9750利用CASTEP模拟计算实例1
- 格式:doc
- 大小:418.50 KB
- 文档页数:5

CASTEP 计算理论总结XBAPRSCASTEP 特点是适合于计算周期性结构,对于非周期性结构一般要将特定的部分作为周期性结构,建立单位晶胞后方可进行计算。
CASTEP 计算步骤可以概括为三步:首先建立周期性的目标物质的晶体;其次对建立的结构进行优化,这包括体系电子能量的最小化和几何结构稳定化。
最后是计算要求的性质,如电子密度分布(Electron density distribution),能带结构(Band structure)、状态密度分布(Densityof states)、声子能谱(Phonon spectrum)、声子状态密度分布(DOS of phonon),轨道群分布(Orbitalpopulations)以及光学性质(Optical properties)等。
本文主要将就各个步骤中的计算原理进行阐述,并结合作者对计算实践经验,在文章最后给出了几个计算事例,以备参考。
CASTEP 计算总体上是基于DFT ,但实现运算具体理论有:离子实与价电子之间相互作用采用赝势来表示;超晶胞的周期性边界条件;平面波基组描述体系电子波函数;广泛采用快速fast Fourier transform (FFT) 对体系哈密顿量进行数值化计算;体系电子自恰能量最小化采用迭带计算的方式;采用最普遍使用的交换-相关泛函实现DFT 的计算,泛函含概了精确形式和屏蔽形式。
一, CASTEP 中周期性结构计算优点与MS 中其他计算包不同,非周期性结构在CASTEP 中不能进行计算。
将晶面或非周期性结构置于一个有限长度空间方盒中,按照周期性结构来处理,周期性空间方盒形状没有限制。
之所以采用周期性结构原因在于:依据Bloch 定理,周期性结构中每个电子波函数可以表示为一个波函数与晶体周期部分乘积的形式。
他们可以用以晶体倒易点阵矢量为波矢一系列分离平面波函数来展开。
这样每个电子波函数就是平面波和,但最主要的是可以极大简化Kohn-Sham 方程。




模型选定我们在进行材料物理模拟所需要做的第一步 (也是很重要的一步) 是模型的选定或建构。
CASTEP 虽然内建了很多功能来预测晶胞参数 (边长,夹角) 与原子位置,但仍然仰赖使用者告诉它 "要进行计算的系统是什么"。
在选定模型时,我们需切记如果系统内原子太多或是超晶胞体积太大,则计算量都会以平面波数的 3 次方增加到计算机难以负荷或使用者难以等待的程度,因此,我时时应考虑设计出一个足以表现出我们所想要研究的物理,而却又能使所有采用的超晶胞越少越好的模型。
如果研究上涉及到一系列原子数不同的大小系统需要做计算,最好能先计算小结构,不要一开始就送入大结构到计算机中。
如果要模拟的系统是含有杂质,则单位晶胞必须进一步放大成超晶胞以便使化学成分里的分数变成整数,因此晶胞会变得很大。
在某些特殊的情况,相互取代的元素种类是很类似的,(即在化学行为上类似),则下一个版本的CASTEP会提供一种叫做虚拟晶体近似 (Virtural Crystal Approximation,VCA) 的方法,则模型里面的原子就可以指定成如0.3A元素加0.7B元素这种样子,因此总可以以最小晶胞来做计算的模型。
但这种方法的精确度通常只适用于合金材料,故要小心使用。
模型的选定有许多人为抉择会含在里面,例如表面计算的层数,因此有些情况也需要进行所谓的收敛性测试。
赝势选择选单VpsMS接口(界面)的设定是选用USP优先于NCP,USP有加速计算与减少内存使用的效果,其精确度也与NCP (norm-conserving potential) 相当。
至于什么时候使用NC呢 (就是在poseudopotential 选项中那些延伸文件名是 .recpot者),使用到NCP的场合有:1.某些CASTEP计算的功能尚未支持到USP,因此需要选用NCP。
2.为了要与已经发展的文献比较或进行验证3.对计算的结果存疑者,能提供『多一种选择』( 注:至于延伸档名是*.psp者也是属于norm-conserving的一种,是TMpotential,在文献上也常被使用,但所需的截止动能较高,因此计算代价较大。
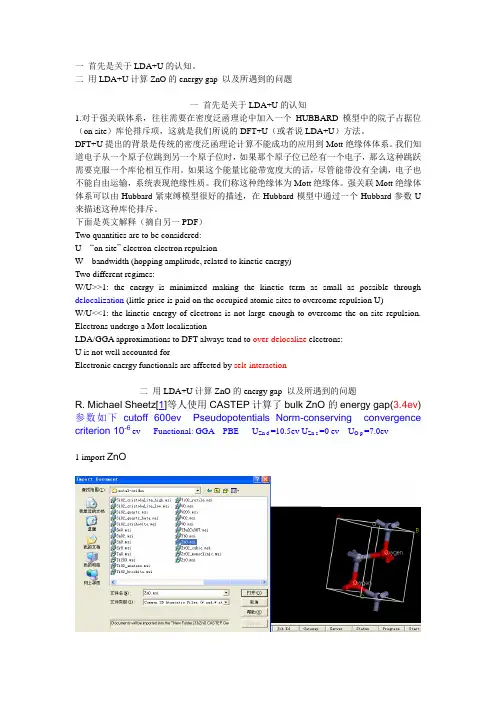
一首先是关于LDA+U的认知。
二用LDA+U计算ZnO的energy gap 以及所遇到的问题一首先是关于LDA+U的认知1.对于强关联体系,往往需要在密度泛函理论中加入一个HUBBARD模型中的院子占据位(on site)库伦排斥项,这就是我们所说的DFT+U(或者说LDA+U)方法。
DFT+U提出的背景是传统的密度泛函理论计算不能成功的应用到Mott绝缘体体系。
我们知道电子从一个原子位跳到另一个原子位时,如果那个原子位已经有一个电子,那么这种跳跃需要克服一个库伦相互作用。
如果这个能量比能带宽度大的话,尽管能带没有全满,电子也不能自由运输,系统表现绝缘性质。
我们称这种绝缘体为Mott绝缘体。
强关联Mott绝缘体体系可以由Hubbard紧束缚模型很好的描述,在Hubbard模型中通过一个Hubbard参数U 来描述这种库伦排斥。
下面是英文解释(摘自另一PDF)Two quantities are to be considered:U “on site” electron-electron repulsionW bandwidth (hopping amplitude, related to kinetic energy)Two different regimes:W/U>>1: the energy is minimized making the kinetic term as small as possible through delocalization (little price is paid on the occupied atomic sites to overcome repulsion U)W/U<<1: the kinetic energy of electrons is not large enough to overcome the on-site repulsion. Electrons undergo a Mott localizationLDA/GGA approximations to DFT always tend to over-delocalize electrons:U is not well accounted forElectronic energy functionals are affected by selt-interaction二用LDA+U计算ZnO的energy gap 以及所遇到的问题R. Michael Sheetz[1]等人使用CASTEP计算了bulk ZnO的energy gap(3.4ev) 参数如下cutoff 600ev Pseudopotentials Norm-conserving convergence criterion 10-6 ev Functional: GGA PBE U Zn d =10.5ev U Zn s =0 ev U O p =7.0ev1 import ZnO2 对bulk ZnO 结构优化(还没有选LDA+U,为了下面比较)结果分析我们可以看到band gap is 0.269ev 与3.4ev 相差很大3. 选上LDA+U 看看结果如何。

关于CASTEPCASTEP是特别为固体材料学而设计的一个现代的量子力学基本程序,其使用了密度泛函(DFT)平面波赝势方法,进行第一原理量子力学计算,以探索如半导体,陶瓷,金属,矿物和沸石等材料的晶体和表面性质。
典型的应用包括表面化学,键结构,态密度和光学性质等研究, CASTEP也可用于研究体系的电荷密度和波函数的3D形式。
此外, CASTEP可用于有效研究点缺陷(空位,间隙和置换杂质)和扩展缺陷(如晶界和位错)的性质。
Material Studio使用组件对话框中的CASTEP选项允许准备,启动,分析和监测CASTEP 服役工作。
计算:允许选择计算选项(如基集,交换关联势和收敛判据),作业控制和文档控制。
分析:允许处理和演示CASTEP计算结果。
这一工具提供加速整体直观化以及键结构图,态密度图形和光学性质图形。
CASTEP的任务CASTEP计算是要进行的三个任务中的一个,即单个点的能量计算,几何优化或分子动力学。
可提供这些计算中的每一个以便产生特定的物理性能。
性质为一种附加的任务,允许重新开始已完成的计算以便产生最初没有提出的额外性能。
在CASTEP计算中有很多运行步骤,可分为如下几组:* 结构定义:必须规定包含所感兴趣结构的周期性的3D模型文件,有大量方法规定一种结构:可使用构建晶体(Build Crystal)或构建真空板(Build Vacuum Stab)来构建,也可从已经存在的的结构文档中引入,还可修正已存在的结构。
注意: CASTEP仅能在3D周期模型文件基础上进行计算,必须构建超单胞,以便研究分子体系。
提示: CASTEP计算所需时间随原子数平方的增加而增加。
因此,建议是用最小的初晶胞来描述体系,可使用Build\Symmetry\Primitive Cell菜单选项来转换成初晶胞。
* 计算设置:合适的3D模型文件一旦确定,必须选择计算类型和相关参数,例如,对于动力学计算必须确定系综和参数,包括温度,时间步长和步数。

CASTEP 计算理论总结XBAPRSCASTEP 特点是适合于计算周期性结构,对于非周期性结构一般要将特定的部分作为周期性结构,建立单位晶胞后方可进行计算。
CASTEP 计算步骤可以概括为三步:首先建立周期性的目标物质的晶体;其次对建立的结构进行优化,这包括体系电子能量的最小化和几何结构稳定化。
最后是计算要求的性质,如电子密度分布(Electron density distribution),能带结构(Band structure)、状态密度分布(Densityof states)、声子能谱(Phonon spectrum)、声子状态密度分布(DOS of phonon),轨道群分布(Orbitalpopulations)以及光学性质(Optical properties)等。
本文主要将就各个步骤中的计算原理进行阐述,并结合作者对计算实践经验,在文章最后给出了几个计算事例,以备参考。
CASTEP 计算总体上是基于DFT ,但实现运算具体理论有:离子实与价电子之间相互作用采用赝势来表示;超晶胞的周期性边界条件;平面波基组描述体系电子波函数;广泛采用快速fast Fourier transform (FFT) 对体系哈密顿量进行数值化计算;体系电子自恰能量最小化采用迭带计算的方式;采用最普遍使用的交换-相关泛函实现DFT 的计算,泛函含概了精确形式和屏蔽形式。
一, CASTEP 中周期性结构计算优点与MS 中其他计算包不同,非周期性结构在CASTEP 中不能进行计算。
将晶面或非周期性结构置于一个有限长度空间方盒中,按照周期性结构来处理,周期性空间方盒形状没有限制。
之所以采用周期性结构原因在于:依据Bloch 定理,周期性结构中每个电子波函数可以表示为一个波函数与晶体周期部分乘积的形式。
他们可以用以晶体倒易点阵矢量为波矢一系列分离平面波函数来展开。
这样每个电子波函数就是平面波和,但最主要的是可以极大简化Kohn-Sham 方程。

CASTEP计算理论总结+实例分析CASTEP 计算理论总结XBAPRSCASTEP 特点就是适合于计算周期性结构,对于非周期性结构一般要将特定的部分作为周期性结构,建立单位晶胞后方可进行计算。
CASTEP 计算步骤可以概括为三步:首先建立周期性的目标物质的晶体;其次对建立的结构进行优化,这包括体系电子能量的最小化与几何结构稳定化。
最后就是计算要求的性质,如电子密度分布(Electron density distribution),能带结构(Band structure)、状态密度分布(Density of states)、声子能谱(Phonon spectrum)、声子状态密度分布(DOS of phonon),轨道群分布(Orbital populations)以及光学性质(Optical properties)等。
本文主要将就各个步骤中的计算原理进行阐述,并结合作者对计算实践经验,在文章最后给出了几个计算事例,以备参考。
CASTEP 计算总体上就是基于DFT,但实现运算具体理论有:离子实与价电子之间相互作用采用赝势来表示;超晶胞的周期性边界条件;平面波基组描述体系电子波函数;广泛采用快速fast Fourier transform (FFT) 对体系哈密顿量进行数值化计算;体系电子自恰能量最小化采用迭带计算的方式;采用最普遍使用的交换-相关泛函实现DFT 的计算,泛函含概了精确形式与屏蔽形式。
一, CASTEP 中周期性结构计算优点与MS 中其她计算包不同,非周期性结构在CASTEP 中不能进行计算。
将晶面或非周期性结构置于一个有限长度空间方盒中,按照周期性结构来处理,周期性空间方盒形状没有限制。
之所以采用周期性结构原因在于:依据Bloch 定理,周期性结构中每个电子波函数可以表示为一个波函数与晶体周期部分乘积的形式。
她们可以用以晶体倒易点阵矢量为波矢一系列分离平面波函数来展开。
这样每个电子波函数就就是平面波与,但最主要的就是可以极大简化Kohn-Sham 方程。

利用CASTEP模拟计算实例一,计算本征半导体硅的能带结构和状态密度等性质计算过程分为三个步骤:首先是建立硅的晶体结构计算模型,这个可以在MS物质结构数据库中调用即可。
在计算时为了节省时间,减少计算量将硅的普通的晶体转化为原胞结构,一个原胞中包含9个原子。
节下来是对晶体原胞结构进行几何结构优化,当然其中也含盖了对体系总能量的最小化。
结构优化过程中的两个图表文档分别表示了优化步骤中体系能量的变化和收敛精度,判断收敛是否成功就要查看最终完成计算后,能量的收敛精度是否达到了事前的设定值。
最后是计算性质,在计算状态密度时可以计算不同原子各个轨道按照角动量分布的偏态密度(PDOS),当体系是自旋极化时,偏态密度(PDOS)中包含了体系多数自旋(majority spin)和少数自旋(minority spin)的偏态密度(PDOS)。
光学性质的计算是模拟中的一个难点,从目前发表的文献来看,影响光学性质计算的因素很多(见光学计算原理部分,对此有详细描述),在研究体系有充足实验数据的条件下,可以对能带采用“剪刀”的工具对能带带隙进行刚性的调整,获得与实验结果符合较好的结论。
但对于初学者而言,这个工具一般是不推荐使用的。
作者对于硅的计算完全按照上述方案完成。
详细的计算结果和计算方法见本文所附带的专门文章。
二,搀杂半导体InP性质计算第三主族和第五主族元素之间形成的半导体,目前越来越受到的重视,在纳米材料中,各种纳米电子器件如场效应晶体管,半导体纳米量子阱,纳米量子点激光器等均广泛采用了诸如AlAS InP等材料,本文对InP能带结构、状态密度以及光学性质进行了计算。
计算步骤与前文描述相同。
详细结果见文章二。
三,FeS2性质计算二硫化亚铁是一种受到广泛研究的窄带隙的半导体,其能带带隙为0.95eV。
肖奇等人也采用CASTEP 对二硫化亚铁整体状态密度和(100)晶面双层超结构状态密度的计算结果进行了对比,发现了表面态对状态密度峰的分裂。
CASTE计算理论总结XBAPRSCASTEP寺点是适合于计算周期性结构,对于非周期性结构一般要将特定的部分作为周期性结构,建立单位晶胞后方可进行计算。
CASTER算步骤可以概括为三步:首先建立周期性的目标物质的晶体;其次对建立的结构进行优化,这包括体系电子能量的最小化和几何结构稳定化。
最后是计算要求的性质,如电子密度分布(Electron density distribution) ,能带结构(Band structure)、状态密度分布(Densityof states)、声子能谱(Pho non spectrum)、声子状态密度分布(DOS of pho non),轨道群分布(Orbital populations) 以及光学性质(Optical properties) 等。
本文主要将就各个步骤中的计算原理进行阐述,并结合作者对计算实践经验,在文章最后给出了几个计算事例,以备参考。
CASTEP十算总体上是基于DFT,但实现运算具体理论有:离子实与价电子之间相互作用采用赝势来表示;超晶胞的周期性边界条件;平面波基组描述体系电子波函数;广泛采用快速fast Fourier tran sform (FFT) 对体系哈密顿量进行数值化计算;体系电子自恰能量最小化采用迭带计算的方式;采用最普遍使用的交换-相关泛函实现DFT的计算,泛函含概了精确形式和屏蔽形式。
一,CASTEF中周期性结构计算优点与MS中其他计算包不同,非周期性结构在CASTEF中不能进行计算。
将晶面或非周期性结构置于一个有限长度空间方盒中,按照周期性结构来处理,周期性空间方盒形状没有限制。
之所以采用周期性结构原因在于:依据Bloch定理,周期性结构中每个电子波函数可以表示为一个波函数与晶体周期部分乘积的形式。
他们可以用以晶体倒易点阵矢量为波矢一系列分离平面波函数来展开。
这样每个电子波函数就是平面波和,但最主要的是可以极大简化Kohn-Sham方程。
CASTEP 计算理论总结XBAPRSCASTEP 特点就是适合于计算周期性结构,对于非周期性结构一般要将特定的部分作为周期性结构,建立单位晶胞后方可进行计算。
CASTEP 计算步骤可以概括为三步:首先建立周期性的目标物质的晶体;其次对建立的结构进行优化,这包括体系电子能量的最小化与几何结构稳定化。
最后就是计算要求的性质,如电子密度分布(Electron density distribution),能带结构(Band structure)、状态密度分布(Density of states)、声子能谱(Phonon spectrum)、声子状态密度分布(DOS of phonon),轨道群分布(Orbital populations)以及光学性质(Optical properties)等。
本文主要将就各个步骤中的计算原理进行阐述,并结合作者对计算实践经验,在文章最后给出了几个计算事例,以备参考。
CASTEP 计算总体上就是基于DFT,但实现运算具体理论有:离子实与价电子之间相互作用采用赝势来表示;超晶胞的周期性边界条件;平面波基组描述体系电子波函数;广泛采用快速fast Fourier transform (FFT) 对体系哈密顿量进行数值化计算;体系电子自恰能量最小化采用迭带计算的方式;采用最普遍使用的交换-相关泛函实现DFT 的计算,泛函含概了精确形式与屏蔽形式。
一, CASTEP 中周期性结构计算优点与MS 中其她计算包不同,非周期性结构在CASTEP 中不能进行计算。
将晶面或非周期性结构置于一个有限长度空间方盒中,按照周期性结构来处理,周期性空间方盒形状没有限制。
之所以采用周期性结构原因在于:依据Bloch 定理,周期性结构中每个电子波函数可以表示为一个波函数与晶体周期部分乘积的形式。
她们可以用以晶体倒易点阵矢量为波矢一系列分离平面波函数来展开。
这样每个电子波函数就就是平面波与,但最主要的就是可以极大简化Kohn-Sham 方程。
欢迎访问,平面点位的坐标计算坐标正算(1)计算公式设测站点平面坐标为(X0,Y0),测站至待定点的平距为D,方位角为a,按下列坐标正算公式计算待定点的坐标为:X=X0+DcosaY=Y0+Dsina程序标识符规定如下:A,B------测站点平面坐标;C---------观测方位角;D---------观测平距;X,Y------待定点平面坐标;程序:(以下用-代表箭头指向)Fix 4:”X0=”?→A:”Y0=”?→B↵Lbl 1:”ALFA=”?→C:”DIST=”?→D:”X=”:A+cos(c) ◢”Y=”:B+Dsin(c)◢Goto 1 ↵欢迎访问,坐标反算(1)计算公式按两个已知点A,B 的平面坐标Xa ,Ya, Xb,Yb,计算两点间的距离D 和方位角a ,又称为“坐标反算”,计算公式为A=arctanYb Ya Xb Xa --()()程序中的标识符规定如下: A,B,C,D ——A,B 点的平面坐标; E,F ——A,B 点的坐标增量; R ——AB 的方位角; S ——AB 的平距。
欢迎访问,(2)程序Fix4:“XA=”?→A:“YA=”?→B↵Lbl1:“XB=”?→C:“YB=”?→D:C-A→E;D-B→F:→S:tan-1(F/E)→R↵If E〈0:ThenR+180→R:Goto2:IfEnd:F〈0⇒R+360—R↵Lbl2:“DIST=”:S◢“ALFA=”:R◢Goto1↵测边交会(1)计算公式设A,B为已知点,其平面坐标为Xa,Ya,Xb,Yb,P为待定点,观测平距D A(a),D B(b).从P点作AB(c)边的垂线,交AB于D点,设AD=e,PD=f.用下式计算辅助线段e,f的长度和待定点P的平面坐标:e=(a2+c2-b2)/2c f=x=xA+ecosaAB+fsinaABy=yA+esinaAB-fcosaAB欢迎访问,程序中的标识符规定如下:A,B,C,D——已知点A,B的平面坐标;P,Q——测边交会的观测边长;X,Y——待定点P的平距坐标。
利用CASTEP模拟计算实例
一,计算本征半导体硅的能带结构和状态密度等性质
计算过程分为三个步骤:首先是建立硅的晶体结构计算模型,这个可以在MS物质结构数据库中调用即可。
在计算时为了节省时间,减少计算量将硅的普通的晶体转化为原胞结构,一个原胞中包含9个原子。
节下来是对晶体原胞结构进行几何结构优化,当然其中也含盖了对体系总能量的最小化。
结构优化过程中的两个图表文档分别表示了优化步骤中体系能量的变化和收敛精度,判断收敛是否成功就要查看最终完成计算后,能量的收敛精度是否达到了事前的设定值。
最后是计算性质,在计算状态密度时可以计算不同原子各个轨道按照角动量分布的偏态密度(PDOS),当体系是自旋极化时,偏态密度(PDOS)中包含了体系多数自旋(majority spin)和少数自旋(minority spin)的偏态密度(PDOS)。
光学性质的计算是模拟中的一个难点,从目前发表的文献来看,影响光学性质计算的因素很多(见光学计算原理部分,对此有详细描述),在研究体系有充足实验数据的条件下,可以对能带采用“剪刀”的工具对能带带隙进行刚性的调整,获得与实验结果符合较好的结论。
但对于初学者而言,这个工具一般是不推荐使用的。
作者对于硅的计算完全按照上述方案完成。
详细的计算结果和计算方法见本文所附带的专门文章。
二,搀杂半导体InP性质计算
第三主族和第五主族元素之间形成的半导体,目前越来越受到的重视,在纳米材料中,各种纳米电子器件如场效应晶体管,半导体纳米量子阱,纳米量子点激光器等均广泛采用了诸如AlAS InP等材料,本文对InP能带结构、状态密度以及光学性质进行了计算。
计算步骤与前文描述相同。
详细结果见文章二。
三,FeS2性质计算
二硫化亚铁是一种受到广泛研究的窄带隙的半导体,其能带带隙为0.95eV。
肖奇等人也采用CASTEP 对二硫化亚铁整体状态密度和(100)晶面双层超结构状态密度的计算结果进行了对比,发现了表面态对状态密度峰的分裂。
作者也首先建立了二硫化亚铁的晶体结构,对优化后的结构也进行了计算,得到能带带隙的较准确结果,但在能带的顶层出现了文献中未出现的新结构,因此还需要其他文献进行证实。
作者也建立了双层的二硫化亚铁(100)晶面的超晶胞结构,但限于计算能力,只对结构进行了分子力场的初步结构优化。
详细结果见文章三。
四,三氧化二铝性质计算
三氧化二铝是广泛用于复合材料中的一种附加材料,在电子工业中用于衬底材料。
作为陶瓷原料更是普遍。
三氧化二铝有多种晶体类型,目前广泛得到研究的是α-三氧化二铝,作者计算了它的能带结构和状态密度分布以及电子密度分布情况,计算结果与实验结果相比较是可靠的,电子密度分布揭示了化学键的性质。
详细结果见文章四。
五,其他几种半导体材料能带结构的计算
作者也计算了几种目前普遍使用的半导体材料的能带结构,晶体结构在计算前是经过了结构优化的,某些计算的能带带隙并不理想,与实验数值相比较,差距较大。
但发现了能带结构和计算晶体结构特别是化学键类型间的关系是密切的。
通过对于几种类型的半导体能带结构和状态密度的计算表明他们与原子轨道杂化类型,原子间成键类型等均有关系,计算几种半导体分别是:本征半导体Si,离子型窄带隙半导体ZnO and Cu2O,搀杂n型半导体BN。
从杂化轨道类型来看,硅为sp3杂化,BN为sp2杂化。
其余两种
是离子型晶体,化学键主要成分是离子键。
从能带结构分析,离子型半导体费米能级以下的部分能带形状平滑,而共价键杂化类型的半导体在费米能级以下部分为抛物线型。
在费米能级以上的部分两者差别不大,均为抛物线型。
状态密
度(DOS)图来看,ZnO and Cu2O型半导体各个分波区分是很明显的,杂化型半导体状态密度各个分波区分并不明显,一般为连续型,原子轨道混合在这些半导体中是很明显的。
六,MnN和MnAs自旋状态密度分布与晶体结构常数间的关系
R. de Paiva1, J. L. A. Alves等人文献中,研究了闪锌矿结构的MnN 和MnAs自旋密度分布随晶体结构常数变化的关系,上述两种物质的闪锌矿结构并不是它们的稳定结构,但在这种结构中Mn以四面体配位性质
在许多二元磁性材料均有体现,人们相信,正是锰元素这种配位环境形成了独特的磁性质。
文献作者采用第一性原理的DFT理论分别用GGA和LDA分别计算了随着晶体尺寸变化,自旋密度分布的变化情况,他们发现上面两种物质自旋密度状态分布结构与晶体尺寸密切相关,当MnN尺寸大于0.490nm,MnAs大于0.571nm
延伸闪锌矿结构是半金属型的,即多数自旋(majority spin)密度分布在费米面处是连续的,少数自旋(minority spin)密度则在费米面处是绝缘体型的。
他们计算中晶体平衡尺寸分别是:
MnN:a o= 4.19 A° (LDA), and 4.30 A° (GGA));
MnAs:the magnetic moments are 2:5¹B and 4:0¹B respectively at the equilibrium lattice parameters ao = 5:32 °A(LDA) and 5.71 °A (GGA).
作者计算均采用GGA,MnN:0.425nm;MnAs:0.5977nm。
自旋密度分布和能带结构基本与文献结果一致,在MnN中,自旋密度在费米能级附近随着尺寸变化是很明显的,但MnAs中则与文献结果不一致。
现将文献结果与作者结果列图如下:
从多数自旋(majority spin)密度和少数自旋(minority spin)密度图中可以明显的得到体系电子电导的单自旋极化现象,半金属特性是
明显的。
但MnAs计算结果与文献差别较大,特别是自旋密度与晶体随尺寸变化的关系不明显,作者计算
了在0.571nm,0.569nm and 0.550nm的情况下的状态密度情况,仍然没有发现自旋密度分布在费米面处明显的变化。
特别是文献中提到的少数自旋(minority spin)密度在晶体尺寸小于0.571nm时会在费米面处产生能隙。