生物竞赛-生物信息学部分-分子进化树
- 格式:ppt
- 大小:2.96 MB
- 文档页数:40

生物信息学进化树进化树是生物信息学中的一项重要工具,用于揭示生物物种之间的进化关系。
通过分析不同物种的基因组序列,可以推断它们之间的亲缘关系和进化历史。
进化树可以帮助我们了解生物的演化过程,揭示不同物种的共同祖先以及它们之间的分支关系。
在构建进化树的过程中,首先需要收集各个物种的基因组数据。
这些数据可以是DNA序列、蛋白质序列或其他形式的生物分子序列。
然后,通过比较这些序列之间的相似性和差异性,可以计算出它们之间的进化距离或相似性分数。
接下来,利用计算机算法可以根据这些进化距离或相似性分数构建进化树。
常见的算法包括最大简约法、邻接法和最大似然法。
这些算法会根据进化距离或相似性分数来确定物种之间的分支关系,从而构建出一棵树状图。
进化树的树枝代表物种的分支演化,而树叶代表当前的物种。
树枝的长度通常表示进化时间的长短,较长的树枝表示较早的分支,较短的树枝表示较晚的分支。
进化树的形态可以有很多种,例如二叉树、无根树和有根树等。
通过观察进化树,我们可以了解到不同物种之间的共同祖先以及它们之间的分支关系。
进化树的分支点代表物种的分裂事件,分支越早代表物种差异越大,分支越近代表物种差异越小。
进化树还可以显示出一些重要的进化事件,例如物种的起源、灭绝、迁移和适应等。
进化树在生物分类学、系统发育学和进化生物学等领域有着广泛的应用。
它可以帮助科学家研究物种的起源和演化过程,揭示生物多样性的来源和演变规律。
进化树还可以用于判断物种的分类和命名,帮助我们更好地理解和研究生物界的多样性。
近年来,随着高通量测序技术的发展,获得大规模的基因组数据变得越来越容易。
这使得构建进化树变得更加准确和可靠。
同时,生物信息学的快速发展也为进化树的构建提供了更多的工具和方法。
例如,基于分子标记的进化树、基于基因组的进化树和基于大数据的进化树等。
生物信息学进化树是一种重要的工具,可以帮助我们揭示生物物种之间的进化关系和演化历史。
通过构建进化树,我们可以了解到不同物种之间的共同祖先以及它们之间的分支关系。

一、关于BLAST简述BLAST搜索的算法思想。
答:BLAST是一种局部最优比对搜索算法,将所查询的序列打断成许多小序列片段,然后小序列逐步与数据库中的序列进行比对,这些小片段被叫做字”word”;当一定长度的的字(W)与检索序列的比对达到一个指定的最低分(T)后,初始比对就结束了;一个序列的匹配度由各部分匹配分数的总和决定,获得高分的序列叫做高分匹配片段(HSP),程序将最好的HSP双向扩展进行比对,直到序列结束或者不再具有生物学显著性,最后所得到的序列是那些在整体上具有最高分的序列,即,最高分匹配片段(MSP),这样,BLAST 既保持了整体的运算速度,也维持了比对的精度。
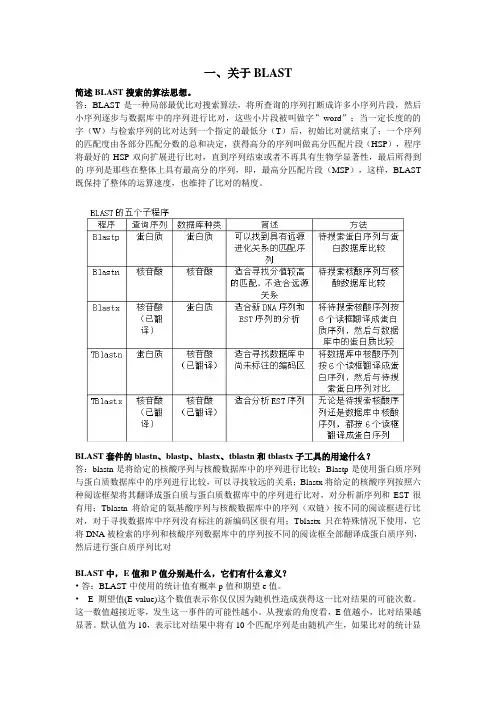
BLAST套件的blastn、blastp、blastx、tblastn和tblastx子工具的用途什么?答:blastn是将给定的核酸序列与核酸数据库中的序列进行比较;Blastp是使用蛋白质序列与蛋白质数据库中的序列进行比较,可以寻找较远的关系;Blastx将给定的核酸序列按照六种阅读框架将其翻译成蛋白质与蛋白质数据库中的序列进行比对,对分析新序列和EST很有用;Tblastn将给定的氨基酸序列与核酸数据库中的序列(双链)按不同的阅读框进行比对,对于寻找数据库中序列没有标注的新编码区很有用;Tblastx只在特殊情况下使用,它将DNA被检索的序列和核酸序列数据库中的序列按不同的阅读框全部翻译成蛋白质序列,然后进行蛋白质序列比对BLAST中,E值和P值分别是什么,它们有什么意义?•答:BLAST中使用的统计值有概率p值和期望e值。
• E期望值(E-value)这个数值表示你仅仅因为随机性造成获得这一比对结果的可能次数。
这一数值越接近零,发生这一事件的可能性越小。
从搜索的角度看,E值越小,比对结果越显著。
默认值为10,表示比对结果中将有10个匹配序列是由随机产生,如果比对的统计显著性值(E值)小于该值(10),则该比对结果将被检出,换句话说,比较低的E值将使搜索的匹配要求更严格,结果报告中随机产生的匹配序列减少。

2024年辽宁省中学生生物学竞赛初赛试卷注意事项:1.请用2B铅笔在机读卡上作答。
2.试题按学科分类,单选和多选混排。
3.答题时间120分钟,共100题,总129分。
第一部分细胞生物学、生物化学、微生物学、生物信息学、生物技术(20题)1.用简便的方法鉴别核糖、葡萄糖、果糖、蔗糖和淀粉时,结果存在错误的一组是(单选)()核糖葡萄糖果糖蔗糖糖原A.碘液----蓝色B.盐酸、间苯二酚绿色淡红色红色--C.斐林试剂红黄色红黄色红黄色--D.溴水褪色褪色---A.A B.B C.C D.D2.华丽硫珠菌是在红树林浅滩中新发现的一种细菌,单个细胞最长可达2cm,其生长的环境富含硫化物。
该细菌形态、结构和部分生理过程如下图。
下列叙述正确的有(多选)()A.该菌基因的转录和翻译都发生在膜囊P内B.大膜囊结构类似植物细胞的液泡可保持菌体形态C.细胞内外硫元素状态表明该菌参与生态系统的硫循环D.大肠杆菌的遗传物质也存在于类似膜囊P的结构中3.某种连续分裂的动物细胞,细胞周期如图所示,包括G1期(8h)、S期(8h)、G2期(6h)和M期(2h)。
现有该种细胞若干,为使所有细胞处于细胞周期同一时相,需按如下步骤操作:①在培养液中添加过量TdR,培养细胞足够时间;②去除TdR,培养细胞10h;③再次添加过量TdR,培养细胞足够时间。
下列叙述错误的是(单选)()A.加入过量TdR,处于S期的细胞立刻被抑制B.操作①后,原处于M期的细胞停滞于G1/S交界处C.操作②后,培养液中还有部分细胞处于S期D.操作③后,所有细胞都停滞于G1/S交界处4.人线粒体呼吸链受损可导致代谢物X的积累,由此引发多种疾病。
动物实验发现,给呼吸链受损小鼠注射适量的酶A和酶B溶液,可发生如图所示的代谢反应,从而降低线粒体呼吸链受损导致的危害。
据图分析错误的是(单选)()A.呼吸链受损会导致有氧呼吸异常,代谢物X是乳酸(C3H6O3)B.过程⑤中酶B为过氧化氢酶,避免过氧化氢对细胞的毒害C.过程④将代谢物X消耗避免代谢产物的积累D.过程①中生成ATP的底物磷酸化需要氧气参加5.羊瘙痒病是感染性蛋白粒子PrP Sc引起的。


生物信息学_复习题及答案(打印)(1)一、名词解释:1.生物信息学:研究大量生物数据复杂关系的学科,其特征是多学科交叉,以互联网为媒介,数据库为载体。
利用数学知识建立各种数学模型; 利用计算机为工具对实验所得大量生物学数据进行储存、检索、处理及分析,并以生物学知识对结果进行解释。
2.二级数据库:在一级数据库、实验数据和理论分析的基础上针对特定目标衍生而来,是对生物学知识和信息的进一步的整理。
3.FASTA序列格式:是将DNA或者蛋白质序列表示为一个带有一些标记的核苷酸或者氨基酸字符串,大于号(>)表示一个新文件的开始,其他无特殊要求。
4.genbank序列格式:是GenBank 数据库的基本信息单位,是最为广泛的生物信息学序列格式之一。
该文件格式按域划分为4个部分:第一部分包含整个记录的信息(描述符);第二部分包含注释;第三部分是引文区,提供了这个记录的科学依据;第四部分是核苷酸序列本身,以“//”结尾。
5.Entrez检索系统:是NCBI开发的核心检索系统,集成了NCBI 的各种数据库,具有链接的数据库多,使用方便,能够进行交叉索引等特点。
6.BLAST:基本局部比对搜索工具,用于相似性搜索的工具,对需要进行检索的序列与数据库中的每个序列做相似性比较。
P947.查询序列(query sequence):也称被检索序列,用来在数据库中检索并进行相似性比较的序列。
P988.打分矩阵(scoring matrix):在相似性检索中对序列两两比对的质量评估方法。
包括基于理论(如考虑核酸和氨基酸之间的类似性)和实际进化距离(如PAM)两类方法。
P299.空位(gap):在序列比对时,由于序列长度不同,需要插入一个或几个位点以取得最佳比对结果,这样在其中一序列上产生中断现象,这些中断的位点称为空位。
P2910.空位罚分:空位罚分是为了补偿插入和缺失对序列相似性的影响,序列中的空位的引入不代表真正的进化事件,所以要对其进行罚分,空位罚分的多少直接影响对比的结果。

生物信息学中的序列比对与进化树构建算法研究序列比对是生物信息学中重要的分析方法之一,通过比对不同生物种类的DNA、RNA或蛋白质序列,可以揭示它们之间的相似性和差异性,并为分析进化关系、功能预测等提供基础。
序列比对的基本思想是将两个或多个序列进行比对,并找出它们之间的相似性。
在序列比对中,常用的方法有全局比对、局部比对和多序列比对。
全局比对方法是将整个序列进行比对,一般采用Needleman-Wunsch算法或Smith-Waterman算法。
这些算法根据序列间的单个碱基或氨基酸之间的匹配、错配和缺失情况,计算出序列的相似度得分。
全局比对方法适用于较短的序列,优点是能够找到完全匹配的区域,但是对长序列不适用,计算复杂度较高。
局部比对方法主要用于比对较长的序列或存在较大插入缺失的序列。
常用的算法有BLAST和FASTA算法。
这些算法采用快速搜索的策略,先找出序列间的高度相似的片段,然后再进行比对和分析。
局部比对方法能够找到较长序列内的相似片段,但可能无法找到全局的最优比对。
多序列比对方法用于比对三个或更多序列,揭示它们之间的共同特征和区别。
常用的方法有多重序列比对和进化树构建。
多重序列比对旨在将多个序列按照匹配和错配的原则进行比对,以找到共同的序列区域。
进化树构建方法基于序列的相似性和进化关系,将多个序列构建成进化树,以揭示它们之间的进化关系。
在序列比对的过程中,常用的比对算法还包括Pairwise比对、局部比对、多重比对等方法。
这些方法都有自己的特点和适用范围,根据具体的研究目的和数据特点选择合适的方法进行序列比对。
进化树构建是生物信息学中的重要研究方向之一,用于揭示不同生物种类之间的进化关系。
进化树是一种图形化的表示方式,能够清晰地展示物种间的分支关系、共同祖先以及进化时间。
进化树的构建主要基于序列的相似性和进化关系。
在进化树构建中,常见的方法包括距离法、最大简约法和最大似然法。
距离法基于序列间的距离矩阵,通过测量序列间的差异程度来构建进化树。

高中生物竞赛难点知识点高中生物竞赛是一项对生物学知识掌握程度和应用能力的综合测试,其中涉及的难点知识点众多。
以下是一些常见的难点知识点:1. 细胞结构与功能:细胞是生物体的基本单位,其结构复杂,功能多样。
难点包括细胞器的分类、功能以及它们之间的相互作用。
2. 遗传学原理:遗传学是生物学中的一个重要分支,涉及到基因的传递、表达和变异。
难点包括孟德尔遗传定律的应用、连锁与基因重组、基因突变和染色体变异等。
3. 生物进化:进化论是生物学的基石之一。
难点包括自然选择、物种形成、进化树的构建以及生物多样性的起源等。
4. 生物化学:生物化学研究生物体内化学过程和物质。
难点包括酶的作用机理、代谢途径、蛋白质结构与功能等。
5. 分子生物学:分子生物学是研究生物分子的结构和功能。
难点包括DNA复制、RNA转录和蛋白质翻译过程,以及基因表达调控机制。
6. 生态学:生态学研究生物与其环境之间的关系。
难点包括生态系统的结构和功能、物种间相互作用、生态平衡以及生物多样性保护等。
7. 生物技术:生物技术是应用生物学原理来创造新产品或改进现有产品。
难点包括基因工程、细胞工程、蛋白质工程等。
8. 生物信息学:生物信息学是生物学与信息技术的交叉领域。
难点包括基因组序列分析、蛋白质结构预测、生物数据库的构建和应用等。
9. 神经生物学:神经生物学研究神经系统的结构和功能。
难点包括神经元的信号传递、神经网络的形成以及大脑功能的调控等。
10. 发育生物学:发育生物学研究生物体从受精卵到成熟个体的发育过程。
难点包括胚胎发育的各个阶段、细胞分化和组织形成等。
掌握这些难点知识点不仅需要扎实的基础知识,还需要能够灵活运用这些知识解决实际问题。
参加生物竞赛的学生应该通过大量的阅读、实验和练习来加深对这些知识点的理解和应用能力。

生物信息学第七章分子进化与系统发育分析(2)同义与非同义的核苷酸替代❒同义替代:编码区的DNA序列,核苷酸的改变不改变编码的氨基酸的组成❒非同义替代:核苷酸改变,从而改变编码氨基酸的组成❒计算方法:进化通径法Kimura两参数法采用密码子替代模型的最大似然法SdS❒Ka/Ks ~ 1: 中性进化❒Ka/Ks << 1: 阴性选择,净化选择❒Ka/Ks >> 1: 阳性选择,适应性进化❒多数基因为中性进化,约1%的基因受到阳性选择->决定物种形成、新功能的产生❒PAML, MEGA等工具:计算Ka/Ks及统计显著性进化通径法:Nei-Gojobori❒首先需要考虑:潜在的同义(S )和非同义位点数(N )❒基本假设:所有核苷酸的替代率相等❒用f i 表示某一个密码子第i 位的核苷酸上发生同义替代的比例;(i=1,2,3)❒所有密码子潜在的同义和非同义替代的位点数定义如下:,n=3-s∑==31i i f s潜在的同义和非同义位点数的估计❒例如对于Phe, 密码子TTT, 第三位T变成C时为同义替代,变成A/G为非同义替代❒因此:❒s=0+0+1/3❒n=3-1/3=8/3❒终止密码子忽略不计;如Cys的TGT, s=0.5整个序列的同义与非同义估计❒和N=3C-S; Sj 为第j 位密码子的s 值,C 为所有密码子的总数❒S+N=3C :所比较的核苷酸的总数∑==C j j S S 1S d 与N d 的计算:进化通径❒当一对密码子仅存在一个差异时,可以立即判断是同义还是非同义,进化通径只有一种可能;例如对于GTT(Val)和GTA(Val),s d =1,n d =0;而对于ATT(I)和ATG(M),s d =0,n d =1❒一对密码子存在两个差异时:两种进化通径(简约法,即最少需要)。
例如:比较TTT(Phe)和GTA(Val): (1) TTT(Phe)<->GTT(Val)<->GTA(Val)(2) TTT(Phe)<->TTA(Leu)<->GTA(Val)❒s d =1/2=0.5,n d =3/2=1.5❒同样,终止密码子不予考虑一对密码子存在三个差异时:六种进化通径。

生物信息学中的序列比对与进化树构建生物信息学是一门涉及生命科学和计算科学的交叉学科,其应用在分子生物学、生物医学、生态学、进化论、生物技术等诸多领域中。
序列比对和进化树构建是生物信息学的重要组成部分,是理解生物学进化的重要途径之一。
一、序列比对序列比对是将两个或多个蛋白质或核酸序列究竟有多少相同、多少不同进行比较的过程。
序列比对在生物学中极其重要,因为它可以帮助科学家确定两个生物物种之间的相似性,进而推断它们之间的亲缘关系以及共同祖先的时间。
序列比对中最基础和常用的方法是全局比对和局部比对。
全局比对试图比较两个序列的完整长度,一般用于比较相似性较高的序列,它最先被应用于分析DNA和蛋白质,是序列比对过程中最古老、最经典的算法方法。
而局部比对则更注重比较两个序列中的相似区域,忽略其中任何间隔,通常用于比较两个较短的序列或者两个相对较不相关的序列。
例如,在核酸序列比对中,这种算法更适用于获取多个剪接变异或者重复序列之间的相似性。
另外,序列比对有一个关键问题,就是如何准确的衡量两条序列的相似性和相异性。
在这方面有很多方法,例如编辑距离、盒子型、PAM矩阵、BLOSUM 矩阵等等,其中都采用了不同的评分标准。
二、进化树构建进化树(Phylogenetic Tree)是用来表示生物物种间亲缘关系的结构,也称演化树或家谱树。
进化树是通过对基于DNA和RNA等生物分子序列进行分析,推导出各物种之间共同祖先的关系构建起来的,同时它也综合了形态、系统和分子信息等其他生物学数据。
进化树的构建过程中涉及许多算法,其中最基础的是贪心算法。
贪心法从序列的最初状态开始,一步步选择最佳的演化路径,最终得到最优的进化树;而Neighborhood-joining (NJ)算法则是以序列之间的 Jukes-Cantor 模型距离或 Kimura 二参数模型距离为基础,使用最小进化步骤(Minimum Evolution,ME)标准构建进化树,是目前应用比较广泛的算法。

分子进化学中的进化树构建方法随着科技的进步和生物技术的广泛应用,分子生物学的研究逐渐深入,成为生物学、生物技术和医药学等领域的重要研究方向。
而分子进化学作为分子生物学中的一个重要分支,研究物种间的分子差异和进化关系。
其中,构建进化树是分子进化学研究中的重要工作,下面我们来了解一下进化树构建的方法。
一、进化树的基本概念进化树是描述不同物种、不同基因或不同蛋白质之间进化关系的图形化表示。
在进化树中,每一个分支代表了一个物种、一个基因或一个蛋白质序列,分支的长度表示了物种、基因或序列的进化距离,而进化距离则是衡量不同物种或不同序列之间关系的基本参数。
而构建进化树的过程则是根据分子序列数据的重构得到物种或基因的进化树。
二、进化树的构建方法构建进化树有多种方法,主要有距离矩阵法、系统发育学法、最大似然法和贝叶斯法等。
下面我们逐一介绍这些方法的基本原理。
1.距离矩阵法距离矩阵法是最早采用的一种构建进化树的方法,它基于序列之间的距离矩阵计算和聚类方法来得到进化树。
该方法首先计算所有分子序列之间的距离(距离可由序列相似性计算得出),然后根据聚类方法构建进化树。
聚类方法包括单链接聚类、均链接聚类和最大链接聚类等。
距离矩阵法的优点是构建速度快、适用性广,但是对于高变异的序列来说,该方法可能会产生误导性的结果。
2.系统发育学法系统发育学法是基于系统学原理,采用系统发生学的理论和方法来构建进化树。
该方法主要是通过分子序列的相似性构建系统发育分析矩阵,然后利用不同的计算方法(如UPGMA、NJ和ML等)推断进化树。
系统发育学法的优点是能够更准确地反映分子序列的演化,并且可以通过不同的方法比较结果,但是该方法需要大量的计算资源和长时间的计算。
3.最大似然法最大似然法是一种统计学上的方法,通过最大化序列数据与观测数据的相似度,来推断出最可能的进化树。
该方法需要整合进化模型和数据,然后计算不同进化模型下数据的似然函数,最终选择似然度最大的进化树。
生物信息学在分子进化中的应用研究生物信息学是一门结合生物学、计算机科学和数学等学科的交叉领域,旨在利用计算机和信息技术来处理生物学实验数据和理论研究,以更深入地了解生物体系的结构和功能,进而应用于医学、农业、工业等领域。
在分子生物学和生物进化学中,生物信息学有着广泛的应用,尤其是在分子进化学中,它的应用研究更是日渐深入。
本文将从分子进化的基础知识入手,探讨生物信息学在分子进化中的应用研究。
一、分子进化的基础知识进化是生物学中的一个关键概念,它是指物种的特征随着时间的推移而发生变化,从而形成新的物种。
分子进化是进化研究的一个分支,它研究的是生物体内分子所发生的变化。
在分子进化学中,主要使用的分子是DNA和蛋白质,因为它们是生物体内的基本分子,其变化可以反映生物体系发生的进化事件。
分子进化的核心理论是演化树,也称作系统发育树或进化树。
演化树是一种用图形化、树状的方式描述不同物种或种群之间进化关系的方法。
它通过生物分子序列的比较,来推测物种之间的近缘关系和进化历史。
演化树的节点表示着共同祖先,唯一的祖先到现在每个物种间的距离表示了它们在进化上的差异程度。
二、生物信息学在分子进化中的应用1、分子进化分析生物信息学被广泛应用于分子进化分析,主要是通过组成各种格式的生物分子序列的比较和构建演化树来研究物种之间的进化关系和进化历史。
分子进化分析中最常用的比较对象是DNA序列和蛋白质序列。
DNA序列的比较可以揭示出生物间的进化关系,包括物种间演化历史、基因家族的起源以及基因本身的起源和演化。
常用的DNA序列比较算法有多序列比对和序列同源性搜索等,这些算法可以将DNA序列的结构和特征与其他物种进行比较,并找出两者之间的相似性和差异。
蛋白质序列的比较则可以推测出分子间的演化历史,揭示出不同的分子结构和功能上的特异性。
常用的蛋白质序列比较算法有比对和聚类等,这些算法可以帮助我们确定蛋白质序列的相似性和差异性,推测分子之间的进化关系,并根据进化树来重建分子的初始状态。
进化树构建参数一、概述进化树构建是生物信息学中的一个重要研究领域,它涉及到许多参数的选择和优化。
进化树构建是基于已知序列的演化关系,通过计算分子进化模型的距离或相似度,从而推断不同物种之间的进化关系。
本文将详细介绍构建进化树时需要考虑的参数。
二、参数种类1. 样本选择:样本选择是构建进化树时必须考虑的第一个因素。
样本数量和种类的选择对于构建出准确可靠的进化树至关重要。
2. 进化模型:不同基因序列在演变过程中所遵循的进化模型是不同的,常见有Jukes-Cantor模型、Kimura 2-parameter模型、HKY85模型等。
3. 距离度量方法:距离度量方法包括无权法(UPGMA)、加权法(WPGMA)、最小演化法(ME)、最大简约法(MP)等。
4. 系统发育假设:系统发育假设包括分子钟假说和非分子钟假说两种,分别应用于有无时间信息两种情况下。
5. 支持率阈值:支持率阈值指代各节点的支持率,通常以Bootstrap值或Bayesian后验概率等指标表示。
支持率阈值越高,节点的可靠性越高,但会导致树的拓扑结构出现偏差。
三、参数选择1. 样本选择:样本应该代表各个物种的演化历史,并且应该包含足够数量的序列以减少噪音和随机误差对结果的影响。
2. 进化模型:进化模型应该选择最适合数据集特征的模型。
可以使用模型比较方法(如AIC、BIC等)来确定最优模型。
3. 距离度量方法:距离度量方法应该根据不同数据集和研究问题进行选择。
UPGMA适用于相对简单的数据集,而ME和MP适用于复杂的数据集。
4. 系统发育假设:系统发育假说应该根据具体情况进行选择。
分子钟假说适用于有时间信息的数据集,而非分子钟假说则适用于无时间信息或时间信息不可靠的数据集。
5. 支持率阈值:支持率阈值应该根据具体情况进行选择。
通常建议设置在70%以上。
四、参数优化1. 交叉验证法:交叉验证法可以用来选择最优的进化模型和距离度量方法。
2. Bootstrap分析:Bootstrap分析可以用来评估节点的支持率阈值,并且可以用来检测树的拓扑结构是否稳定。
分⼦进化树构建的简要步骤(以蛋⽩为例)PhyML利⽤氨基酸序列建树步骤(核酸建树也可以作为参考)前⾔:本⽂阅读对象适合建树新⼿,⽣物信息学⾼⼿请勿嘲笑,其中有什么错误还恳请指点。
为什么要建树及其你要解决什么问题这⾥不做讨论,只是⼀个纯粹的建树过程,前期的序列收集过程⾃⼰费⼼,根据⾃⼰的需要来做。
这⾥主要是最⼤似然法来建树,NJ法像mega这些软件中都有集成,最新的mega7也集成ML法,不过模型及各种参数不⼀定适合你,所以学习多种多种⽅法也是有⽤的,PhyML速度较慢,如果数列数量较多、步长检验次数多,等待时间会很长,有可能达到⼏⼗⼩时,也与电脑配置有关,⼀般时间都是以⼩时计数,所以要有⼼理准备,如果数据量⼤,推荐⽤RaxML或其他⽅法建树,它处理速度要⽐PhyML 快,不过RaxML是纯命令操作,对不熟悉命令及参数意义的⼈有⼀定难度,我只在linux 下操作过,在win下没有使⽤过。
本⽂是⽤氨基酸建树过程,如果你是⽤核酸序列建树,也可以参考这个过程,核酸替代模型请⽤jmodeltest或其他同功软件计算。
由于PhyML计算过程⽐较长,做⼀遍⽐较耗时,推荐你⽤其他软件⽤NJ法先⾏试验建树,看看你选择的序列是否有效及符合你的预期结果,调整好序列后再⽤PhyML跑⼀遍看结果是否符合⾃⼰的要求。
PhyML有线上版本,只需要提交序列⽐对结果,设置模型参数,留下邮箱等待就会给你返回结果,不过时间不可控,根据⾃⾝情况选择线上还是本地⾃⼰建树。
⽔平有限,如有错误遗漏恳请各位指点。
如果在⽂库不能下载,可以去⽹盘下载,见⽂末。
●建树过程:序列准备-模型选择-建树及树的验证。
●环境准备:电脑^-^Windows或者Linux都可以(没试过mac,如果是mac环境,请参考具体的操作⼿册)、ProtTest、PhyMl及序列⽐对的软件,线上或本地都可以。
1.序列准备:在⾃⼰熟悉的数据库中(我⾃⼰⽐较熟悉Ncbi)上做blast,选取跟要建树蛋⽩同源的各物种序列,下载到本地,整合到⼀个fasta⽂件中,注意修改物种名称,字数最好不要太长,序列⽐对后.phy格式⽂件对⽂件名长度有限制(这个可能跟软件有关系,只要⾃⼰知道是什么物种,不⾄于混淆就⾏),注意规范性,fasta⽂件中最好除了>头标,字母及下划线不要有其他不相关的字符,因为如果后⾯你要⽤软件分析.phy⽂件的时候这些软件对.phy的格式要求⽐较变态,有其他多余字符它都会报错的(你如果在dos 下⽤命令合并⽂件请注意⽂件中最后⼀⾏的字符,请删除)。