2015版药典检测方法分类
- 格式:xlsx
- 大小:13.64 KB
- 文档页数:4

2015版中国药典四部微生物限度中国药典是我国医药行业的重要规范,其中微生物限度是其中一个关键内容。
微生物限度是指药品中允许存在的微生物数量的上限。
它对药品的质量和安全性提出了严格要求。
2015版中国药典中明确了药品微生物限度的标准和方法,以保障药品的质量和使用的安全性。
一、微生物限度的定义和意义微生物限度是指在药品中存在的微生物数量的上限。
它是衡量药品质量和安全性的重要指标之一。
由于微生物对人体健康有潜在的危害,药品中过多的微生物污染可能导致药品的变质和降解,甚至引起严重的药品安全问题。
因此,微生物限度的控制是确保药品质量和安全性的关键步骤之一。
二、微生物的限度标准和分类微生物限度的标准和分类在2015版中国药典中得到了详细说明。
根据药品的特性和用途不同,微生物限度标准和分类也有所差异。
常见的微生物限度分类包括细菌总数、大肠菌群、霉菌和酵母菌等。
各类微生物的限度标准和方法在药典中都有详细描述,以确保药品的质量和安全性。
三、微生物限度的检测方法为了准确检测微生物限度,2015版中国药典提供了一系列的微生物检测方法。
常见的方法包括菌落总数的测定、大肠菌群的测定、霉菌和酵母菌的测定等。
这些方法要求检测人员具备专业的技术和操作能力,确保检测结果的准确性和可靠性。
四、微生物限度的控制措施为了保证药品的质量和安全性,生产企业需要采取一系列的控制措施来控制微生物限度。
这些措施包括原料和辅料的检验、生产环境的控制、生产工艺的严格执行和产品的质量控制等。
通过全面有效的控制措施,企业可以确保药品的微生物限度在合理的范围内,从而保证药品的质量和安全性。
五、微生物限度的意义和前景微生物限度的控制是保障药品质量和安全性的重要手段,它直接影响着人们的生命健康。
随着人们对药品质量和安全性的要求越来越高,对微生物限度的控制也越来越严格。
中国药典不断完善微生物限度标准和方法,为药品生产和使用提供了规范和指导。
未来,微生物限度的研究和控制将成为药品质量控制的重要方向。

0832 水分测定法第一法(费休氏法)1. 容量滴定法本法是根据碘和二氧化硫在吡啶和甲醇溶液中与水定量反应的原理来测定水分。
所用仪器应干燥,并能避免空气中水分的侵入;测定应在干燥处进行。
费休氏试液的制备与标定(1)制备称取碘(置硫酸干燥器内48小时以上)110g,置干燥的具塞锥形瓶(或烧瓶)中,加无水吡啶160ml,注意冷却,振摇至碘全部溶解,加无水甲醇300ml,称定重量,将锥形瓶(或烧瓶)置冰浴中冷却,在避免空气中水分侵入的条件下,通入干燥的二氧化硫至重量增加72g,再加无水甲醇使成1000ml,密塞,摇匀,在暗处放置24小时。
也可以使用稳定的市售费休氏试液。
市售的费休氏试液可以是不含吡啶的其他碱化试剂,或不含甲醇的其他伯醇类等制成;也可以是单一的溶液或由两种溶液临用前混合而成。
本试液应遮光,密封,阴凉干燥处保存。
临用前应标定滴定度。
(2)标定精密称取纯化水10~30mg,用水分测定仪直接标定;或精密称取纯化水10~30mg,置干燥的具塞锥形瓶中,除另有规定外,加无水甲醇适量,在避免空气中水分侵入的条件下,用费休氏试液滴定至溶液由浅黄色变为红棕色,或用电化学方法[如永停滴定法(通则0701)等]指示终点;另做空白试验,按下式计算:F=WA−B式中F为每1ml费休氏试液相当于水的重量,mg;W为称取纯化水的重量,mg;A为滴定所消耗费休氏试液的容积,ml;B为空白所消耗费休氏试液的容积,ml。
测定法精密称取供试品适量(约消耗费休氏试液1~5ml),除另有规定外,溶剂为无水甲醇,用水分测定仪直接测定。
或精密称取供试品适量,置干燥的具塞锥形瓶中,加溶剂适量,在不断振摇(或搅拌)下用费休氏试液滴定至溶液由浅黄色变为红棕色,或用永停滴定法(通则0701)指示终点;另做空白试验,按下式计算:×100%供试品中水分含量(%)=(A−B)FW式中A为供试品所消耗费休氏试液的体积,ml;B为空白所消耗费休氏试液的体积,ml;F为每1ml费休氏试液相当于水的重量,mg;W为供试品的重量,mg。

3302 外源病毒因子检查法病毒类制品在毒种选育和生产过程中,经常使用动物或细胞基质培养,因此,有可能造成外源因子的污染。
为了保证制品质量,需要对毒种和对照细胞进行外源病毒因子的检测。
对病毒主种子批或工作种子批,应抽取足够检测试验需要量的供试品进行外源病毒因子检测。
根据病毒的特性,有些检测需要在试验前中和病毒。
病毒中和时尽可能不稀释,但当中和抗体不能有效中和病毒而需要进行稀释病毒时,应选择可被中和的最大病毒量,但至少不得超过生产接种时毒种的稀释倍数。
进行病毒中和时,应采用非人源和非猴源(特殊情况除外)的特异性抗体中和本病毒,为降低样品中外源病毒被中和的可能性,最好采用单克隆抗体,中和过程不应干扰外源病毒的检测。
制备抗血清(或单克隆抗体)所用的免疫原应采用与生产疫苗(或制品)不同种而且无外源因子污染的细胞(或动物)制备。
如果病毒曾在禽类组织或细胞中繁殖过,则抗体不能用禽类来制备。
若用鸡胚,应来自SPF 鸡群。
病毒种子批外源因子检查⒈动物试验法⑴小鼠试验法取15~20g小鼠至少10只,取病毒种子批或经抗血清中和后的病毒悬液,每只脑内接种0.03ml,同时腹腔接种0.5ml,至少观察21天。
解剖每只在试验24小时后死亡或有患病体征的小鼠,直接肉眼观察其病理改变,并将有病变的相应的组织制成悬液通过脑内和腹腔接种另外至少5只小鼠,并观察21天。
接种24小时内小鼠死亡超过20%,试验无效。
在观察期内最初接种的乳鼠以及每个盲传组的小鼠至少有80%健存,且小鼠未出现与待测毒种无关的可传播性因子或其他病毒感染,为符合要求。
⑵乳鼠试验法取出生24小时内的乳鼠至少10只,取病毒种子批或经抗血清中和后的病毒悬液,脑内接种0.01ml,同时腹腔接种至少0.1ml。
每天观察至少14天。
解剖每只在试验24小时后死亡或有患病体征的乳鼠,直接肉眼观察其病理改变,并取有病变的相应的组织和脑、脾制备成悬液通过脑内和腹腔接种另外至少5只乳鼠,并每天观察至接种后14天。
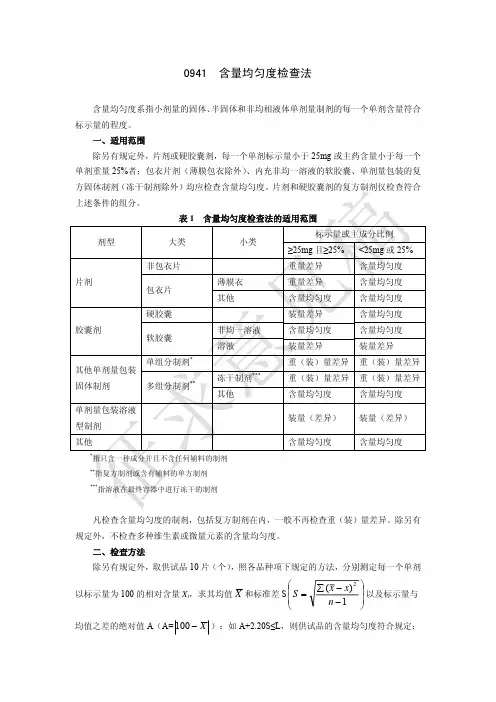
0941含量均匀度检查法含量均匀度系指小剂量的固体、 半固体和非均相液体单剂量制剂的每一个单剂含量符合 标示量的程度。
一、适用范围 除另有规定外,片剂或硬胶囊剂,每一个单剂标示量小于 25mg 或主药含量小于每一个 单剂重量 25%者;包衣片剂(薄膜包衣除外) 、内充非均一溶液的软胶囊、单剂量包装的复 方固体制剂(冻干制剂除外)均应检查含量均匀度。
片剂和硬胶囊剂的复方制剂仅检查符合 上述条件的组分。
表1 剂型 大类 非包衣片 片剂 包衣片 硬胶囊 胶囊剂 软胶囊 单组分制剂* 多组分制剂** 冻干制剂*** 其他 非均一溶液 溶液 薄膜衣 其他 含量均匀度检查法的适用范围 小类 标示量或主成分比例 ≥25mg 且≥25% 重量差异 重量差异 含量均匀度 装量差异 含量均匀度 装量差异 重(装)量差异 重(装)量差异 含量均匀度 装量(差异) 含量均匀度指只含一种成分并且不含任何辅料的制剂 指复方制剂或含有辅料的单方制剂 指溶液在最终容器中进行冻干的制剂<25mg 或 25% 含量均匀度 含量均匀度 含量均匀度 含量均匀度 含量均匀度 装量差异 重(装)量差异 重(装)量差异 含量均匀度 装量(差异) 含量均匀度其他单剂量包装 固体制剂 单剂量包装溶液 型制剂 其他******凡检查含量均匀度的制剂,包括复方制剂在内,一般不再检查重(装)量差异。
除另有 规定外,不检查多种维生素或微量元素的含量均匀度。
二、检查方法 除另有规定外,取供试品 10 片(个) ,照各品种项下规定的方法,分别测定每一个单剂⎛ 以标示量为 100 的相对含量 Xi,求其均值 X 和标准差 S ⎜ S = ⎜ ⎝∑ ( x − x) n −12⎞ ⎟ 以及标示量与 ⎟ ⎠均值之差的绝对值 A(A= 100 − X ):如 A+2.20S≤L,则供试品的含量均匀度符合规定;若 A+S>L,则不符合规定;若 A+2.20S>L,且 A+S≤L,则应另取 20 片(个)复试。

2321 铅、镉、砷、汞、铜测定法一、原子吸收分光光度法本法系采用原子吸收分光光度法测定中药中的铅、镉、砷、汞、铜,所用仪器应符合使用要求(通则0406)。
除另有规定外,按下列方法测定。
1.铅的测定(石墨炉法)测定条件参考条件:波长283. 3nm,干燥温度100~120 ℃,持续20秒;灰化温度400~750℃,持续20~25秒;原子化温度1700~2100℃,持续4~5秒。
银标准贮备液的制备精密量取铅单元素标准溶液适量,用2%硝酸溶液稀释,制成每lml含铅(Pb)lug 的溶液,即得(0~5℃贮存)。
标准曲线的制备分别精密量取铅标准贮备液适量,用2%硝酸溶液制成每lml分别含铅0ng、5ng、20ng、40ng、60ng、80ng的溶液。
分别精密量取lml,精密加含1%磷酸二氢铵和0.2%硝酸镁的溶液0 .5 ml,混匀,精密吸取20ul注人石墨炉原子化器,测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
供试品溶液的制备A法取供试品粗粉0.5g,精密称定,置聚四氯乙烯消解罐内,加硝酸3~5ml,混匀,浸泡过夜,盖好内盖,旋紧外套,置适宜的微波消解炉内,进行消解(按仪器规定的消解程序操作)。
消解完余后,取消解内罐置电热板上缓缓加热至红棕色蒸气挥尽,并继续缓缓浓缩至2~3ml,放冷,用水转入25ml量瓶中,并稀释至刻度,摇勻,即得。
同法同时制备试剂空白溶液。
B法取供试品粗粉1g , 精密称定,置凯氏烧瓶中,加硝酸-高氣酸(4:1 )混合溶液5~10ml,混勻,瓶口加一小漏斗,浸泡过夜。
置电热板上加热消解,保持微沸,若变棕黑色,再加硝酸-髙氣酸(4:1)混合溶液适量,持续加热至溶液澄明后升高温度,继续加热至冒浓烟,直至白烟散尽,消解液呈无色透明或略带黄色,放冷,转入50ml量瓶中,用2%硝酸溶液洗涤容器,洗液合并于量瓶中,并稀释至刻度,摇匀,即得。
同法同时制备试剂空白溶液。
C法取供试品粗粉0 .5g,精密称定,置瓷坩埚中,于电热板上先低温炭化至无烟,移人高温炉中,于500℃灰化5~6小时(若个别灰化不完全,加硝酸适童,于电热板上低温加热,反复多次直至灰化完全),取出冷却,加10%硝酸溶液5ml使溶解,转人25ml量瓶中,用水洗涤容器,洗液合并于量瓶中,并稀释至刻度,摇匀,即得。


1143 细菌内毒素检查法本法系利用鲎试剂来检测或量化由革兰阴性菌产生的细菌内毒素,以判断供试品中细菌内毒素的限量是否符合规定的一种方法。
细菌内毒素检查包括两种方法,即凝胶法和光度测定法,后者包括浊度法和显色基质法。
供试品检测时,可使用其中任何一种方法进行试验。
当测定结果有争议时,除另有规定外,以凝胶限度试验结果为准。
本试验操作过程应防止内毒素的污染。
细菌内毒素的量用内毒素单位(EU)表示,1EU与1个内毒素国际单位(IU)相当。
细菌内毒素国家标准品系自大肠埃希菌提取精制而成,用于标定、复核、仲裁鲎试剂灵敏度、标定细菌内毒素工作标准品的效价,干扰试验及检查法中编号B和C溶液的制备、凝胶法中鲎试剂灵敏度复核试验、光度测定法中标准曲线可靠性试验。
细菌内毒素工作标准品系以细菌内毒素国家标准品为基准标定其效价,用于干扰试验及检查法中编号B和C溶液的制备、凝胶法中鲎试剂灵敏度复核试验、光度测定法中标准曲线可靠性试验。
细菌内毒素检查用水应符合灭菌注射用水标准,其内毒素含量小于0.015EU/ml(用于凝胶法)或0.005EU/ml(用于光度测定法),且对内毒素试验无干扰作用。
试验所用的器皿需经处理,以去除可能存在的外源性内毒素。
耐热器皿常用干热灭菌法(250℃、30分钟以上)去除,也可采用其他确证不干扰细菌内毒素检查的适宜方法。
若使用塑料器皿,如微孔板和与微量加样器配套的吸头等,应选用标明无内毒素并且对试验无干扰的器具。
供试品溶液的制备某些供试品需进行复溶、稀释或在水性溶液中浸提制成供试品溶液。
必要时,可调节被测溶液(或其稀释液)的pH值,一般供试品溶液和鲎试剂混合后溶液的pH值在6.0~8.0的范围内为宜,可使用适宜的酸、碱溶液或缓冲溶液调节pH值。
酸或碱溶液须用细菌内毒素检查用水在已去除内毒素的容器中配制。
缓冲液必须经过验证不含内毒素和干扰因子。
内毒素限值的确定药品、生物制品的细菌内毒素限值(L)一般按以下公L=K/M式中L为供试品的细菌内毒素限值,一般以EU/ml、EU/mg或EU/U(活性单位)表示;K为人每千克体重每小时最大可接受的内毒素剂量,以EU/(kg·h)表示,注射剂K=5 EU/(kg·h),放射性药品注射剂K=2.5 EU/(kg·h),鞘内用注射剂K=0.2 EU/(kg·h);M为人用每千克体重每小时的最大供试品剂量,以ml/(kg·h)、mg/(kg·h)或U/(kg·h)表示,人均体重按60kg计算,人体表面积按1.62㎡计算。
0832 水分测定法第一法(费休氏法)1. 容量滴定法本法是根据碘和二氧化硫在吡啶和甲醇溶液中与水定量反应的原理来测定水分。
所用仪器应干燥,并能避免空气中水分的侵入;测定应在干燥处进行。
费休氏试液的制备与标定(1)制备称取碘(置硫酸干燥器内48小时以上)110g,置干燥的具塞锥形瓶(或烧瓶)中,加无水吡啶160ml,注意冷却,振摇至碘全部溶解,加无水甲醇300ml,称定重量,将锥形瓶(或烧瓶)置冰浴中冷却,在避免空气中水分侵入的条件下,通入干燥的二氧化硫至重量增加72g,再加无水甲醇使成1000ml,密塞,摇匀,在暗处放置24小时。
也可以使用稳定的市售费休氏试液。
市售的费休氏试液可以是不含吡啶的其他碱化试剂,或不含甲醇的其他伯醇类等制成;也可以是单一的溶液或由两种溶液临用前混合而成。
本试液应遮光,密封,阴凉干燥处保存。
临用前应标定滴定度。
(2)标定精密称取纯化水10~30mg,用水分测定仪直接标定;或精密称取纯化水10~30mg,置干燥的具塞锥形瓶中,除另有规定外,加无水甲醇适量,在避免空气中水分侵入的条件下,用费休氏试液滴定至溶液由浅黄色变为红棕色,或用电化学方法[如永停滴定法(通则0701)等]指示终点;另做空白试验,按下式计算:F=WA−B式中F为每1ml费休氏试液相当于水的重量,mg;W为称取纯化水的重量,mg;A为滴定所消耗费休氏试液的容积,ml;B为空白所消耗费休氏试液的容积,ml。
测定法精密称取供试品适量(约消耗费休氏试液1~5ml),除另有规定外,溶剂为无水甲醇,用水分测定仪直接测定。
或精密称取供试品适量,置干燥的具塞锥形瓶中,加溶剂适量,在不断振摇(或搅拌)下用费休氏试液滴定至溶液由浅黄色变为红棕色,或用永停滴定法(通则0701)指示终点;另做空白试验,按下式计算:×100%供试品中水分含量(%)=(A−B)FW式中A为供试品所消耗费休氏试液的体积,ml;B为空白所消耗费休氏试液的体积,ml;F为每1ml费休氏试液相当于水的重量,mg;W为供试品的重量,mg。

0904 可见异物检查法可见异物系指存在于注射剂、眼用液体制剂和无菌原料药中,在规定条件下目视可以观测到的不溶性物质,其粒径或长度通常大于50μm。
注射剂、眼用液体制剂应在符合药品生产质量管理规范(GMP)的条件下生产,产品在出厂前应采用适宜的方法逐一检查并同时剔除不合格产品。
临用前,需在自然光下目视检查(避免阳光直射),如有可见异物,不得使用。
可见异物检查法有灯检法和光散射法。
一般常用灯检法,也可采用光散射法。
灯检法不适用的品种,如用深色透明容器包装或液体色泽较深(一般深于各标准比色液7号)的品种可选用光散射法;混悬型、乳状液型注射液和滴眼液不能使用光散射法。
实验室检测时应避免引人可见异物。
当制备注射用无菌粉末和无菌原料药供试品溶液时,或供试品的容器不适于检查(如透明度不够、不规则形状容器等),需转移至适宜容器中时,均应在B级的洁净环境(如层流净化台)中进行。
用于本试验的供试品,必须按规定随机抽样。
第一法(灯检法)灯检法应在暗室中进行。
检查装置如下图所示。
ABCD图灯检法示意A.带有遮光板的日光灯光源(光照度可在1000~4000lx范围内调节);B.不反光的黑色背景;C.不反光的白色背景和底部(供检査有色异物);D.反光的白色背景(指遮光板内侧)。
检查人员条件远距离和近距离视力测验,均应为4.9及以上(矫正后视力应为5.0及以上);应无色盲。
检査法按以下各类供试品的要求,取规定量供试品,除去容器标签,擦净容器外壁,必要时将药液转移至洁净透明的适宜容器内,将供试品置遮光板边缘处,在明视距离(指供试品至人眼的清晰观测距离,通常为25cm),手持容器颈部,轻轻旋转和翻转容器(但应避免产生气泡),使药液中可能存在的可见异物悬浮,分别在黑色和白色背景下目视检查,重复观察,总检查时限为20秒。
供试品装量每支(瓶)在10ml及10ml以下的,每次检查可手持2支(瓶)。
50ml或50ml以上大容量注射液按直、横、倒三步法旋转检视。

药典三部(2015版)-通则-1143细菌内毒素检查法本法系利用鲎试剂来检测或量化由革兰阴性菌产生的细菌内毒素,以判断供试品中细菌内毒素的限量是否符合规定的一种方法。
细菌内毒素检查包括两种方法,即凝胶法和光度测定法,后者包括浊度法和显色基质法。
供试品检测时,可使用其中任何一种方法进行试验。
当测定结果有争议时,除另有规定外,以凝胶限度试验结果为准。
本试验操作过程应防止内毒素的污染。
细菌内毒素的量用内毒素单位(EU)表示,1EU与1个内毒素国际单位(IU)相当。
细菌内毒素国家标准品系自大肠埃希菌提取精制而成,用于标定、复核、仲裁鲎试剂灵敏度、标定细菌内毒素工作标准品的效价,干扰试验及检查法中编号B和C溶液的制备、凝胶法中鲎试剂灵敏度复核试验、光度测定法中标准曲线可靠性试验。
细菌内毒素工作标准品系以细菌内毒素国家标准品为基准标定其效价,用于干扰试验及检查法中编号B和C溶液的制备、凝胶法中鲎试剂灵敏度复核试验、光度测定法中标准曲线可靠性试验。
细菌内毒素检查用水应符合灭菌注射用水标准,其内毒素含量小于0.015EU/ml(用于凝胶法)或0.005EU/ml(用于光度测定法),且对内毒素试验无干扰作用。
试验所用的器皿需经处理,以去除可能存在的外源性内毒素。
耐热器皿常用干热灭菌法(250℃、30分钟以上)去除,也可采用其他确证不干扰细菌内毒素检查的适宜方法。
若使用塑料器皿,如微孔板和与微量加样器配套的吸头等,应选用标明无内毒素并且对试验无干扰的器具。
供试品溶液的制备某些供试品需进行复溶、稀释或在水性溶液中浸提制成供试品溶液。
必要时,可调节被测溶液(或其稀释液)的pH值,一般供试品溶液和鲎试剂混合后溶液的pH值在6.0~8.0的范围内为宜,可使用适宜的酸、碱溶液或缓冲溶液调节pH值。
酸或碱溶液须用细菌内毒素检查用水在已去除内毒素的容器中配制。
缓冲液必须经过验证不含内毒素和干扰因子。
内毒素限值的确定药品、生物制品的细菌内毒素限值(L)一般按以下公L=K/M式中L为供试品的细菌内毒素限值,一般以EU/ml、EU/mg或EU/U(活性单位)表示;K为人每千克体重每小时最大可接受的内毒素剂量,以EU/(kg·h)表示,注射剂K=5EU/(kg·h),放射性药品注射剂K=2.5EU/(kg·h),鞘内用注射剂K=0.2EU/(kg·h);M为人用每千克体重每小时的最大供试品剂量,以ml/(kg·h)、mg/(kg·h)或U/(kg·h)表示,人均体重按60kg计算,人体表面积按1.62㎡计算。
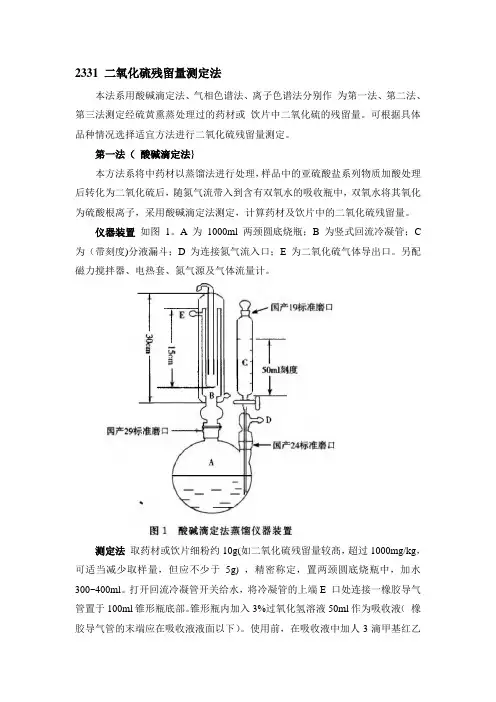
2331 二氧化硫残留量测定法本法系用酸碱滴定法、气相色谱法、离子色谱法分别作为第一法、第二法、第三法测定经硫黄熏蒸处理过的药材或饮片中二氧化硫的残留量。
可根据具体品种情况选择适宜方法进行二氧化硫残留量测定。
第一法(酸碱滴定法}本方法系将中药材以蒸馏法进行处理,样品中的亚硫酸盐系列物质加酸处理后转化为二氧化硫后,随氮气流带入到含有双氧水的吸收瓶中,双氧水将其氧化为硫酸根离子,采用酸碱滴定法测定,计算药材及饮片中的二氧化硫残留量。
仪器装置如图1。
A为1000ml两颈圆底烧瓶;B 为竖式回流冷凝管;C 为(带刻度)分液漏斗;D为连接氮气流入口;E为二氧化硫气体导出口。
另配磁力搅拌器、电热套、氮气源及气体流量计。
测定法取药材或饮片细粉约10g(如二氧化硫残留量较髙,超过1000mg/kg,可适当减少取样量,但应不少于5g) ,精密称定,置两颈圆底烧瓶中,加水300~400ml。
打开回流冷凝管开关给水,将冷凝管的上端E 口处连接一橡胶导气管置于100ml锥形瓶底部。
锥形瓶内加入3%过氧化氢溶液50ml作为吸收液(橡胶导气管的末端应在吸收液液面以下)。
使用前,在吸收液中加人3滴甲基红乙醇溶液指示剂(2.5mg /ml) ,并用0.01mol/L氢氧化钠滴定液滴定至黄色(即终点;如果超过终点,则应舍弃该吸收溶液)。
开通氮气,使用流量计调节气体流量至约0.2L/min;打开分液漏斗C的活塞,使盐酸溶液(6mol/L)10ml流入蒸馏瓶,立即加热两颈烧瓶内的溶液至沸,并保持微沸;烧瓶内的水沸腾1 .5小时后,停止加热。
吸收液放冷后,置于磁力搅拌器上不断搅拌,用氢氧化钠滴定液(0.01mol/L) 滴定,至黄色持续时间20秒不褪,并将滴定的结果用空白实验校正。
照下式计算:供试品中二氧化硫残留量(ug/g)=()WcBA610032.0⨯⨯⨯-式中A 为供试品溶液消耗氢氧化钠滴定液的体积,ml;B 为空白消耗氢氧化钠滴定液的体积,ml;c 为氢氧化钠滴定液摩尔浓度,mol/L;0. 032为lml氢氧化钠滴定液(l mol/L )相当的二氧化硫的质量,g;W 为供试品的重量,g。
0711 乙醇量测定法—、气相色谱法本法系采用气相色谱法(通则0521) 测定各种含乙醇制剂中在20℃时乙醇(C2H5OH )的含量(%) (ml /ml ) 。
除另有规定外,按下列方法测定。
第一法(毛细管柱法)色谱条件与系统适用性试验采用(6% )氰丙基苯基- (94%)二甲基聚硅氧烷为固定液的毛细管柱;起始温度为40℃,维持2分钟,以每分钟3℃的速率升温至65℃,再以每分钟25℃的速率升温至200℃,维持10分钟;进样口温度200℃;检测器(FID )温度220℃;采用顶空分流进样,分流比为1:1;顶空瓶平衡温度为85℃,平衡时间为20分钟。
理论板数按乙醇峰计算应不低于10000,乙醇峰与正丙醇峰的分离度应大于2 .0。
校正因子测定精密量取恒温至20℃的无水乙醇5ml,平行两份;置100ml量瓶中,精密加入恒温至20的正丙醇(内标物质)5ml,用水稀释至刻度,摇匀,精密量取该溶液lml ,置100ml量瓶中,用水稀释至刻度,摇匀(必要时可进一步稀释),作为对照品溶液。
精密量取3ml,置10ml顶空进样瓶中,密封,顶空进样,每份对照品溶液进样3次,测定峰面积,计算平均校正因子,所得校正因子的相对标准偏差不得大于2.0% 。
测定法精密量取恒温至20的供试品适量(相当于乙醇约5 ml ) ,置100ml量瓶中,精密加入恒温至20 ℃的正丙醇5 ml,用水稀释至刻度,摇匀,精密量取该溶液lml ,置100ml 量瓶中,用水稀释至刻度,摇匀(必要时可进一步稀释),作为供试品溶液。
精密量取3 ml ,置10ml顶空进样瓶中,密封,顶空进样,测定峰面积,按内标法以峰面积计算,即得。
【附注】毛细管柱建议选择大口径、厚液膜色谱柱,规格为30m×0.53mm×3.00um。
第二法(填充柱法)色谱条件与系统适用性试验用直径为0.18~0.25mm的二乙烯苯-乙基乙烯苯型高分子多孔小球作为载体,柱温为120~150℃。
0904 可见异物检查法可见异物系指存在于注射剂、眼用液体制剂和无菌原料药中,在规定条件下目视可以观测到的不溶性物质,其粒径或长度通常大于50μm。
注射剂、眼用液体制剂应在符合药品生产质量管理规范(GMP)的条件下生产,产品在出厂前应采用适宜的方法逐一检查并同时剔除不合格产品。
临用前,需在自然光下目视检查(避免阳光直射),如有可见异物,不得使用。
可见异物检查法有灯检法和光散射法。
一般常用灯检法,也可采用光散射法。
灯检法不适用的品种,如用深色透明容器包装或液体色泽较深(一般深于各标准比色液7号)的品种可选用光散射法;混悬型、乳状液型注射液和滴眼液不能使用光散射法。
实验室检测时应避免引人可见异物。
当制备注射用无菌粉末和无菌原料药供试品溶液时,或供试品的容器不适于检查(如透明度不够、不规则形状容器等),需转移至适宜容器中时,均应在B级的洁净环境(如层流净化台)中进行。
用于本试验的供试品,必须按规定随机抽样.第一法(灯检法)灯检法应在暗室中进行。
检查装置如下图所示。
ABCD图灯检法示意A。
带有遮光板的日光灯光源(光照度可在1000~4000lx范围内调节);B.不反光的黑色背景;C。
不反光的白色背景和底部(供检査有色异物);D.反光的白色背景(指遮光板内侧).检查人员条件远距离和近距离视力测验,均应为4.9及以上(矫正后视力应为5。
0 及以上);应无色盲.检査法按以下各类供试品的要求,取规定量供试品,除去容器标签,擦净容器外壁,必要时将药液转移至洁净透明的适宜容器内,将供试品置遮光板边缘处,在明视距离(指供试品至人眼的清晰观测距离,通常为25cm),手持容器颈部,轻轻旋转和翻转容器(但应避免产生气泡),使药液中可能存在的可见异物悬浮,分别在黑色和白色背景下目视检查,重复观察,总检查时限为20秒。
供试品装量每支(瓶)在10ml及10ml以下的,每次检查可手持2支(瓶)。
50ml或50ml以上大容量注射液按直、横、倒三步法旋转检视。
2351 黄曲霉毒素测定法第一法本法系用高效液相色谱法(通则0512)测定药材、饮片及制剂中的黄曲霉毒素B1、黄曲霉毒素B2、黄曲霉毒素G1、黄曲霉毒素G2总量计),除另有规定外,按下列方法测定。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以%甲醇-乙腈-水(40:18:42)为流动相;采用柱后衍生法检测,①碘衍生法:衍生溶液为0.05% 的碘溶液(取碘0.5g,加入甲醇100ml使溶解,用水稀释至1000ml制成),衍生化泵流速每分钟0.3ml,衍生化温度70℃;②光化学衍生法:光化学衍生器(254nm);以荧光检测器检测,激发波长λex = 360nm(或365nm),发射波长λex =450nm。
两个相邻色谱峰的分离度应大于1.5。
混合对照品溶液的制备精密量取黄曲霉毒素混合对照品溶液(黄曲霉毒素B1、黄曲霉毒素B2、黄曲霉毒素G1、黄曲霉毒素G2标示浓度分别为1.0ug/ml、0.3ug/ml、1.0ug/ml、0.3ug/ml)0.5ml,置10ml 量瓶中,用甲醇稀释至刻度,作为贮备溶液。
精密量取贮备溶液lml,置25ml量瓶中,用甲醇稀释至刻度,即得。
供试品溶液的制备取供试品粉末约15g(过二号筛),精密称定,置于均质瓶中,加人氯化钠3g,精密加入70%甲醇溶液75ml,高速搅拌2分钟(搅拌速度大于11000转/分钟),离心5分钟(离心速度2500转/分钟),精密量取上清液15ml,置50ml量瓶中,用水稀释至刻度,摇匀,用微孔滤膜(0.45um)滤过,量取续滤液20.0ml,通过免疫亲合柱,流速每分钟3ml,用水20ml洗脱,洗脱液弃去,使空气进入柱子,将水挤出柱子,再用适量甲醇洗脱,收集洗脱液,置2ml量瓶中,并用甲醇稀释至刻度,摇匀,即得。
测定法分别精密吸取上述混合对照品溶液5ul、l0ul、15ul、20ul、25ul,注人液相色谱仪,测定峰面积,以峰面积为纵坐标,进样量为横坐标,绘制标准曲线。