通过离心将DNA片段吸附到胶柱上。
c.倒掉废液,并使用洗涤缓冲液洗脱胶柱上的残驻琼脂糖凝胶和其他杂质。
重复此步骤1-2次以确保彻底洗脱。
4.DNA洗脱:a.将洗柱放回空的收集管中。
b.加入预热的碱性ELUTION缓冲液(65℃),封闭试管盖,并放入电子式试剂盒设备中。
c.按照设备操作说明进行启动,并设置相应的洗脱温度和时间。
d.在洗脱完成后,将洗脱液转移到新的离心管中。
可以使用适当的方法(如乙醇沉淀或其他纯化方法)进一步纯化DNA。
5.质量检测:a.使用电泳进行质量检测,以确认回收的DNA片段大小和纯度。
b.如果需要,可以定量将DNA溶液保存在冰箱中以备后续实验使用。
三、注意事项1.使用前确保熟悉试剂盒的组成和使用步骤。
2.使用过程中务必佩戴手套和面具,确保操作环境的洁净。
3.严格按照试剂盒的说明进行操作,并遵循实验室的安全操作规程。
4.需要注意的是,琼脂糖凝胶切割和回收过程极易受到外源DNA污染,因此要确保实验环境的洁净,并尽量避免不必要的接触。
5.回收的DNA片段应尽快进行下一步的实验处理,以避免DNA降解和污染。
2006年《分子生物学实验技术》实验内容一、RT-PCR(一)总RNA的提取实验安排:每两人抽提一管。
为了使操作同步以节省时间,各组样品请一起离心。
操作步骤:1、将100μl液体样品加入1.5ml Ep管中,再加入900μl冰预冷的LS-Biotragents TM(苯酚和异硫氰酸胍的混合物)。
2、将样品剧烈混合后,在室温放置5min。
3、加入200μl氯仿,颠倒Ep管混和两次,并剧烈振荡混和10s。
4、在4℃条件下,以10000×g离心15min。
5、将上层水相转移到一个新的Ep管中,加入等体积的异丙醇(Isopropanol)并混匀,然后在4℃放置至少10min。
6、在4℃条件下,以10000×g离心15min后,小心并尽可能地去除全部上清夜。
7、用1ml 75%乙醇洗涤RNA沉淀和管壁。
8、将RNA沉淀进行干燥(不能完全干燥)处理后,用10μl无RNase污染的水(RNase-Free Water)将RNA溶解并于-20℃保存。
注意事项:1、所有的玻璃器皿均应在使用前于180℃的高温下干烤6hr或更长时间。
2、所用的塑料材料,如吸头、离心管等需用0.1% DEPC水浸泡过夜。
3、配制的溶液应尽可能用0.1% DEPC,在37℃处理12hr以上。
然后用高压灭菌除去残留的DEPC。
不能高压灭菌的试剂,应当用DEPC处理过的无菌双蒸水配制,然后经0.22μm滤膜过滤除菌。
4、操作人员需在超净工作台上操作,并戴一次性口罩、手套,实验过程中手套要勤换。
(二)反转录实验安排:每人做一管。
反应体系(20μl):按下列顺序加样反应条件:42℃ 1h注意事项:1、加样时,一般从体积大的开始加,样品最后加。
如在一般的PCR反应体系中,应先加水、Buffer、dNTPs、引物,最后加酶和模板。
2、液体应直接加到管底,且每加一种试剂后应更换新的吸头。
3、加完所有试剂后,应用手指轻弹混匀,然后低速离心数秒以收集管壁上沾有的液体。
产品简介:碧云天2005年底最新推出的DNA凝胶回收试剂盒(DNA Gel Extraction Kit),是世界上最先进的DNA凝胶回收试剂盒之一。
本产品采用了融胶液,可以使琼脂糖凝胶完全融化。
同时采用了一种新型的离子交换柱,在特定条件下,使DNA能在离心过柱的瞬间,结合到DNA纯化柱上,在一定条件下又能将DNA充分洗脱,从而实现DNA的快速纯化。
无需酚氯仿抽提,无需酒精沉淀,通常12个样品只需不足20分钟即可完成。
DNA纯化柱的容量约为15微克。
适用于PCR反应后去除引物,酶,矿物油,甘油,盐等杂质;也同样适用于酶切,连接,磷酸化,补平或切平,随机引物等反应后的DNA纯化。
所得的DNA可直接用于酶切,连接,转化细菌,测序,PCR,杂交等后续操作。
本试剂盒适用于纯化100bp-10kb DNA。
长至30个碱基的引物均可被完全去除。
DNA回收效率通常为60-90%。
接近100bp或10kb的DNA片断回收效率要略低一些,大于10kb的DNA回收效率迅速下降。
另外如果样品中DNA含量特别低也会导致回收效率下降。
每个试剂盒足够用于50个平均重量不超过400微克的凝胶样品。
包装清单:保存条件:室温保存,一年有效。
注意事项:第一次使用前在溶液II (洗涤液)中加入39ml无水乙醇,混匀,并在瓶上做好标记。
溶液I对人体有刺激性,操作时请小心,并注意适当防护。
本试剂盒所有操作均在室温进行,操作时无需冰浴。
所有离心也均在室温进行。
废液收集管在一次抽提中需多次使用,切勿中途丢弃。
为了您的安全和健康,请穿实验服并戴一次性手套操作。
使用说明:1. 切胶回收后,称重,将胶切碎或在离心管内用1ml枪头捣碎。
2. 加入等体积的溶液I(如胶为100mg,则加100微升溶液I),vortex或颠倒混匀。
3. 50-60℃水浴加热约10分钟至胶全融,期间,需vortex或颠倒混匀3-4次,以加速凝胶融解。
若果胶碎片较小,3-5分钟即可全融;凝胶碎片较大则需较长时间,胶全融后至少再在50-60℃水浴加热2分钟。
操作流程1. 使用TAE缓冲液或TBE缓冲液制作琼脂糖凝胶,然后对目的DNA进行琼脂糖凝胶电泳。
2. 在紫外灯下切出含有目的DNA的琼脂糖凝胶,用纸巾吸尽凝胶表面的液体。
此时应注意尽量切除不含目的DNA部分的凝胶,尽量减小凝胶体积,提高DNA回收率。
胶块超过300 mg时,请使用多个Column进行回收,否则严重影响收率。
注)切胶时请注意不要将DNA 长时间暴露于紫外灯下,以防止DNA损伤。
3. 切碎胶块。
胶块切碎后可以加快操作步骤6的胶块溶解时间,提高DNA回收率。
4. 称量胶块重量,计算胶块体积。
计算胶块体积时,以1 mg=1 μl 进行计算。
5. 向胶块中加入胶块溶解液Buffer GM,Buffer GM的加量如下表:凝胶浓度Buffer GM使用量1.0%3个凝胶体积量1.0%~1.5%4个凝胶体积量1.5%~2.0%5个凝胶体积量6. 均匀混合后室温15-25℃溶解胶块(胶浓度较大或比较难溶时可以在37℃加热)。
此时应间断振荡混合,使胶块充分溶解(约5~10分钟)。
7. 当凝胶完全溶解后,观察溶胶液的颜色,如果溶胶液颜色由黄色变为橙色或粉色,向上述胶块溶解液中加入3 M醋酸钠溶液(pH5.2)10 μl,均匀混合至溶液恢复黄色。
当分离小于400 bp的DNA片段时,应在此溶液中再加入终浓度为20%的异丙醇。
8. 将试剂盒中的Spin Column安置于Collection Tube上。
9. 将上述操作步骤7的溶液转移至Spin Column中,12,000 rpm离心1分钟,弃滤液。
注)如将滤液再加入Spin Column中离心一次,可以提高DNA的回收率。
10. 将700 μl 的Buffer WB加入Spin Column中,室温12,000 rpm离心30秒钟,弃滤液。
注)请确认Buffer WB中已经加入了指定体积的100%乙醇。
11. 重复操作步骤10。
12. 将Spin Column安置于Collection Tube上,室温12,000 rpm离心1分钟。
琼脂糖凝胶回收试剂盒E.Z.N.A Gel Extraction Kit
适合于D2501-**,D2500-**&D2502-**
准备工作
1. 浓缩的SPW Buffer需用乙醇按如下稀释:
D2500-00&D2501-00&D2502-00 加入20ml 100%的乙醇;
D2500-01/02&D2501-01/02&D2502-01/02 每瓶加入100ml 100%的乙醇;
注意:稀释后的DNA Wash Buffer需室温保存;所有步骤必须在室温下进行.
操作方案
配制琼脂糖EB凝胶,电泳以分离DNA片段.任何类型或等级的琼脂糖都可以使用.
我们强烈的推荐使用新鲜的TAE/TBE电泳缓冲液.不要重复使用电泳缓冲液,旧的电泳缓冲液PH会增加而降低DNA的回收产量;
2. 电泳足够时间后,在紫外灯下小心地把所需的DNA片段切下来.并尽量去除多余的凝胶.
注意:DNA在紫外灯下的曝光的时间不要超过30秒,同时在紫外灯下操作的时候一定要戴保护眼镜.
3.称取空离心管的重量,切下带目的片段的凝胶装在1.5ml离心管中并称其重量,求出凝胶块的重量,近似地确定其体积.一般情况下,凝胶的密度为1g/ml,于是凝胶的体积与重量的关系可按下面换算:凝胶薄片的重量为0.2g 则其体积为0.2ml;加入等倍凝胶体积的Binding Buffer,把混合物置于55℃~65℃水浴中温浴7min至凝胶完全融化,其间每隔2-3分钟混匀一次;
重要提醒:在凝胶完全溶解之后,注意凝胶-Binding Buffer混合物的pH值.如果其pH值大于8的话,DNA的产量将大大减少.观察混合物的颜色,如果是橙色或红色,则要加入5μl 浓度为5 M,pH为5.2的醋酸钠,以调低其pH值.经过这一调节,该混合物的颜色将恢复为正常的浅黄色.一般情况下,使用新鲜的电泳缓冲液,凝胶-Binding buffer混合物的PH值的不会升高;
4.转移700μl的DNA-琼脂糖溶液到一个HiBindTM DNA柱子,并把柱子装在一个干净的2ml收集管内,室温下,10,000×g离心1min,弃去液体.
一个HiBind DNA柱子最多可容纳700μl的溶液,如果DNA-琼脂糖混合物的体积大于700μl,可先转移700μl溶液至柱子,离心完后,将余下的溶液继续加上柱子上.但是每一个HiBindTM柱子最多可以结合25~30μg DNA.如果预期产量较大,则把样品分别加到合适数目的柱中.
5. 将柱子重新套回收集管中,加300μl Binding Buffer至HiBind DNA 柱子中;室温下,10,000×g离心1分钟,去弃滤出液;这一步相当关键,不要忽略此步.
6.将柱子重新套回收集管中,加入700μl SPW Wash buffer至HiBind DNA柱子中,室温下,10,000×g离心1分钟,去弃滤出液;注:SPW Wash buffer在使用前必须按瓶子标鉴要求用无水乙醇进行稀释.
7. 将柱子重新套回收集管中,重复加入700μl SPW Wash buffer至HiBind DNA柱子中,室温下,10,000×g离心1分钟, 弃去滤出液;,将空柱子重新套回收集管中,10,000×g离心1min以甩干柱基质残余的液体.
这步可以去除柱子基质上残余的乙醇,不要省略此步―――对得到好的DNA产量是十分重要的.
8. 把柱子装在一个干净的1.5ml离心管上,加入30~50μl洗脱液或灭菌水上柱子膜上,10,000×g离心1分钟,离心管中的溶液就是纯化的DNA产物,保存于-20度.
如果再洗脱一次的话可以把残余的DNA洗脱出来,不过那样的浓度就会较低.
DNA的产量及质量:把纯化产物的样品稀释一定的倍数后,分别在260nm和280nm下测其光吸收值,回收得到的DNA的浓度可按以下公式来计算: DNA浓度=光吸收260×50×稀释倍数μg/ml长度大于500bp的片段通常能纯化得到80%的产量. 而50bp~500bp的带则可达到55%~80%的回收率.(光吸收260/光吸收280)的比率是核酸纯度的一个标记.如果此值1.8,则意味着核酸的纯度>90%.另一方面,如果纯化产物的产量较低时,可以用琼脂糖EB电泳估算产物的浓度.载体。