PCR设计引物时酶切位点的保护
- 格式:doc
- 大小:321.50 KB
- 文档页数:5

引物设计原则及酶切位点选择和设计:最初的时候,由于害怕设计酶切位点最后且不开,所以经常采用最通用的方法,用[整理]载体克隆解决问题,但后来发现她也有问题,就是浓度提不上去,你需要体大量的载体来T连入质粒中的重要目的酶切,所以感到还是直接扩增好一点。
但这就需要你仔细设计引物。
扩增出靶基因的时候在核就是进行酶切和连接,当然首先就是在想要合成或者是进行PCR可以在质粒的图谱说明书上酸的两端接入酶切位点,酶切位点是与你的质粒的特点相关的,找取相应的位点,进行设计。
(一)设计引物前应做的准备工作:准备载体图谱,大致准备把片断插在那个部分对片断进行酶切分析,确定一下那些酶切位点不能用准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用(二)设计引物所要考虑的问题往往导致两个,所连接片断上没有这两个位点,且距离不能太近,两个位点应是载体上的,除非恰好是与上面两个酶在一起的酶切位点。
只能切一个,酶都切不好。
因此,紧挨在一起,还有一种情况是:不能有碱基的交叉,比如promega的说明书上说,最好隔四个。
我看AGATCTTAAG,这样的位点比较难切。
两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。
最好使用酶切效率高的。
的酶。
最好使用双酶切有共同buffer最好使用自己实验室有的酶,这样可,ecor1等),最好使用较常用的酶(如hind3,bamh1以省钱。
的问题,很多的战友都有疑惑。
其实园子里有很多的解释了。
的计算,关于TmTm大家可以理解,双链溶解所需的温度。
即是DNA叫溶解温度(melting temperature, Tm),Tm因此,的溶解是没有作用的。
而不互补的区域对DNA 这个温度是由互补的DNA区域决定的,(除时,只计算互补的区域Tm才有贡献。
计算Tm只有和模板互补的区域对对于引物的Tm,过低,是因为他们误把保护碱。
不少战友设计的引物都Tm 非你的酶切位点也与模板互补)反应的诸多困难。



经常有战友对一些常见问题在丁香园反复问答了很多遍,所以希望园子中一些战友,特别是低分与0分战友,能将好的帖子归纳总结了一下,并结合自己的经验整理,一方面这是个学习提高的过程,另一方面也能帮助大家解决这方面的问题。
同时如有不当或不完善的地方,希望各位战友不断补充,争取有朝一日我们能把园子里战友的经验系统整理,给大家以帮助。
我先把设计引物如何设计酶切位点这方面的帖子整理一下,因为昨天一下子看到三个相似问题。
原帖如下:我想向你求教一个问题,假如说我想把胰岛素基因和腺病毒载体连接起来,如何确定设计目的基因PCR时的引物呢?和相应的限制性核酸内切酶呢?谢谢老师能给予讲解,谢谢[整理]:最初的时候,由于害怕设计酶切位点最后切不开,所以经常采用最通用的方法,用T载体克隆解决问题,但后来发现她也有问题,就是浓度提不上去,你需要体大量的载体来酶切,所以感到还是直接扩增好一点。
但这就需要你仔细设计引物。
连入质粒中的重要目的就是进行酶切和连接,当然首先就是在想要合成或者是进行PCR扩增出靶基因的时候在核酸的两端接入酶切位点,酶切位点是与你的质粒的特点相关的,可以在质粒的图谱说明书上找取相应的位点,进行设计。
(一)设计引物前应做的准备工作:准备载体图谱,大致准备把片断插在那个部分对片断进行酶切分析,确定一下那些酶切位点不能用准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用(二)设计引物所要考虑的问题两个位点应是载体上的,,所连接片断上没有这两个位点,且距离不能太近,往往导致两个酶都切不好。
因此,紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点。
我看promega的说明书上说,最好隔四个。
还有一种情况是:不能有碱基的交叉,比如AGATCTTAAG,这样的位点比较难切。
两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。
最好使用酶切效率高的。
最好使用双酶切有共同buffer的酶。

酶切位点保护碱基-PCR引物设计用于限制性内切酶酶切反应来源:easylabs 发布时间:2009-11-08 查看次数:12704本文给出了分子克隆中常用限制性内切酶的保护碱基序列,如AccI,A flIII,AscI,AvaI,BamHI,BglII,BssHII,BstEII,BstXI,ClaI,E coRI,HaeIII,HindIII,KpnI,MluI,NcoI,NdeI,NheI,NotI,N siI,PacI,PmeI,PstI,PvuI,SacI,SacII,SalI,ScaI,SmaI,S peI,SphI,StuI,XbaI,XhoI,XmaI,为什么要添加保护碱基?在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
该如何添加保护碱基?添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
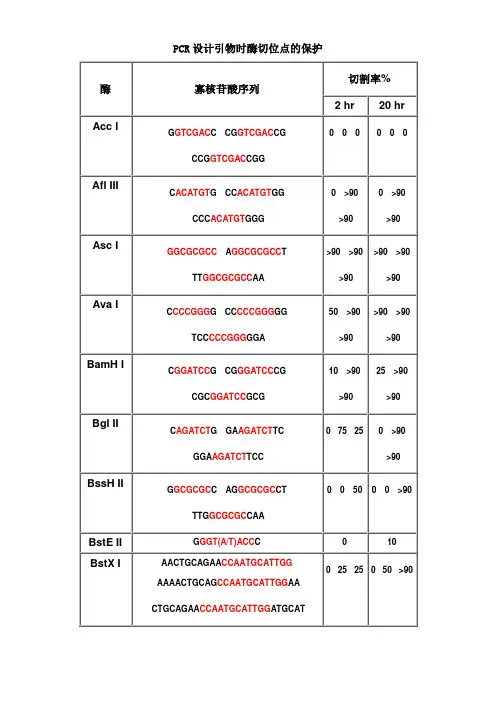
为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A2 60单位的寡核苷酸。

PCR引物设计酶切位点保护酶切位点保护是指在PCR引物的设计中避免引物与目标DNA片段上可能的酶切位点发生内切。
内切是DNA酶切酶将DNA序列切割为两段的过程。
如果酶切位点在PCR引物的靶序列上,PCR扩增后的产物可能会被酶切导致短小片段的产生。
这种情况下会影响PCR的特异性和扩增效率。
因此,为了保护酶切位点,我们需要在设计PCR引物时注意以下几点:1.避免引物包含酶切位点:在设计引物的时候,可以使用一些基因分析软件或数据库来查找可能的酶切位点。
如果目标序列上存在多个酶切位点,应尽量避免引物与这些位点发生重叠。
可以通过调整引物的位置或序列来避免重叠。
2.引物设计时考虑位点长度:不同的酶切位点具有不同的长度要求。
在设计引物时,应考虑不同位点的酶切长度,并确保引物的长度远大于所考虑的位点长度。
这样可以避免在PCR扩增过程中引物与位点发生内切。
3.考虑酶切位点的限制性:有些酶切位点具有特定的限制性,即只在特定的序列上发挥作用。
在引物设计中,如果目标序列上的酶切位点是特定限制性的,可以选择避免引物与这些位点发生重叠,或者调整引物的位置使其尽量远离位点。
4.引物序列的选择和优化:在设计引物时,可以使用引物设计软件来评估引物的特异性和自身的稳定性。
通过优化引物的碱基组成、GC含量、熔解温度等参数,可以增加引物的特异性和PCR扩增的效率。
总之,PCR引物设计中的酶切位点保护是确保PCR扩增特异性和效率的重要考虑因素。
通过避免引物与目标DNA上的酶切位点重叠、考虑位点长度和限制性、优化引物序列等方法,可以有效地保护酶切位点,提高PCR扩增的可靠性和准确性。

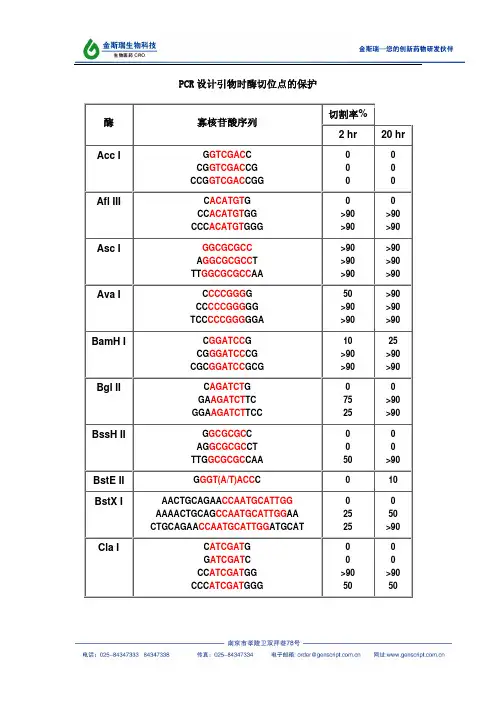
本文给出了分子克隆中常用限制性内切酶的保护碱基序列,如AccI,AflIII,AscI,AvaI,BamHI,BglII,BssHII,BstEII,BstXI,ClaI,EcoRI,HaeIII,HindIII,KpnI,MluI,NcoI,NdeI,NheI,NotI,NsiI,PacI,PmeI,PstI,PvuI,SacI,SacII,SalI,ScaI,SmaI,SpeI,SphI,StuI,XbaI,XhoI,XmaI,为什么要添加保护碱基?在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
该如何添加保护碱基?添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A26单位的寡核苷酸。
取1µg已标记了的寡核苷酸与20单位的内切酶,在2 00°C条件下分别反应2小时和20小时。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments (oligonucleotides))为什么要添加保护碱基?在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
该如何添加保护碱基?添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
单位的寡实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260核苷酸。
取1 µg已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCl,25 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments (oligonucleotides))为什么要添加保护碱基?在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
该如何添加保护碱基?添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
单位的寡实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260核苷酸。
取1 µg已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCl,25 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
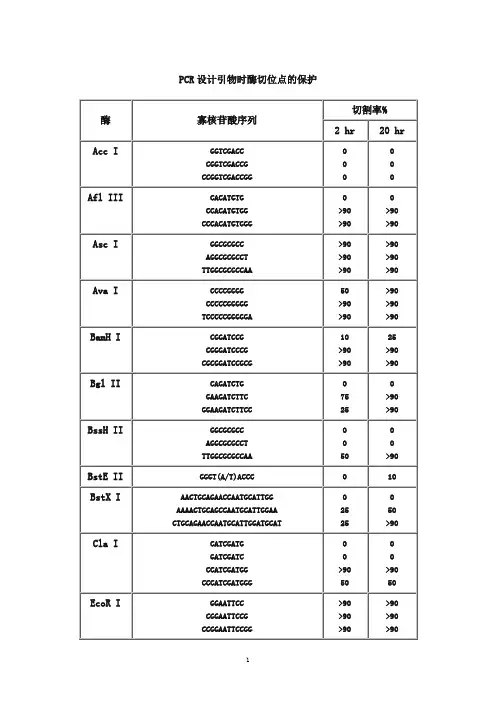
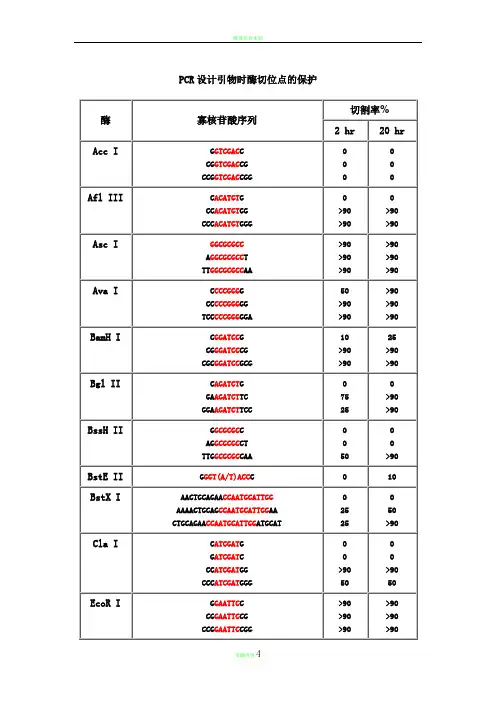
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments (oligonucleotides))为了解不同内切酶对识别位点以外最少保护碱基数目的要求, NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA 末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上 6 个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在 T4 多聚核苷酸激酶的作用下标记 0.1A260单位的寡核苷酸。
取 1 μg已标记了的寡核苷酸与 20 单位的内切酶,在 20°C 条件下分别反应 2 小时和 20 小时。
反应缓冲液含 70 mM Tris-HCl (pH , 10 mM MgCl2 , 5 mM DTT 及适量的 NaCl 或 KCl(视酶的具体要求而定)。
20%的 PAGE(7 M 尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
DNA 合成,新链的延伸方向是5→ 3 因此,需要在 5 端加上酶切位点的识别位点之外,还需多几个无需特异性的碱基提供一个platform,因为内切酶除了有特异让它可以结合上去,否则会掉下来. 引物的结构就是(5→ 3):保护碱基+酶切位点+原来的引物序列首先要看目的基因中是否含有该酶切位点,只有没有的才可以选 (小虾米酶切位点分析 )。
其次,如果需要做表达,需要考虑起始密码子,防止移码突变切割率 %酶寡核苷酸序列链长2 hr 20 hrAcc I GGTCGACC 8 0 0CGGTCGACCG 10 0 0CCGGTCGACCGG 12 0 0Afl III CACATGTG 8 0 0CCACATGTGG 10 >90 >90CCCACATGTGGG 12 >90 >90Asc I GGCGCGCC 8 >90 >90AGGCGCGCCT 10 >90 >90TTGGCGCGCCAA 12 >90 >90Ava I CCCCGGGG 8 50 >90CCCCCGGGGG 10 >90 >90TCCCCCGGGGGA 12 >90 >90BamH IBgl II BssH IIBstE II BstX ICla I EcoR I Hae III Hind III Kpn I Mlu I Nco ICGGATCCG 8 10 25CGGGATCCCG 10 >90 >90CGCGGATCCGCG 12 >90 >90CAGATCTG 8 0 0GAAGATCTTC 10 75 >90GGAAGATCTTCC 12 25 >90GGCGCGC 8 0 0AGGCGCGCCT 10 0 0TTGGCGCGCCAA 12 50 >90GGGT(A/T)ACCC 9 0 10AACTGCAGAACCAATGCATTGG 22 0 0 AAAACTGCAGCCAATGCATTGGAA 24 25 50 CTGCAGAACCAATGCATTGGATGCAT 27 25 >90CATCGATG 8 0 0GATCGATC 8 0 0CCATCGATGG 10 >90 >90CCCATCGATGGG 12 50 50GGAATTCC 8 >90 >90CGGAATTCCG 10 >90 >90CCGGAATTCCGG 12 >90 >90GGGGCCCC 8 >90 >90AGCGGCCGCT 10 >90 >90TTGCGGCCGCAA 12 >90 >90CAAGCTTG 8 0 0CCAAGCTTGG 10 0 0CCCAAGCTTGGG 12 10 75GGGTACCC 8 0 0GGGGTACCCC 10 >90 >90CGGGGTACCCCG 12 >90 >90GACGCGTC 8 0 0CGACGCGTCG 10 25 50CCCATGGG 8 0 0 CATGCCATGGCATG 14 50 75Nde INhe I Not INsi I Pac I Pme I Pst I Pvu ISac I Sac IISal ICCATATGG 8 0 0CCCATATGGG 10 0 0CGCCATATGGCG 12 0 0 GGGTTTCATATGAAACCC 18 0 0GGAATTCCATATGGAATTCC 20 75 >90GGGAATTCCATATGGAATTCCC 22 75 >90GGCTAGCC 8 0 0CGGCTAGCCG 10 10 25CTAGCTAGCTAG 12 10 50TTGCGGCCGCAA 12 0 0ATTTGCGGCCGCTTTA 16 10 10 AAATATGCGGCCGCTATAAA 20 10 10 ATAAGAATGCGGCCGCTAAACTAT 24 25 90 AAGGAAAAAAGCGGCCGCAAAAGGAAAA 28 25 >90TGCATGCATGCA 12 10 >90 CCAATGCATTGGTTCTGCAGTT 22 >90 >90TTAATTAA 8 0 0GTTAATTAAC 10 0 25CCTTAATTAAGG 12 0 >90GTTTAAAC 8 0 0GGTTTAAACC 10 0 25GGGTTTAAACCC 12 0 50 AGCTTTGTTTAAACGGCGCGCCGG 24 75 >90GCTGCAGC 8 0 0 TGCACTGCAGTGCA 14 10 10 AACTGCAGAACCAATGCATTGG 22 >90 >90AAAACTGCAGCCAATGCATTGGAA 24 >90 >90 CTGCAGAACCAATGCATTGGATGCAT 26 0 0CCGATCGG 8 0 0ATCGATCGAT 10 10 25TCGCGATCGCGA 12 0 10CGAGCTCG 8 10 10GCCGCGG 8 0 0 TCCCCGCGGGGA 12 50 >90GTCGACGTCAAAAGGCCATAGCGGCCGC 28 0 0 GCGTCGACGTCTTGGCCATAGCGGCCGCGG 30 10 50 ACGCGTCGACGTCGGCCATAGCGGCCGCGGAA 32 10 75Sca I Sma ISpe ISph I Stu I Xba I Xho I Xma IGAGTACTC 8 10 25 AAAAGTACTTTT 12 75 75CCCGGG 6 0 10 CCCCGGGG 8 0 10 CCCCCGGGGG 10 10 50 TCCCCCGGGGGA 12 >90 >90GACTAGTC 8 10 >90 GGACTAGTCC 10 10 >90 CGGACTAGTCCG 12 0 50 CTAGACTAGTCTAG 14 0 50GGCATGCC 8 0 0 CATGCATGCATG 12 0 25 ACATGCATGCATGT 14 10 50AAGGCCTT 8 >90 >90 GAAGGCCTTC 10 >90 >90 AAAAGGCCTTTT 12 >90 >90CTCTAGAG 8 0 0 GCTCTAGAGC 10 >90 >90 TGCTCTAGAGCA 12 75 >90 CTAGTCTAGACTAG 14 75 >90CCTCGAGG 8 0 0 CCCTCGAGGG 10 10 25 CCGCTCGAGCGG 12 10 75CCCCGGGG 8 0 0 CCCCCGGGGG 10 25 75 CCCCCCGGGGGG 12 50 >90 TCCCCCCGGGGGGA 14 >90 >90DNA 合成,新链的延伸方向是5→ 3 因此,需要在 5 端加上酶切位点,因为内切酶除了有特异的识别位点之外,还需多几个无需特异性的碱基提供一个platform 让它可以结合上去,否则会掉下来 . 引物的结构就是(5→ 3):保护碱基 +酶切位点 +原来的引物序列。
注释:
1.如果要加在序列的5‘端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3‘端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。
2.切割率:正确识别并酶切的效率
3。
加保护碱基时最好选用切割率高时加的相应碱基。
为什么要添加保护碱基?
在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
该如何添加保护碱基?
添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠
近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1?g已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70mM Tris-HCl (pH 7.6), 10 mM MgCl2 , 5 mMDTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。