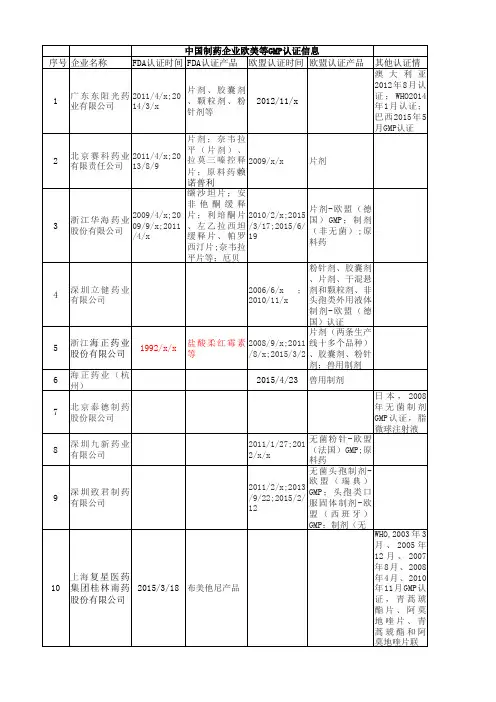
中国药企制剂国际认证情况2015版
- 格式:xls
- 大小:57.00 KB
- 文档页数:37

根据《中华⼈民共和国药品管理法》、《国务院关于改⾰药品医疗器械审评审批制度的意见》(国发〔2015〕44号)等有关规定,为解决药品注册申请积压问题,提⾼药品审评审批质量和效率,经国务院同意,实⾏如下药品注册审评审批政策。
现予以公告: ⼀、提⾼仿制药审批标准 仿制药按与原研药质量和疗效⼀致的原则受理和审评审批。
其中,对已在中国境外上市但尚未在境内上市药品的仿制药注册申请,应与原研药进⾏⽣物等效性研究并按国际通⾏技术要求开展临床试验,所使⽤的原研药由企业⾃⾏采购,向国家⾷品药品监督管理总局申请⼀次性进⼝;未能与原研药进⾏对⽐研究的,应按照创新药的技术要求开展研究。
已经受理的仿制药注册申请,实⾏分类处理: (⼀)中国境内已有批准上市原研药,申请注册的仿制药没有达到与原研药质量和疗效⼀致的,不予批准。
(⼆)中国境外已上市但境内没有批准上市原研药,申请仿制药注册的企业可以选择按原规定进⾏审评审批,但在药品批准上市3年内需按照国发〔2015〕44号⽂件规定进⾏质量和疗效⼀致性评价,未通过⼀致性评价的注销药品批准⽂号;企业也可以选择撤回已申报的注册申请,改按与原研药质量和疗效⼀致的标准完善后重新申报。
对上述重新申报的注册申请实⾏优先审评审批,批准上市后免于进⾏质量和疗效⼀致性评价。
对申报上市的仿制药注册申请,⾸先审查药学研究的⼀致性,药学研究未达到要求的,不再对其他研究资料进⾏审查,直接作出不予批准决定。
⼆、规范改良型新药的审评审批 对改变原研药剂型、酸根、碱基和给药途径等的药品注册申请,申请⼈需证明其技术创新性且临床价值与原品种⽐较具有明显优势;⽆法证明具备上述优势的,不予批准。
改变剂型和规格的⼉童⽤药注册申请除外。
三、优化临床试验申请的审评审批 对新药的临床试验申请,实⾏⼀次性批准,不再采取分期申报、分期审评审批的⽅式;审评时重点审查临床试验⽅案的科学性和对安全性风险的控制,保障受试者的安全。
加强临床试验申请前及过程中审评⼈员与申请⼈的沟通交流,及时解决注册申请和临床试验过程中的问题。



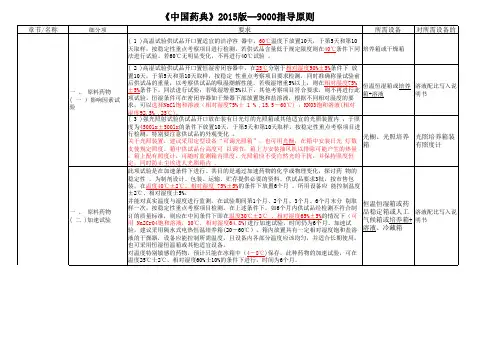
《中国药典》2015年版制剂通则变化⽐较《中国药典》2015年版制剂通则变化⽐较2015年版《中国药典》⽆论是在品种收载、标准增修订幅度、检验⽅法完善、检测限度设定,还是在标准体系的系统完善、质控⽔平的整体提升都上了⼀个新的台阶。
通过学习、熟悉2015 年版《中国药典》制剂通则的变化,了解⽬前我国⽤药⽔平、制药⽔平和监管⽔平现状,解读未来我国药品⾏业的趋势。
学习重点:熟悉药典制剂通则,掌握其标准的提升。
第⼀部分制剂通则⽐较2015年版《中国药典》已于2015年6⽉5⽇由国家⾷品药品监督管理总局正式颁布,于2015年12⽉1⽇正式实施。
新版药典是国家药品标准的组成部分,是国家药品标准体系的核⼼。
按照党中央提出的“四个最严”要求,新版药典的制修订始终坚持“科学、先进、实⽤、规范”的原则,依据试验数据、研究结果、专家评估,体现药典编制的科学性和严谨性,以持续改进提⾼药品质量。
新版药典的颁布标志着我国⽤药、制药以及监管⽔平的全⾯提升,将促进药品质量的整体提⾼,对于保障公众⽤药安全有效意义重⼤。
《中国药典》2015年版进⼀步扩⼤药品品种的收载和修订,共收载品种5608种。
其中,⼀部收载品种2598种,⼆部收载品种2603种,三部收载品种137种。
本版药典是将三部药典的附录合⼀,加强共性的系统化、完善化及规范化,新版《中国药典》的附录调整为凡例、通则与⽅法、指导原则、药⽤辅料等单独成卷,为第四部。
第四部的名称为“《中国药典》2015 年版总则”,包括现有药典⼀部、⼆部、三部的附录(现改为“通则”)内容和药⽤辅料品种正⽂。
⼀、《中国药典》2015 年版总则(四部)项⽬组成:1、前⾔2、第⼗届药典委员会委员名单3、⽬录4、中国药典沿⾰5、品种及通则变化名单6、凡例(三部合⼀)7、品名⽬次(保留笔画索引,品种正⽂拟改为按拼⾳排序)8、通则(原药典附录内容):包括导引图、制剂通则、通⽤⽅法/检测⽅法及指导原则9、附表:包括原⼦量表、国际单位换算表及新旧附录/通则编码对照表10、药⽤辅料品种正⽂(原收载于药典⼆部正⽂品种第⼆部分)11、总索引(中英⽂索引)⼆、修订说明1、使分类更加清晰明确,药典标准更加系统化、规范化将上⼀版药典中中药、化学药、⽣物制品三部分别收载的附录(凡例、制剂通则、分析⽅法、指导原则、药⽤辅料等)三合⼀,独⽴成卷作为第四部。

《中国药典》2015版四部9101药品质量标准分析⽅法验证指导原则《中国药典》2015版四部9101药品质量标准分析⽅法验证指导原则药品质量标准分析⽅法验证的⽬的是证明采⽤的⽅法适合于相应检测要求。
在建⽴药品质量标准时,分析⽅法需经验证;在药品⽣产⼯艺变更、制剂的组分变更、原分析⽅法进⾏修订时,则质量标准分析⽅法也需进⾏验证。
⽅法验证理由、过程和结果均应记载在药品质量标准起草说明或修订说明中。
⽣物制品质量控制中采⽤的⽅法包括理化分析⽅法和⽣物学测定⽅法,其中理化分析⽅法的验证原则与化学药品基本相同,所以可参照本指导原则进⾏,但在进⾏具体验证时还需要结合⽣物制品的特点考虑;相对于理化分析⽅法⽽⾔,⽣物学测定⽅法存在更多的影响因素,因此本指导原则不涉及⽣物学测定⽅法验证的内容。
验证的分析项⽬有:鉴别试验、限量或定量检查、原料药或制剂中有效成分含量测定,以及制剂中其他成分(如防腐剂等,中药中其他残留物、添加剂等)的测定。
药品溶出度、释放度等检查中,其溶出量等的测定⽅法也应进⾏必要验证。
验证指标有:准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐⽤性。
在分析⽅法验证中,须采⽤标准物质进⾏试验。
由于分析⽅法具有各⾃的特点,并随分析对象⽽变化,因此需要视具体⽅法拟订验证的指标。
表1中列出的分析项⽬和相应的验证指标可供参考。
表1检验项⽬和验证指标项⽬鉴别杂质测定含量测定及校正因⼦内容定量限度溶出量测定准确度-+-++精密度重复性-+-++中间精密度-+①-+①+专属性②+++++检测限--③+--定量限-+--+线性-+-++范围-+-++耐⽤性+++++①巳有重现性验证,不需验证中间精密度。
②如⼀种⽅法不够专属,可⽤其他分析⽅法予以补充。
③视具体情况予以验证。
⼀、准确度准确度系指采⽤该⽅法测定的结果与真实值或参考值接近的程度,⼀般⽤回收率(%)表⽰。
准确度应在规定的范围内测定。

2015版中国药典第一部中药质量标准全面提升《中国药典》一部主要收载中药材、饮片、提取物和中成药的质量标准,也就是大家常说的中药标准。
日前正式发行的《中国药典》2015年版一部目录中共收载药材和饮片618个(不含收载在品种下的饮片标准)、植物油脂和提取物47个、成方制剂和单味制剂1493个。
较2010年版药典,2015年版药典的中药标准在安全性、有效性等方面均有所提升。
正如国家药典委员会秘书长张伟所说的那样:“药典是引领产业结构调整和产品质量升级的‘导航仪’。
”由于中药多数来源于天然物质,不仅所含成分非常复杂,而且受气候、生态环境、栽培(生产)技术等因素影响很大。
中药质量标准是随着中药所含成分的不断阐明、对中药安全性认识的不断提升,以及中药市场出现的新问题和现代分析方法的不断出现而不断提高的,因此每版药典一部的标准水平都有较大程度的提升。
据介绍,每一版药典在开始工作前,都需要成立新一届药典委员会,规划新版药典工作,并形成编制大纲。
中药的安全性、有效性和质量可控性是《中国药典》2015年版编制大纲的“规定动作”。
安全性:变单一指标为“组合拳”近年来,随着药品监管理念的不断成熟,加强对中药中有害物质的控制成为国家药品标准工作满足药品监管需求,保障公众用药安全的重要任务。
在国家食品药品监管总局统一部署下,国家药典委员会在《中国药典》2015年版编制工作中,组织有关单位和专家,通过科学研究,参照国际发达国家地区的限度标准,并结合我国中药发展现阶段的实际情况,突出风险控制重点,按照“成熟一个,修订一个,公示一个”的原则,将研究成果应用于《中国药典》制定和修订中。
《中国药典》2015年版在2010年版的基础上,重点加强和完善了安全性控制技术的应用,主要涵盖八个方面:二氧化硫残留、重金属及有害元素、农药残留、真菌毒素(黄曲霉毒素)、色素、内源性有害物质、微生物、致病菌控制。
“新版药典在凡例、通则以及品种的标准中分别增加了对中药安全性检查的总体要求、技术指导则、检测方法和具体品种限度规定,改变了过去单纯强调设置检测项目和指标的做法。

附件2确认与验证第一章范围第一条本附录适用于在药品生产质量管理过程中涉及的所有确认与验证活动。
第二章原则第二条企业应当确定需要进行的确认或验证工作,以证明有关操作的关键要素能够得到有效控制。
确认和验证的范围和程度应根据风险评估的结果确认。
确认与验证应当贯穿于产品生命周期的全过程。
第三章验证总计划第三条所有的确认与验证活动都应当事先计划。
确认与验证的关键要素都应在验证总计划或同类文件中详细说明。
第四条验证总计划应当至少包含以下信息:(一)确认与验证的基本原则;(二)确认与验证活动的组织机构及职责;(三)待确认或验证项目的概述;(四)确认或验证方案、报告的基本要求;(五)总体计划和日程安排;(六)在确认与验证中偏差处理和变更控制的管理;(七)保持持续验证状态的策略,包括必要的再确认和再验证;(八)所引用的文件、文献。
第五条对于大型和复杂的项目,可制订单独的项目验证总计划。
第四章文件第六条确认与验证方案应当经过审核和批准。
确认与验证方案应当详述关键要素和可接受标准。
第七条供应商或第三方提供验证服务的,企业应当对其提供的确认与验证的方案、数据或报告的适用性和符合性进行审核、批准。
第八条确认或验证活动结束后,应当及时汇总分析获得的数据和结果,撰写确认或验证报告。
企业应当在报告中对确认与验证过程中出现的偏差进行评估,必要时进行彻底调查,并采取相应的纠正措施和预防措施;变更已批准的确认与验证方案,应当进行评估并采取相应的控制措施。
确认或验证报告应当经过书面审核、批准。
第九条当确认或验证分阶段进行时,只有当上一阶段的确认或验证报告得到批准,或者确认或验证活动符合预定目标并经批准后,方可进行下一阶段的确认或验证活动。
上一阶段的确认或验证活动中不能满足某项预先设定标准或偏差处理未完成,经评估对下一阶段的确认或验证活动无重大影响,企业可对上一阶段的确认或验证活动进行有条件的批准。
第十条当验证结果不符合预先设定的可接受标准时,应当进行记录并分析原因。

[转载]国际GMP认证与PIC/S简介提要:中国药品制剂出口的瓶颈在于国际认证,药品制剂要想进入国际主流市场,首先要过认证关。
本文简要地介绍了国际认证组织之一的国际医药品检查协约计划(PIC/S)及其GMP认证覆盖的国家,介绍PIC/S-GMP,和进行GMP认证之前的《制药工厂基本资料编写指南(Site Master File)》。
并通过介绍台湾如何普及PIC/S-GMP认证,以为我国制药企业提升GMP水平与国际出轨提供借鉴。
PIC/S简介:国际药品监查合作计划(The Pharmaceutical Inspection Co-operation Scheme, 简称PIC/S )成立于1995年11月,为世界上唯一的由各国GMP检查权责机关组成的国际合作组织,成立的宗旨为了消除药品贸易中的障碍,提高药品获取许可的一致性,确保药品质量,促进国际GMP法规标准之协和及GMP检查质量的一致化。
PIC/S现有33个会员分属32个国家,如澳大利亚、奥地利、比利时、加拿大、捷克、丹麦、芬兰、法国、德国、希腊、匈牙利、冰岛、爱尔兰、意大利、拉脱维亚、列支敦士登、马来西亚、荷兰、挪威、波兰、葡萄牙、罗马尼亚、新加坡、斯洛伐克、西班牙、瑞典、瑞士和英国等等。
该组织在全球享有较高声誉,其内部检查官均来自各成员国的相关专业权威人士,同时身兼参与的官方(监管机构)的代表。
组织内会员国拥有一致的GMP 规范与检查系统,且相互承认检查結果,该组织颁发的GMP证书在PIC/S组织成员国之间相互认可,通过某一成员国的PIC/S-GMP认证也就意味着跨进了32个国家的第一道门槛,所以,是进入国际市场的快捷通道之一。
PIC/S前身则为创立于1970年的「Pharmaceutical Inspection Convention,简称PIC」,PIC的组织章程不同于PIC/S,PIC为一个以国家为会员单位共同签署成立的正式国际组织,该组织直运作到1995年欧盟成立时,PIC受限于欧盟体制而无法与其它想加入的国家签署协议,因而衍生出另一个非正式、更有弹性的国际合作组织——PIC/S。

2015版药典主要的七个方面变化七大变化提升总体水平张伟表示,2015版药典主要有七个方面的变化:一是收载品种增幅达到27.4%。
2015版药典拟收载5800个品种,比2010版药典增加1200多个,修订品种751个。
二是通过药典凡例、通则、总论的全面增修订,从整体上进一步提升了对药品质量控制的要求,完善了药典标准的技术规定,使药典标准更加系统化、规范化。
三是健全了药品标准体系。
特别是药用辅料品种增加至260个,新增相关指导原则;在归纳、验证和规范的基础上实现了《中国药典》各部共性检测方法的协调统一。
四是2015版药典附录(通则)、辅料独立成卷,构成《中国药典》四部的主要内容。
五是药用辅料品种收载数量显著增加。
拟新增128个,共计260个,增长率高达97%。
六是安全性控制项目大幅提升。
中药:制定了中药材及饮片中二氧化硫残留量限度标准,推进建立和完善重金属及有害元素、黄曲霉毒素、农药残留量等物质的检测限度标准;加强对重金属以及中药材的有毒有害物质的控制等。
化学药:有关物质加强了杂质定性和定量测定方法的研究,实现对已知杂质和未知杂质的区别控制,优化抗生素聚合物测定方法,设定合理的控制限度,整体上进一步提高有关物质项目的科生物制品:增加相关总论的要求,严格生物制品全过程质量控制要求,以保证产品的安全有效性,同时增订“生物制品生产用原辅材料质量控制通用性技术要求”,加强源头控制,最大限度降低安全性风险等。
七是进一步加强有效性控制。
中药材加强了专属性鉴别和含量测定项设定。
化学药适当增加了控制制剂有效性的指标,研究建立科学合理的检查方法。
生物制品进一步提高效力测定检测方法的规范性,加强体外法替代体内法效力测定方法的研究与应用,保证效力测定方法的准确性和可操作性。
2015版<药典>中药材标准的变化新药典变化概述:1.药典一部收载药材和饮片、植物油脂和提取物、成方制剂和单味制剂等,品种共计2598种,其中新增440种,修订517种,不收载7种。
行业研究/2016年06月05日总局发布《2015年度药品检查报告》监管加强利好品牌药企[Table_Summary]投资要点:事件:●2016年6月3日,总局发布《2015年度药品检查报告》,对全年药品检查情况及检查发现主要问题进行了阐述。
点评:●总局药品检查力度超预期,制药产业监管趋严,升级整合迹象显现。
2015年,国家食品药品监督管理总局食品药品审核查验中心秉持客观、公正、廉洁、高效的原则,依法依规、科学严格开展药品检查。
共检查企业/品种数698家,派出检查组数682次,派出人次达到2082次。
对检查发现的违法违规行为进行严厉查处,药品GMP认证整改率、飞行检查不通过率均明显提高,显示总局监管趋严。
我们认为,药品生产质量监管力度仍将继续加强,“小散乱差”的药企将逐渐退出,产业升级整合迹象显现。
(图1)●药品GMP认证检查数量同比降低,但监管力度明显加强,整改率提高近一倍。
2015年总局共接收药品GMP认证申报资料221份,涉及201家药企,认证检查任务较2013、2014年数量明显降低,但整改复核17家,占7.7%,同比提高近一倍,总局监管力度明显加强。
(图2)2015年对114个品种进行药品注册生产现场检查,较2014年小幅降低,包括化学原料药31个、化学制剂56个、中药8个、生物制品15个、血液制品4个。
最终完成审核件108个,其中通过103个,不通过5个。
2015年药品GMP跟踪检查计划271家次,全年实际检查181家次,以注射剂和预防类生物制品为主。
●飞行检查力度空前,通过率仅32%,中药饮片成重灾区。
全年共派出49个检查组对59个企业进行了飞检,其中中药饮片14组19家企业,银杏叶提取物及制剂5组8家企业。
(图3)现场检查通过的19家,不通过39家,1家待处理,通过率仅32%。
共有22家被收回药品GMP证书,6家被吊销GMP证书,3家立案查处。
(图4)●中药材生产规范化、规模化和现代化仍需提升。
[转载]国际GMP认证与PIC/S简介提要:中国药品制剂出口的瓶颈在于国际认证,药品制剂要想进入国际主流市场,首先要过认证关。
本文简要地介绍了国际认证组织之一的国际医药品检查协约计划(PIC/S)及其GMP认证覆盖的国家,介绍PIC/S-GMP,和进行GMP认证之前的《制药工厂基本资料编写指南(Site Master File)》。
并通过介绍台湾如何普及PIC/S-GMP认证,以为我国制药企业提升GMP水平与国际出轨提供借鉴。
PIC/S简介:国际药品监查合作计划(The Pharmaceutical Inspection Co-operation Scheme, 简称PIC/S )成立于1995年11月,为世界上唯一的由各国GMP检查权责机关组成的国际合作组织,成立的宗旨为了消除药品贸易中的障碍,提高药品获取许可的一致性,确保药品质量,促进国际GMP法规标准之协和及GMP检查质量的一致化。
PIC/S现有33个会员分属32个国家,如澳大利亚、奥地利、比利时、加拿大、捷克、丹麦、芬兰、法国、德国、希腊、匈牙利、冰岛、爱尔兰、意大利、拉脱维亚、列支敦士登、马来西亚、荷兰、挪威、波兰、葡萄牙、罗马尼亚、新加坡、斯洛伐克、西班牙、瑞典、瑞士和英国等等。
该组织在全球享有较高声誉,其内部检查官均来自各成员国的相关专业权威人士,同时身兼参与的官方(监管机构)的代表。
组织内会员国拥有一致的GMP规范与检查系统,且相互承认检查結果,该组织颁发的GMP证书在PIC/S组织成员国之间相互认可,通过某一成员国的PIC/S-GMP认证也就意味着跨进了32个国家的第一道门槛,所以,是进入国际市场的快捷通道之一。
PIC/S前身则为创立于1970年的「Pharmaceutical Inspection Convention,简称PIC」,PIC的组织章程不同于PIC/S,PIC为一个以国家为会员单位共同签署成立的正式国际组织,该组织直运作到1995年欧盟成立时,PIC受限于欧盟体制而无法与其它想加入的国家签署协议,因而衍生出另一个非正式、更有弹性的国际合作组织——PIC/S。
0100本制剂通则中原料药物系指用于制剂制备的活性物质,包括中药、化学药、生物制品原料药物。
中药原料药物系指 饮片、植物油脂、提取物、有效成分或有效部位》化学药原料药物系指化学合成、或来源于天然物质或采用生物技术获 得的有效成分(即原料药);生物制品原料药物系指生物制品原液或将生物制品原液干燥后制成的原粉。
本制剂通则中各剂型、亚剂型并不适用于所有原料药物,而应取决于原料药物特性、临床给药需求以及药品的安 全性、有效性和稳定性等。
本制剂通则适用于中药、化学药和治疗用生物制品(包 括血液制品、免疫血清、细胞因子、单克隆抗体、免疫调节 剂、微生态制剂等)。
预防类生物制品,应符合本版药典三部相应品种项下的有关要求。
除另有规定外,生物制品应于2〜8X:避光贮存和运输。
片剂系指原料药物或与适宜的辅料制成的圆形或异形的 片状固体制剂。
中药还有浸膏片、半浸膏片和全粉片等。
片剂以口服普通片为主,另有含片、舌下片、口腔貼 片、咀嚼片、分散片、可溶片、泡腾片、阴道片、阴道泡腾 片、缓释片、控释片、肠溶片与口崩片等。
含片系指含于口腔中缓慢溶化产生局部或全身作用的片剂。
含片中的原料药物一般是易溶性的,主要起局部消炎、杀菌、收敛、止痛或局部麻醉等作用。
舌下片系指置于舌下能迅速溶化,药物经舌下黏膜吸 收发挥全身作用的片剂。
舌下片中的原料药物应易于直接吸收,主要适用于急症 的治疗。
口腔貼片系指粘贴于口腔,经黏膜吸收后起局部或全身作用的片剂。
口腔貼片应进行溶出度或释放度(通则0931)检查。
咀嚼片系指于口腔中咀嚼后吞服的片剂。
咀嚼片一般应选择甘露醇、山梨醉、蔗糖等水溶性辅料作填充剂和黏合剂。
咀嚼片的硬度应适宜。
分散片系指在水中能迅速崩解并均勻分散的片剂。
分散片中的原料药物应是难溶性的。
分散片可加水分散 后口服,也可将分散片含于口中吮服或吞服。
分散片应进行溶出度(通则0931)和分散均匀性检查。
可溶片系指临用前能溶解于水的非包衣片或薄膜包衣片剂。
【7A版】《中国药典》2015年版四部凡例详解总则一、《中华人民共和国药典》简称《中国药典》,依据《中华人民共和国药品管理法》组织制定和颁布实施。
《中国药典》一经颁布实施,其同品种的上版标准或其原国家标准即同时停止使用。
《中国药典》由一部、二部、三部、四部及其增补本组成。
一部收载中药,二部收载化学药品,三部收载生物制品,四部收载通则和药用辅料。
除特别注明版次外,《中国药典》均指现行版《中国药典》。
本部为《中国药典》四部。
二、国家药品标准由凡例与正文及其引用的通则共同构成。
本部药典收载的凡例与通则对未载入本部药典的其他药品标准具同等效力。
三、凡例是为正确使用《中国药典》进行药品质量检定的基本原则,是对《中国药典》正文、通则与药品质量检定有关的共性问题的统一规定。
四、凡例和通则中采用“除另有规定外”这一用语,表示存在与凡例或通则有关规定不一致的情况时,则在正文中另作规定,并按此规定执行。
五、正文中引用的药品系指本版药典收载的品种,其质量应符合相应的规定。
六、正文所设各项规定是针对符合《药品生产质量管理规范》GoodManufacturingPractices,GMP)的产品而言。
任何违反GMP或有未经批准添加物质所生产的药品,即使符合《中国药典》或按照《中国药典》没有检出其添加物质或相关杂质,亦不能认为其符合规定。
七、《中国药典》的英文名称为PharmacopoeiaofthePeople’sRepublicofChina;英文简称为ChinesePharmacopoeia;英文缩写为ChP。
正文八、《中国药典》各品种项下收载的内容为标准正文。
正文系根据药物自身的理化与生物学特性,按照批准的处方来源、生产工艺、贮藏运输条件等所制定的、用以检测药品质量是否达到用药要求并衡量其质量是否稳定均一的技术规定。
九、药用辅料标准正文内容一般包括:(1)品名(包括中文名、汉语拼音与英文名);(2)有机物的结构式;(3)分子式、分子量与CAS编号;(4)来源;(5)制法;(6)性状;(7)鉴别;(8)理化检查;(9)含量测定;(10)类别;(11)贮藏;(12)标示等。