如何找同源基因并用DNAman比对序列
- 格式:ppt
- 大小:2.15 MB
- 文档页数:18
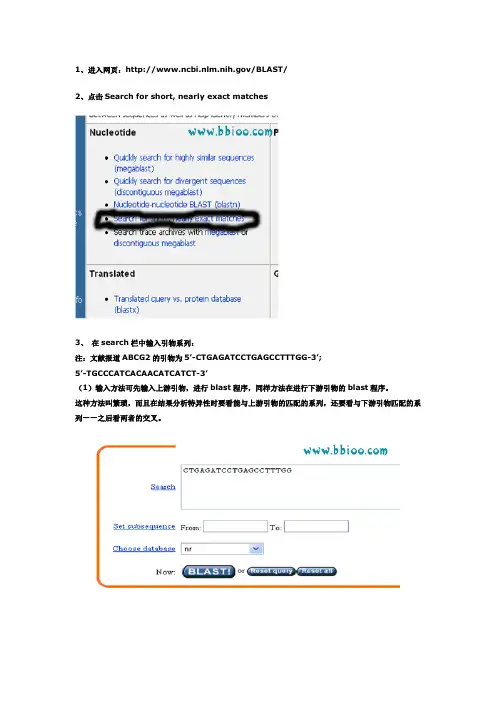
1、进入网页:/BLAST/2、点击Search for short, nearly exact matches3、在search栏中输入引物系列:注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’;5’-TGCCCATCACAACATCATCT-3’(1)输入方法可先输入上游引物,进行blast程序,同样方法在进行下游引物的blast程序。
这种方法叫繁琐,而且在结果分析特异性时要看能与上游引物的匹配的系列,还要看与下游引物匹配的系列——之后看两者的交叉。
(2)简便的做法是同时输入上下游引物:有以下两种方法。
输入上下游引物系列都从5’——3’。
A、输入上游引物空格输入下游引物B、输入上游引物回车输入下游引物4、在options for advanced blasting中:select from 栏通过菜单选择Homo sapiensExpect后面的数字改为105、在format中:select from 栏通过菜单选择Homo sapiens Expect后面的数字填上0 106、点击网页中最下面的“BLAST!”7、出现新的网页,点击Format!8、等待若干秒之后,出现results of BLAST的网页。
该网页用三种形式来显示blast的结果。
(1)图形格式:图中①代表这些序列与上游引物匹配、并与下游引物互补的得分值都位于40~50分图中②代表这些序列与上游引物匹配的得分值位于40~50分,而与下游引物不互补图中③代表这些序列与下游引物互补的得分值小于40分,而与上游引物不匹配通过点击相应的bar可以得到匹配情况的详细信息。
(2)结果信息概要:从左到右分别为:A、数据库系列的身份证:点击之后可以获得该序列的信息B、系列的简单描述C、高比值片段对(high-scoring segment pairs, HSP)的字符得分。
按照得分的高低由大到小排列。
得分的计算公式=匹配的碱基×2+0.1。


dnaman使用方法使用DNAMAN的方法DNAMAN是一款功能强大的生物信息学软件,广泛应用于DNA 和蛋白质序列分析、比对、编辑和可视化等领域。
本文将介绍DNAMAN的使用方法,帮助读者快速上手并熟练运用该软件。
一、安装和启动DNAMAN1. 下载软件安装包:在DNAMAN官方网站上下载适用于您的操作系统的安装包,并保存到本地。
2. 安装软件:双击安装包,按照提示完成软件的安装过程。
3. 启动软件:安装完成后,双击桌面上的DNAMAN图标,即可启动软件。
二、导入和编辑序列1. 导入序列:在DNAMAN的主界面,点击"文件"菜单,选择"导入序列",然后选择您要导入的序列文件(支持FASTA、GenBank 等格式)。
2. 编辑序列:在序列编辑界面,您可以对序列进行添加、删除、替换等操作。
点击"编辑"菜单,选择相应的编辑功能,然后在弹出的对话框中进行操作。
三、序列比对和分析1. 序列比对:点击"工具"菜单,选择"序列比对",然后选择比对算法和参数设置,点击"开始比对"按钮即可进行序列比对。
2. 序列分析:DNAMAN提供了丰富的序列分析工具,如ORF预测、限制酶切位点分析、引物设计等。
点击"工具"菜单,选择相应的分析工具,按照提示进行操作。
四、序列可视化和输出1. 序列可视化:DNAMAN提供了多种序列可视化方式,如线性图、环形图、比对图等。
点击"视图"菜单,选择相应的可视化方式,即可查看序列的结构和特征。
2. 输出结果:完成序列分析后,您可以将结果导出为文本文件或图像文件。
点击"文件"菜单,选择"导出结果",然后选择输出格式和保存路径,点击"保存"按钮即可导出结果。
五、保存和管理项目1. 保存项目:在使用DNAMAN进行序列分析时,建议您保存项目以便后续操作。


序列分析软件DNAMAN 的使用方法简介DNAMAN 是一种常用的核酸序列分析软件。
由于它功能强大,使用方便,已成为一种普遍使用的DNA 序列分析工具。
本文以DNAMAN 5.2.9 Demo version 为例,简单介绍其使用方法。
打开DNAMAN,可以看到如下界面::第一栏为主菜单栏。
除了帮助菜单外,有十个常用主菜单,如下所示第二栏为工具栏:如下所示:第三栏为浏览器栏:如下所示:在浏览器栏下方的工作区左侧,可见Channel 工具条,DNAMAN 提供20 个Channel,如左所示:点击Channel 工具条上相应的数字,即可击活相应的Channel。
每个Channel 可以装入一个序列。
将要分析的序列(DNA 序列或氨基酸序列)放入Channel 中可以节约存取序列时间,加快分析速度。
此版本DNAMAN 提供自动载入功能,用户只需激活某个Channel ,然后打开一个序列文件,则打开的序列自动载入被激活的Channel 中。
本文以具体使用DNAMAN 的过程为例来说明如何使用DNAMAN 分析序列。
1.将待分析序列装入Channel(1)通过File|Open 命令打开待分析序列文件,则打开的序列自动装入默认Channel。
(初始为channel1)可以通过激活不同的channel(例如:channel5)来改变序列装入的Channel。
(2)通过Sequence|Load Sequence 菜单的子菜单打开文件或将选定的部分序列装入Channel。
可以通过Sequence|Current Sequence|Analysis Defination 命令打开一个对话框,通过此对话框可以设定序列的性质(DNA 或蛋白质),名称,要分析的片段等参数。
2.以不同形式显示序列通过Sequence|Display Sequence 命令打开对话框,如下图所示:根据不同的需要,可以选择显示不同的序列转换形式。


序列分析软件DNAMAN的使用方法中文演示文稿第一部分:软件介绍1.DNAMAN是什么?-DNAMAN是一款用于DNA和蛋白质序列分析的软件。
-它提供多种功能,包括序列比对、引物设计、限制酶分析和进化树构建等。
2.DNAMAN的应用领域-DNAMAN广泛应用于生物学、生物技术和医药领域。
-它可以帮助研究人员进行序列分析、设计实验方案和解读实验结果。
第二部分:基本序列比对1.创建新项目-打开DNAMAN软件,点击“新建”按钮创建新项目。
-输入项目名称和序列信息,保存并打开该项目。
2.导入序列-点击“导入”按钮,选择需要比对的序列文件,点击“确定”导入。
-系统会自动将序列导入到项目中。
3.序列比对-选择需要比对的序列,点击“比对”按钮进行序列比对。
-系统会自动比对序列并生成比对结果。
4.结果解读-比对结果以图形和文本形式展示。
-可以通过选择不同的比对算法和调整参数来优化比对结果。
第三部分:引物设计1.创建新项目-打开DNAMAN软件,点击“新建”按钮创建新项目。
-输入项目名称和序列信息,保存并打开该项目。
2.导入标记序列-点击“导入”按钮,选择需要设计引物的标记序列文件,点击“确定”导入。
-系统会自动将标记序列导入到项目中。
3.引物设计-点击“引物设计”按钮,选择设计引物的参数和算法。
-系统会根据所选参数和算法自动生成引物设计结果。
4.结果解读-引物设计结果以图形和文本形式展示。
-可以通过选择不同的参数和算法来优化引物设计结果。
第四部分:限制酶分析1.创建新项目-打开DNAMAN软件,点击“新建”按钮创建新项目。
-输入项目名称和序列信息,保存并打开该项目。
2.导入限制酶序列-点击“导入”按钮,选择需要分析的限制酶序列文件,点击“确定”导入。
-系统会自动将限制酶序列导入到项目中。
3.限制酶分析-点击“限制酶分析”按钮,选择分析参数和算法。
-系统会根据所选参数和算法自动进行限制酶分析。
4.结果解读-限制酶分析结果以图形和文本形式展示。


dnaman基因序列的比对方法
DNAMAN是用于多序列比对、PCR引物设计、限制性酶切分析、质粒绘图、蛋白质分析等的高度集成化的分子生物学综合应用软件。
以下是使用DNAMAN进行基因序列比对的步骤:
1. 打开DNAMAN,点击“Sequence-Alignment-Multiple sequence alignment”,进入比对页面。
2. 点击“File”,上传序列文件(fasta格式),选择序列类型,点击“Next”。
3. 这一步和下一步默认即可。
4. 参数默认即可,点击“Finish”,即可得到比对结果。
5. 若需要导出图,点击“Output-Graphic file”,保存EMF格式图片。
随后在画图工具中另存为需要的照片格式即可。
以上步骤仅供参考,建议查阅DNAMAN软件使用说明或咨询专业人士,
获取更准确的信息。

步一步教你使用N C B I查找D N A、m R N A、c D N A--------------------------------------------------------------------------作者: _____________--------------------------------------------------------------------------日期: _____________一步一步教你使用 NCBI 查找DNA、mRNA、cDNA、...最近看到很多战友在论坛上询问如何查询基因序列、如何进行引物设计、如何使用BLAST 进行序列比对……,这些问题在 NCBI 上都可以方便的找到答案。
现在我就结合我自己使用 NCBI的一些经历(经验)跟大家交流一下 BCBI 的使用。
希望大家都能发表自己的使用心得,让我们共同进步!我分以下几个部分说一下 NCBI 的使用:Part one 如何查找基因序列、mRNA、PromoterPart two 如何查找连续的 mRNA、cDNA、蛋白序列Part three 运用 STS 查找已经公布的引物序列Part four 如何运用 BLAST 进行序列比对、检验引物特异性特别感谢本版版主,将这个帖子置顶!从发帖到现在,很多战友对该帖给与了积极的关注,在此向给我投票的(以及想给我投票却暂时不能投票的)各位战友表示真诚的感谢,谢谢各位战友!请大家对以下我发表的内容提出自己的意见。
关于NCBI 其他方面的使用也请水平较高的战友给予补充First of all,还是让我们从查找基因序列开始。
第一部分利用Map viewer 查找基因序列、mRNA 序列、启动子(Promoter)下面以人的 IL6(白细胞介素 6)为例讲述一下具体的操作步骤1.打开Map viewer 页面,网址为:在 search 的下拉菜单里选择物种,for 后面填写你的目的基因。
dnaman比对序列结果解读
根据您提供的信息,"dnaman"应该指的是DNA比对软件"DnaMAN"。
DNA比对是一种将两个或多个DNA序列进行比较的分析方法,以确定它们之间的相似性和差异性。
"DnaMAN"软件的结果解读通常包括以下几个方面:
1. 序列相似性:该软件会计算DNA序列之间的相似性分数,通常以百分比表示。
较高的相似性分数表示两个序列之间更相似。
2. 序列对齐:软件会根据相似性算法将输入的DNA序列进行配对对齐,以使相同的碱基对齐在一起,从而揭示其相似性和差异性。
3. 碱基差异:比对结果可能会指示在两个序列之间存在的碱基差异,包括插入、缺失或替换等。
4. 序列特征:软件还可以鉴定序列中的重复序列、基因、启动子或其他相关特征,并提供这些信息。
总之,"DnaMAN"比对序列的结果解读将提供关于DNA序列相似性、差异、对齐和特征的相关信息,以帮助研究者进一步理解DNA序列之间的关系和功能。
如何用DNAMAN对比核酸序列在生物学的研究中,有一个常用的方法,就是通过比较分析获取有用的信息和知识。
想当年,达尔文爷爷正是研究比较了达尔文雀(galapagos finches)同其它一些物种的形态学特征,从而提出了自然选择学说。
在科技飞速发展的今天,对两个物种进行全基因组序列比较已经不再是一个梦想。
达尔文爷爷泉下有知,一定会喜极而泣!介绍如何使用DNAMAN 8作核酸多序列比对。
工具/原料•电脑•软件 DNAMAN方法/步骤1.下载好 DNAMAN 8 的软件并打开2.:第一栏为主菜单栏,除了帮助菜单外,有十个常用主菜单;第二栏为工具栏;第三栏为浏览器栏。
打开File-New,将序列粘贴到弹出的窗口中,点击File-save,保存到指定的文件夹。
3.END方法/步骤21.将所需比对的序列保存好以后,选中Sequence—Aligment—Multiple aligment sequence进行多序列比较。
2.在弹出的窗口Sequence&Files中加载序列,File、Fold、channel、Database分别表示从文件、文件夹、channel和数据库中获取序列。
勾选窗口中的“DNA”,点击“下一步”。
3.在弹出的窗口Method中,“optimalaligment”最佳比对方式中有四个高大上的选项:Full Alignment(完全比对)、Prosile Aligment(轮廓比对)、 New Swquence on Profile (轮廓上的新序列)、Fast Alignment(快速比对),本文选择了Fast Alignment,并且勾选了Try both strands(尝试使用双链)。
其他项目不需修改,默认就OK,点击“下一步”。
4.“Duang”,结果就出来啦!点击“Option”可以选择要显示的内容。
END方法/步骤31.保存:点击File-output-Graphic(EMF)File,该序列比对的结果窗口可以图片格式保存。
dnaman比对序列结果解读(原创实用版)目录1.Dnaman 比对序列结果2.结果解读3.结论正文在生物信息学领域,Dnaman 是一款广泛使用的 DNA 比对工具,能够帮助研究人员快速准确地比较两个或多个 DNA 序列。
本文将基于 Dnaman 比对序列结果,对其进行解读。
首先,我们来看 Dnaman 比对序列结果。
Dnaman 将输入的 DNA 序列进行比对,并生成比对结果文件。
该文件包含了比对过程中各碱基之间的匹配程度、插入和删除情况等详细信息。
通过对比结果文件,研究人员可以直观地了解序列之间的相似性和差异性。
接下来,我们来解读比对结果。
在 Dnaman 的比对结果中,颜色代表了不同类型的信息。
例如,红色表示不匹配的碱基,蓝色表示匹配的碱基,绿色表示插入的碱基,橙色表示删除的碱基。
通过观察这些颜色,我们可以了解序列之间的相似性。
如果两个序列之间的匹配程度较高,那么它们之间的颜色将以蓝色为主。
反之,如果序列之间的差异较大,那么将会看到大量的红色、绿色和橙色。
此外,我们还可以通过比对结果文件中的数字信息来了解插入和删除的具体情况。
在解读比对结果时,我们需要注意以下几点。
首先,比对结果可能会受到序列长度、质量等因素的影响,因此在分析结果时需要综合考虑这些因素。
其次,对于某些特定的序列,可能需要采用其他的比对方法和参数设置,以获得更准确的结果。
最后,在解读结果时,我们需要将比对结果与生物学背景相结合,从而得出更有意义的结论。
综上所述,Dnaman 比对序列结果的解读对于研究 DNA 序列的相似性和差异性具有重要意义。
通过对比结果文件的观察和分析,我们可以了解序列之间的匹配程度、插入和删除情况等信息。
然而,在解读结果时,我们需要注意比对方法的局限性,并结合生物学背景进行综合分析。
mane transcript确定方法在生物学研究领域,对mane transcript(马转录组)的确定方法探讨有着重要的意义。
本文将详细介绍几种常用的方法,以帮助研究人员准确确定mane transcript。
一、基于序列相似性的方法1.同源比对法:通过将待研究的马转录组序列与已知基因库(如NCBI、Ensembl等)中的序列进行比对,寻找相似度较高的序列。
若相似度超过一定阈值(如80%以上),则可认为该转录组与已知基因具有同源性。
2.跨物种比对法:由于马与其它哺乳动物的基因序列具有较高的保守性,研究人员可以将马转录组序列与其它物种的已知基因序列进行比对,从而确定mane transcript。
二、基于功能注释的方法1.基因本体(GO)注释:通过将马转录组序列进行GO注释,可以了解其可能的功能。
若与已知基因的GO注释结果相似,则可认为这些转录组可能具有相同的功能。
2.信号肽预测:对马转录组序列进行信号肽预测,可判断其是否为分泌蛋白。
若与已知基因的信号肽预测结果一致,则有助于确定mane transcript。
三、基于表达谱的方法1.转录组测序:通过高通量测序技术(如RNA-seq)对马转录组进行测序,获得其表达谱。
将表达谱与已知基因的表达谱进行比对,可找到表达模式相似的基因。
2.实时荧光定量PCR:利用实时荧光定量PCR技术,对马转录组在不同组织、发育阶段或处理条件下的表达水平进行定量分析。
若与已知基因的表达模式一致,则有助于确定mane transcript。
四、综合方法1.聚类分析:将马转录组序列与已知基因序列进行聚类分析,根据聚类结果判断其可能的同源基因。
2.系统进化分析:构建马转录组与已知基因的系统进化树,分析其进化关系。
若与已知基因的进化关系相近,则有助于确定mane transcript。
总之,确定mane transcript的方法多种多样,研究人员可根据实际研究需求选择合适的方法。